
S100 Proteins in Fatty Liver Disease and Hepatocellular Carcinoma

 ,
,
Abstract
:1. Introduction
2. Structure, Expression and Regulation of S100 Proteins
2.1. Protein Structure
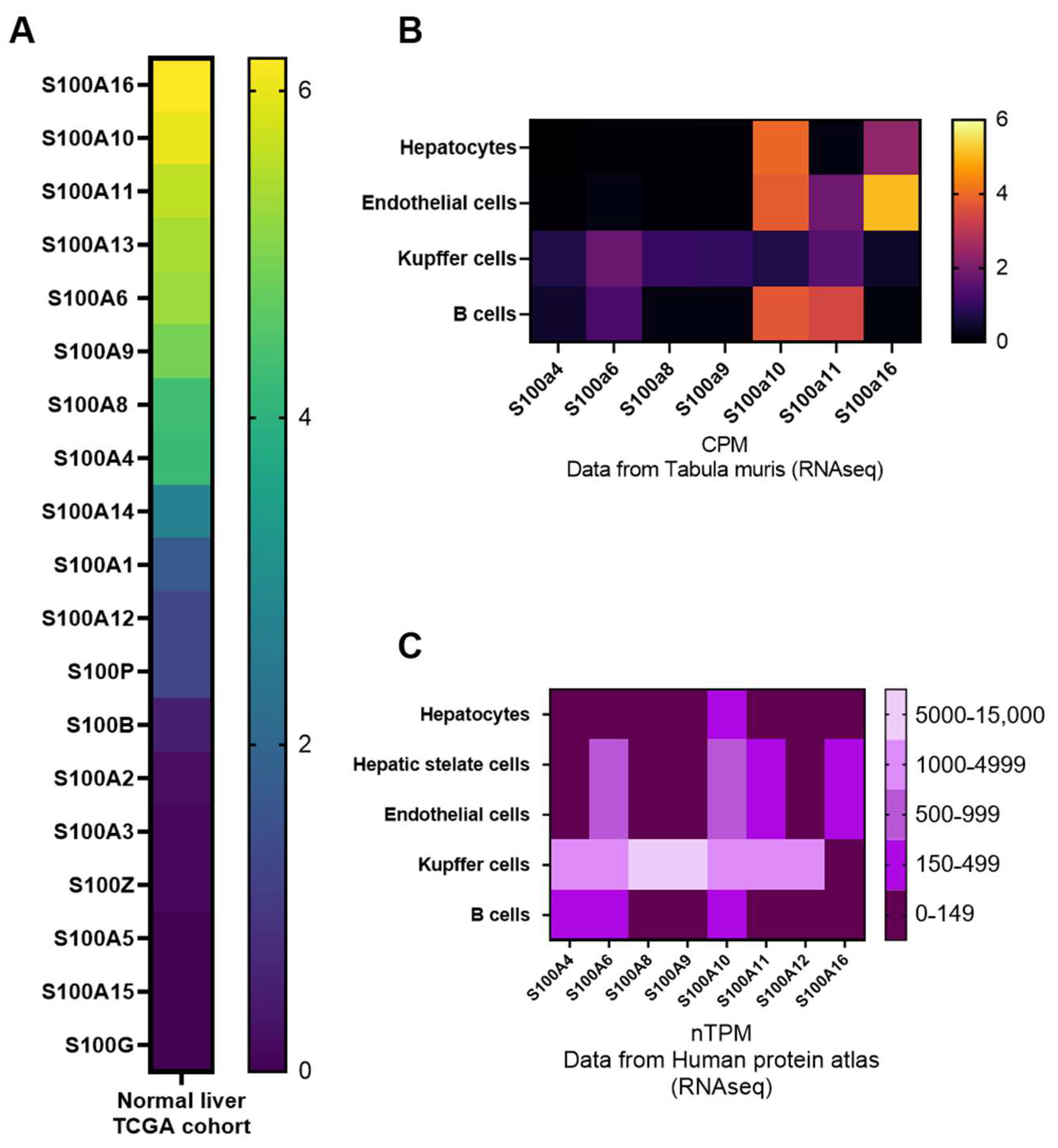
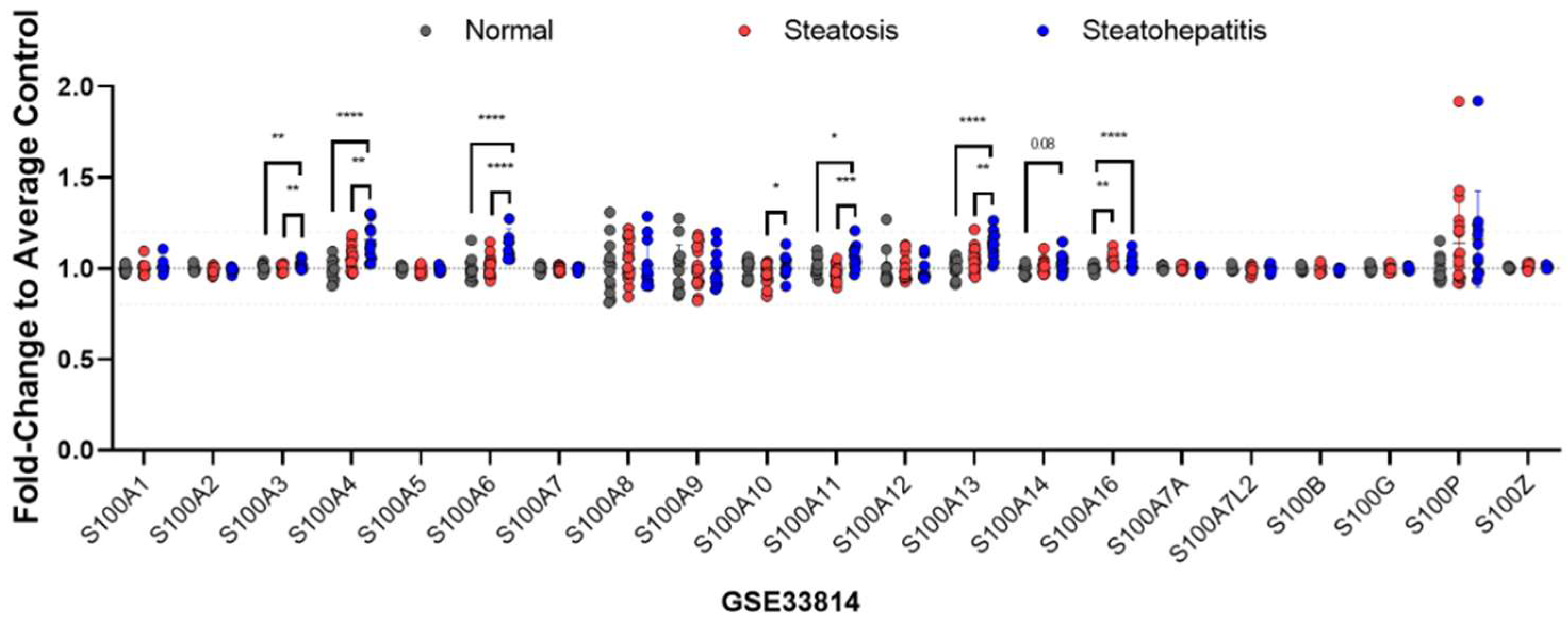
2.2. Expression Patterns of S100 Proteins in the Liver
2.3. Regulation of S100 Expression and Activity
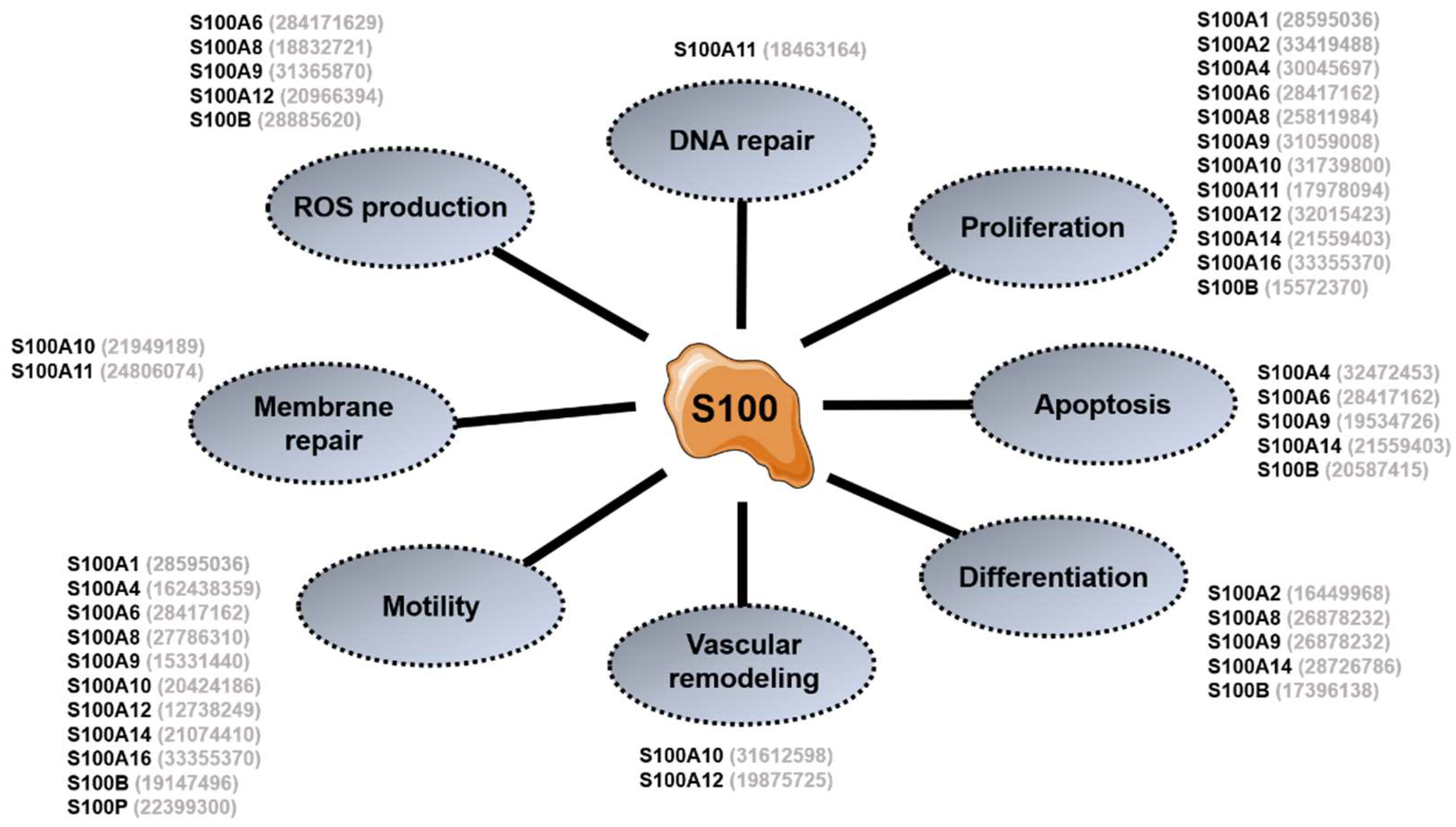
3. General Overview of S100 Proteins Functions
3.1. Intracellular Functions
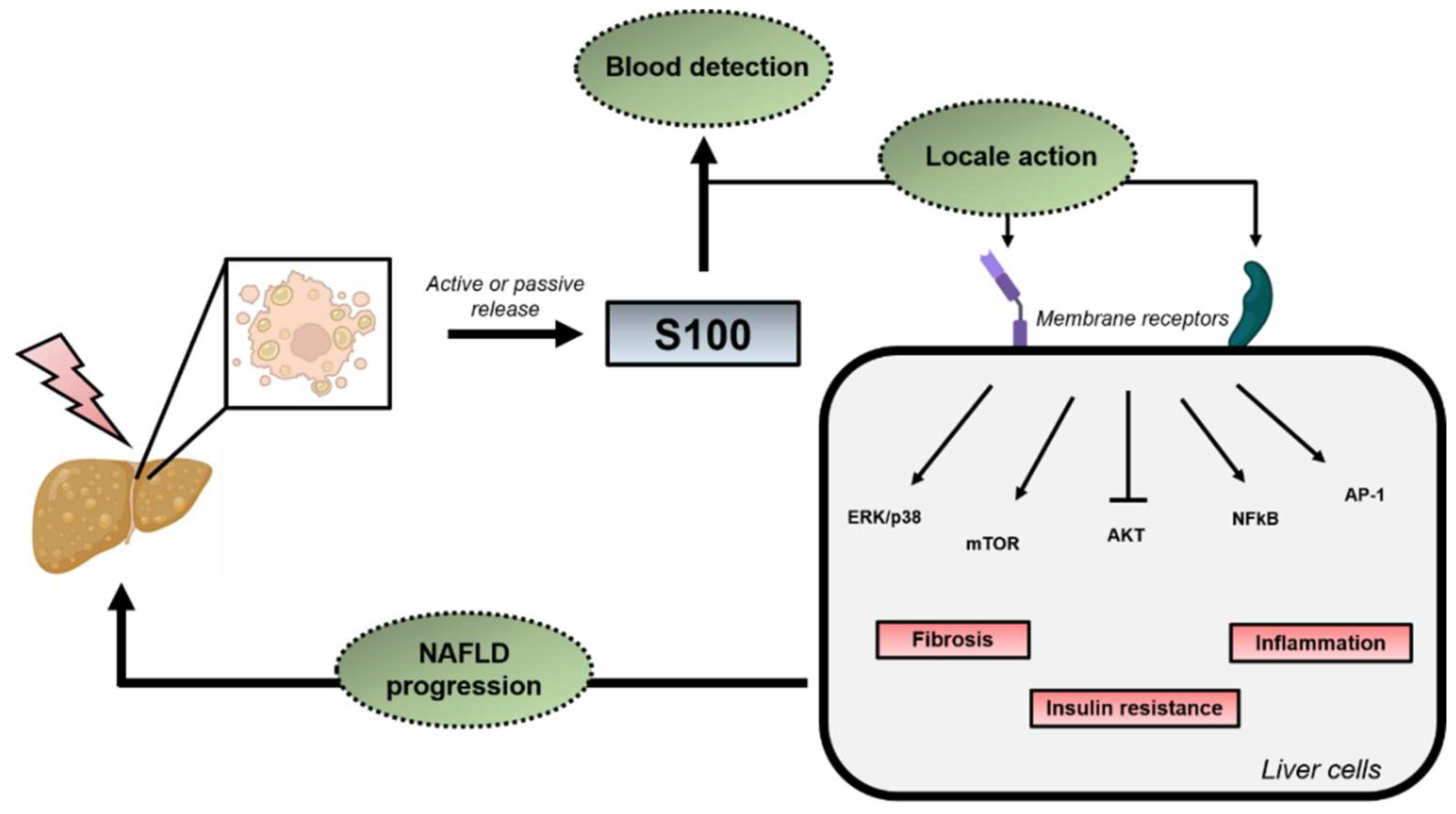
3.2. Extracellular Functions
4. S100 Proteins in NAFLD/NASH and HCC Development
4.1. Steatosis and Insulin Resistance (IR)
4.2. From Simple Steatosis to NASH
4.3. Hepatic Fibrosis
5. Implication of S100 Proteins in the Occurrence of HCC
5.1. S100A4
5.2. S100A8/A9
5.3. S100A10
5.4. S100A11
6. S100 Proteins as Potential Biomarkers and Therapeutic Targets in NAFLD/NASH and HCC
6.1. S100 Proteins as Potential Biomarkers in NAFLD/NASH and HCC
6.2. S100 Proteins as Therapeutic Targets for NAFLD/NASH and HCC
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Cotter, T.G.; Rinella, M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Blissett, D.; Blissett, R.; Henry, L.; Stepanova, M.; Younossi, Y.; Racila, A.; Hunt, S.; Beckerman, R. