
Review of the Current State of Freely Accessible Web Tools for the Analysis of 16S rRNA Sequencing of the Gut Microbiome


Abstract
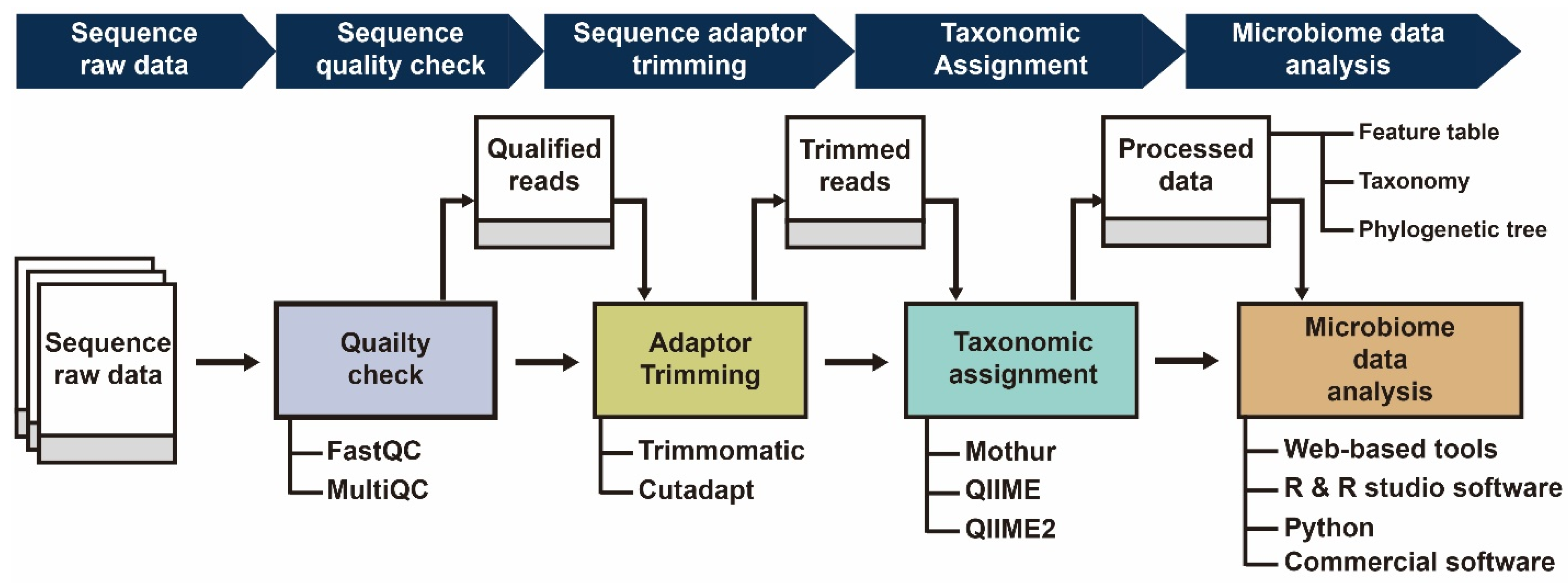
:1. Introduction
2. Freely Accessible Web-Based Tools for Microbiome Analysis
2.1. Visualization and Analysis of Microbial Population Structures (VAMPS)
2.2. MicrobiomeAnalyst
2.3. Mian
2.4. Global Catalogue of Metagenomics (gcMeta)
2.5. Microbiome Toolbox
3. Comparison of the Web Tools Using Gut Microbiome Dataset
Limitations of Web-Based Tools for Microbiome Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jin, B.T.; Xu, F.; Ng, R.T.; Hogg, J.C. Mian: Interactive web-based microbiome data table visualization and machine learning platform. Bioinformatics 2021, 38, 1176–1178. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Ravel, J. The vocabulary of microbiome research: A proposal. Microbiome 2015, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef] [PubMed]
- Prados-Bo, A.; Casino, G. Microbiome research in general and business newspapers: How many microbiome articles are published and which study designs make the news the most? PLoS ONE 2021, 16, e0249835. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. Next-generation sequencing platforms. Annu. Rev. Anal. Chem. 2013, 6, 287–303. [Google Scholar] [CrossRef]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef]
- Ju, F.; Zhang, T. 16S rRNA gene high-throughput sequencing data mining of microbial diversity and interactions. Appl. Microbiol. Biotechnol. 2015, 99, 4119–4129. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Mahurkar, A.; Rahnavard, G.; Crabtree, J.; Orvis, J.; Hall, A.B.; Brady, A.; Creasy, H.H.; McCracken, C.; Giglio, M.G.; et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 2017, 550, 61–66. [Google Scholar] [CrossRef]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G.; et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef]
- Shi, W.; Qi, H.; Sun, Q.; Fan, G.; Liu, S.; Wang, J.; Zhu, B. gcMeta: A Global Catalogue of Metagenomics platform to support the archiving, standardization and analysis of microbiome data. Nucleic Acids Res. 2019, 47, D637–D648. [Google Scholar] [CrossRef]
- Methé, B.A.; Nelson, K.E.; Pop, M.; Creasy, H.H.; Giglio, M.G.; Huttenhower, C.; Mannon, P.J. A framework for human microbiome research. Nature 2012, 486, 215–221. [Google Scholar]
- Shreiner, A.B.; Kao, J.Y.; Young, V.B. The gut microbiome in health and in disease. Curr. Opin. Gastroenterol. 2015, 31, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Frame, L.A.; Costa, E.; Jackson, S.A. Current explorations of nutrition and the gut microbiome: A comprehensive evaluation of the review literature. Nutr. Rev. 2020, 78, 798–812. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, 67, 1716–1725. [Google Scholar] [CrossRef]
- Alkan, C.; Sajjadian, S.; Eichler, E.E. Limitations of next-generation genome sequence assembly. Nat. Methods 2010, 8, 61–65. [Google Scholar] [CrossRef]
- Expósito, R.R.; Galego-Torreiro, R.; González-Domínguez, J. Sequal: Big data tool to perform quality control and data pre-processing of large NGS datasets. IEEE Access 2020, 8, 146075–146084. [Google Scholar] [CrossRef]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef]
- Yang, L.-A.; Chang, Y.-J.; Chen, S.-H.; Lin, C.-Y.; Ho, J.-M. SQUAT: A Sequencing Quality Assessment Tool for data quality assessments of genome assemblies. BMC Genom. 2019, 19, 238. [Google Scholar] [CrossRef]