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology 2016, 64, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Gramlich, T.; Matteoni, C.A.; Boparai, N.; McCullough, A.J. Nonalcoholic fatty liver disease in patients with type 2 diabetes. Clin. Gastroenterol. Hepatol. 2004, 2, 262–265. [Google Scholar] [CrossRef]
- Loomba, R.; Abraham, M.; Unalp, A.; Wilson, L.; Lavine, J.; Doo, E.; Bass, N.M. Association between diabetes, family history of diabetes, and risk of nonalcoholic steatohepatitis and fibrosis. Hepatology 2012, 56, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Powell, E.E.; Wong, V.W.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Brunt, E.M.; Wong, V.W.; Nobili, V.; Day, C.P.; Sookoian, S.; Maher, J.J.; Bugianesi, E.; Sirlin, C.B.; Neuschwander-Tetri, B.A.; Rinella, M.E. Nonalcoholic fatty liver disease. Nat. Rev. Dis. Primers 2015, 1, 15080. [Google Scholar] [CrossRef]
- Mundi, M.S.; Velapati, S.; Patel, J.; Kellogg, T.A.; Abu Dayyeh, B.K.; Hurt, R.T. Evolution of NAFLD and Its Management. Nutr. Clin. Pract. 2020, 35, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Adams, L.A. The 20% Rule of NASH Progression: The Natural History of Advanced Fibrosis and Cirrhosis Caused by NASH. Hepatology 2019, 70, 1885–1888. [Google Scholar] [CrossRef]
- Brunner, S.F.; Roberts, N.D.; Wylie, L.A.; Moore, L.; Aitken, S.J.; Davies, S.E.; Sanders, M.A.; Ellis, P.; Alder, C.; Hooks, Y.; et al. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature 2019, 574, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Lee, J.M. Primary malignant tumours in the non-cirrhotic liver. Eur. J. Radiol. 2017, 95, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Poh, Z.; Goh, B.B.; Chang, P.E.; Tan, C.K. Rates of cirrhosis and hepatocellular carcinoma in chronic hepatitis B and the role of surveillance: A 10-year follow-up of 673 patients. Eur. J. Gastroenterol. Hepatol. 2015, 27, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef]
- Postic, C.; Girard, J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: Lessons from genetically engineered mice. J. Clin. Investig. 2008, 118, 829–838. [Google Scholar] [CrossRef]
- Santoleri, D.; Titchenell, P.M. Resolving the Paradox of Hepatic Insulin Resistance. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 447–456. [Google Scholar] [CrossRef]
- James, D.E.; Stöckli, J.; Birnbaum, M.J. The aetiology and molecular landscape of insulin resistance. Nat. Rev. Mol. Cell. Biol. 2021, 22, 751–771. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Bessone, F.; Razori, M.V.; Roma, M.G. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell Mol. Life Sci. 2019, 76, 99–128. [Google Scholar] [CrossRef] [PubMed]