- Del Fabbro, C.; Scalabrin, S.; Morgante, M.; Giorgi, F. An Extensive Evaluation of Read Trimming Effects on Illumina NGS Data Analysis. PLoS ONE 2013, 8, e85024. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lewinski, R.A.; Oakley, B.B.; Parks, D.H.; Sahl, J.W.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for de-scribing and comparing microbial communities. Appl. Environ. Microb. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzalez Peña, A.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Vincent, A.T.; Derome, N.; Boyle, B.; Culley, A.I.; Charette, S.J. Next-generation sequencing (NGS) in the microbiological world: How to make the most of your money. J. Microbiol. Methods 2017, 138, 60–71. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef]
- Huse, S.M.; Welch, D.B.M.; Voorhis, A.; Shipunova, A.; Morrison, H.G.; Eren, A.M.; Sogin, M.L. VAMPS: A website for visualization and analysis of microbial population structures. BMC Bioinform. 2014, 15, 41. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Aßhauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data: Fig. 1. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Costello, E.K.; Berg-Lyons, D.; Gonzalez, A.; Stombaugh, J.; Knights, D.; Gajer, P.; Ravel, J.; Fierer, N.; et al. Moving pictures of the human microbiome. Genome Biol. 2011, 12, R50. [Google Scholar] [CrossRef] [PubMed]
- Muegge, B.D.; Kuczynski, J.; Knights, D.; Clemente, J.C.; González, A.; Fontana, L.; Henrissat, B.; Knight, R.; Gordon, J.I. Diet Drives Convergence in Gut Microbiome Functions Across Mammalian Phylogeny and Within Humans. Science 2011, 332, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Costello, E.K.; Lauber, C.L.; Hamady, M.; Fierer, N.; Gordon, J.I.; Knight, R. Bacterial Community Variation in Human Body Habitats Across Space and Time. Science 2009, 326, 1694–1697. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Meehan, C.J.; Koenig, J.E.; Dhanani, A.S.; Rose, R.A.; Howlett, S.E.; Beiko, R.G. Microbial shifts in the aging mouse gut. Microbiome 2014, 2, 50. [Google Scholar] [CrossRef] [PubMed]
- Dogra, S.K.; Banjac, J.; Sprenger, N. Microbiome Toolbox: Methodological approaches to derive and visualize microbiome trajectories. bioRxiv, 2022. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The Effect of Diet on the Human Gut Microbiome: A Metagenomic Analysis in Humanized Gnotobiotic Mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef]
- Ho, N.T.; Li, F.; Lee-Sarwar, K.A.; Tun, H.M.; Brown, B.; Pannaraj, P.S.; Bender, J.M.; Azad, M.B.; Thompson, A.L.; Weiss, S.T.; et al. Meta-analysis of effects of exclusive breastfeeding on infant gut microbiota across populations. Nat. Commun. 2018, 9, 4169. [Google Scholar] [CrossRef]
- Behjati, S.; Tarpey, P.S. What is next generation sequencing? Arch. Dis. Child. Educ. Pract. 2013, 98, 236–238. [Google Scholar] [CrossRef]
- Azimirad, M.; Jo, Y.J.; Kim, M.S.; Jeong, M.S.; Shahrokh, S.; Aghdaei, H.A.; Zali, M.R.; Lee, S.J.; Yadegar, A.; Shin, J.H. Alterations and Prediction of Functional Profiles of Gut Microbiota After Fecal Microbiota Transplantation for Iranian Recurrent Clos-tridioides difficile Infection with Underlying Inflammatory Bowel Disease: A Pilot Study. J. Inflamm. Res. 2022, 15, 105. [Google Scholar] [CrossRef]
- Jung, Y.; Tagele, S.B.; Son, H.; Ibal, J.C.; Kerfahi, D.; Yun, H.; Lee, B.; Park, C.Y.; Kim, E.S.; Kim, S.-J.; et al. Modulation of Gut Microbiota in Korean Navy Trainees following a Healthy Lifestyle Change. Microorganisms 2020, 8, 1265. [Google Scholar] [CrossRef]