- Kitade, H.; Chen, G.; Ni, Y.; Ota, T. Nonalcoholic Fatty Liver Disease and Insulin Resistance: New Insights and Potential New Treatments. Nutrients 2017, 9, 387. [Google Scholar] [CrossRef]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Aspects Med. 2019, 65, 37–55. [Google Scholar] [CrossRef]
- Ezhilarasan, D.; Sokal, E.; Najimi, M. Hepatic fibrosis: It is time to go with hepatic stellate cell-specific therapeutic targets. Hepatobiliary Pancreat. Dis. Int. 2018, 17, 192–197. [Google Scholar] [CrossRef]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology 2004, 127, S35–S50. [Google Scholar] [CrossRef]
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73 (Suppl. S1), 4–13. [Google Scholar] [CrossRef]
- Clark, T.; Maximin, S.; Meier, J.; Pokharel, S.; Bhargava, P. Hepatocellular Carcinoma: Review of Epidemiology, Screening, Imaging Diagnosis, Response Assessment, and Treatment. Curr. Probl. Diagn. Radiol. 2015, 44, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Willatt, J.; Ruma, J.A.; Azar, S.F.; Dasika, N.L.; Syed, F. Imaging of hepatocellular carcinoma and image guided therapies—How we do it. Cancer Imaging 2017, 17, 9. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, A. Changing role of histopathology in the diagnosis and management of hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 4000–4013. [Google Scholar] [CrossRef] [PubMed]