- Bahr, S.M.; Tyler, B.; Wooldridge, N.; Butcher, B.; Burns, T.L.; Teesch, L.M.; Oltman, C.L.; Azcarate-Peril, M.A.; Kirby, J.R.; Calarge, C.A. Use of the second-generation antipsychotic, risperidone, and secondary weight gain are associated with an altered gut microbiota in children. Transl. Psychiatry 2015, 5, e652. [Google Scholar] [CrossRef]
- Yun, Y.; Kim, H.-N.; Kim, S.E.; Heo, S.G.; Chang, Y.; Ryu, S.; Shin, H.; Kim, H.-L. Comparative analysis of gut microbiota associated with body mass index in a large Korean cohort. BMC Microbiol. 2017, 17, 151. [Google Scholar] [CrossRef]
- Liu, B.; Lin, W.; Chen, S.; Xiang, T.; Yang, Y.; Yin, Y.; Xu, G.; Liu, Z.; Liu, L.; Pan, J.; et al. Gut microbiota as an objective measurement for auxiliary diagnosis of insomnia disorder. Front Microbial 2019, 10, 1770. [Google Scholar] [CrossRef] [PubMed]
- Gharaibeh, R.Z.; Jobin, C. Microbiota and cancer immunotherapy: In search of microbial signals. Gut 2018, 68, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Li, Y.; Wang, X.; Wang, S.; Bi, D. The impact of Lactobacillus plantarum on the gut microbiota of mice with DSS-induced colitis. BioMed Res. Int. 2019, 10, 3291310–3921395. [Google Scholar]
- Saito, K.; Koido, S.; Odamaki, T.; Kajihara, M.; Kato, K.; Horiuchi, S.; Adachi, S.; Arakawa, H.; Yoshida, S.; Akasu, T.; et al. Metagenomic analyses of the gut microbiota associated with colorectal adenoma. PLoS ONE 2019, 14, e0212406. [Google Scholar] [CrossRef]
- Li, K.; Dan, Z.; Gesang, L.; Wang, H.; Zhou, Y.; Du, Y.; Ren, Y.; Shi, Y.; Nie, Y. Comparative Analysis of Gut Microbiota of Native Tibetan and Han Populations Living at Different Altitudes. PLoS ONE 2016, 11, e0155863. [Google Scholar] [CrossRef]
- Bishop, C.M.; Nasrabadi, N.M. Pattern Recognition and Machine Learning; Springer: New York, NY, USA, 2006; Volume 4, No. 4. [Google Scholar]
- Yu, L.; Liu, H. Feature selection for high-dimensional data: A fast correlation-based filter solution. In Proceedings of the 20th international conference on machine learning (ICML-03), Washington, DC, USA, 21–24 August 2003; pp. 856–863. [Google Scholar]
- Thompson, J.; Johansen, R.; Dunbar, J.; Munsky, B. Machine learning to predict microbial community functions: An analysis of dissolved organic carbon from litter decomposition. PLoS ONE 2019, 14, e0215502. [Google Scholar] [CrossRef]
- Statnikov, A.; Aliferis, C.F.; Tsamardinos, I.; Hardin, D.; Levy, S. A comprehensive evaluation of multicategory classification methods for microarray gene expression cancer diagnosis. Bioinformatics 2004, 21, 631–643. [Google Scholar] [CrossRef]
- Aryal, S.; Alimadadi, A.; Manandhar, I.; Joe, B.; Cheng, X. Machine Learning Strategy for Gut Microbiome-Based Diagnostic Screening of Cardiovascular Disease. Hypertension 2020, 76, 1555–1562. [Google Scholar] [CrossRef]
- Ai, D.; Pan, H.; Han, R.; Li, X.; Liu, G.; Xia, L.C. Using Decision Tree Aggregation with Random Forest Model to Identify Gut Microbes Associated with Colorectal Cancer. Genes 2019, 10, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galkin, F.; Mamoshina, P.; Aliper, A.; Putin, E.; Moskalev, V.; Gladyshev, V.N.; Zhavoronkov, A. Human Gut Microbiome Aging Clock Based on Taxonomic Profiling and Deep Learning. iScience 2020, 23, 101199. [Google Scholar] [CrossRef]
- Xia, Y.; Sun, J.; Chen, D.G. Statistical Analysis of Microbiome Data with R; Springer: Singapore, 2018; Volume 847. [Google Scholar]
- Tong, W.M.; Chan, Y. GenePiper, a Graphical User Interface Tool for Microbiome Sequence Data Mining. Microbiol. Resour. Announc. 2020, 9, e01119–e01195. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-A.; Park, J.; Natsume-Kitatani, Y.; Kawashima, H.; Mohsen, A.; Hosomi, K.; Tanisawa, K.; Ohno, H.; Konishi, K.; Murakami, H.; et al. MANTA, an integrative database and analysis platform that relates microbiome and phenotypic data. PLoS ONE 2020, 15, e0243609. [Google Scholar] [CrossRef] [PubMed]
- Baldini, F.; Heinken, A.; Heirendt, L.; Magnusdottir, S.; Fleming, R.M.T.; Thiele, I. The Microbiome Modeling Toolbox: From microbial interactions to personalized microbial communities. Bioinformatics 2018, 35, 2332–2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Upload and Function | VAMPS | MicrobiomeAnalyst | Mian | gcMeta | Microbiome Toolbox | |
|---|---|---|---|---|---|---|
| File format | Edited FASTA | BIOM | BIOM | Edited OTU table | ||
| Database | SILVA, Greengenes | SILVA, Greengenes | NA | NA | ||
| Common analysis | Rarefaction curve | X | O | O | X | |
| Bar/stack analysis | O | O | O | X | ||
| Pie chart | O | O | X | X | ||
| Core microbiome analysis | X | O | X | X | ||
| Phylogenetic tree | O | O | X | X | ||
| α-Diversity | Shannon index | O | O | O | X | |
| Simpson index | O | O | O | X | ||
| Richness index | X | O | X | X | ||
| Chao1 index | O | O | X | X | ||
| ACE index | O | X | X | X | ||
| Evenness index | X | X | X | X | ||
| β-Diversity | Bray-Custis dissimilarity | O | O | O | O | |
| Jaccard distance | X | O | X | X | ||
| Unweighted UniFrac | X | O | O | X | ||
| Weighted UniFrac | X | O | O | X | ||
| NMDS | X | O | O | X | ||
| CCA analysis | X | O | X | X | ||
| RDA analysis | X | O | X | X | ||
| Correlation and Clustering analysis | Heatmap | O | O | O | O | |
| Correlation plot | O | O | O | O | ||
| DEseq2 | X | O | X | X | ||
| Network analysis | X | O | X | X | ||
| Functional gene prediction | PICRUSt | X | O | X | X | |
| Tax4Fun | X | O | X | X | ||
| Comparative analysis | LEfSe | X | O | O | X | |
| Random Forest | X | O | O | X | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibal, J.C.; Park, Y.-J.; Park, M.-K.; Lee, J.; Kim, M.-C.; Shin, J.-H. Review of the Current State of Freely Accessible Web Tools for the Analysis of 16S rRNA Sequencing of the Gut Microbiome. Int. J. Mol. Sci. 2022, 23, 10865. https://doi.org/10.3390/ijms231810865
Ibal JC, Park Y-J, Park M-K, Lee J, Kim M-C, Shin J-H. Review of the Current State of Freely Accessible Web Tools for the Analysis of 16S rRNA Sequencing of the Gut Microbiome. International Journal of Molecular Sciences. 2022; 23(18):10865. https://doi.org/10.3390/ijms231810865
Chicago/Turabian StyleIbal, Jerald Conrad, Yeong-Jun Park, Min-Kyu Park, Jooeun Lee, Min-Chul Kim, and Jae-Ho Shin. 2022. "Review of the Current State of Freely Accessible Web Tools for the Analysis of 16S rRNA Sequencing of the Gut Microbiome" International Journal of Molecular Sciences 23, no. 18: 10865. https://doi.org/10.3390/ijms231810865
APA StyleIbal, J. C., Park, Y. -J., Park, M. -K., Lee, J., Kim, M. -C., & Shin, J. -H. (2022). Review of the Current State of Freely Accessible Web Tools for the Analysis of 16S rRNA Sequencing of the Gut Microbiome. International Journal of Molecular Sciences, 23(18), 10865. https://doi.org/10.3390/ijms231810865