- Foerster, F.; Gairing, S.J.; Müller, L.; Galle, P.R. NAFLD-driven HCC: Safety and efficacy of current and emerging treatment options. J. Hepatol. 2022, 76, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Austermann, J.; Spiekermann, C.; Roth, J. S100 proteins in rheumatic diseases. Nat. Rev. Rheumatol. 2018, 14, 528–541. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.L.; Garrie, K.; Turner, M.D. Role of S100 proteins in health and disease. Biochim. Biophys. Acta Mol. Cell. Res. 2020, 1867, 118677. [Google Scholar] [CrossRef]
- Xiao, X.; Yang, C.; Qu, S.L.; Shao, Y.D.; Zhou, C.Y.; Chao, R.; Huang, L.; Zhang, C. S100 proteins in atherosclerosis. Clin. Chim. Acta 2020, 502, 293–304. [Google Scholar] [CrossRef]
- Riuzzi, F.; Chiappalupi, S.; Arcuri, C.; Giambanco, I.; Sorci, G.; Donato, R. S100 proteins in obesity: Liaisons dangereuses. Cell Mol. Life Sci. 2020, 77, 129–147. [Google Scholar] [CrossRef]
- Allgöwer, C.; Kretz, A.L.; von Karstedt, S.; Wittau, M.; Henne-Bruns, D.; Lemke, J. Friend or Foe: S100 Proteins in Cancer. Cancers 2020, 12, 2037. [Google Scholar] [CrossRef]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.F.; Huang, H.; Jiang, F.; Ni, R.Z.; Xiao, M.B. S100 family signaling network and related proteins in pancreatic cancer (Review). Int. J. Mol. Med. 2014, 33, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Du, G.; Wang, D. The S100 protein family in lung cancer. Clin. Chim. Acta 2021, 520, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.L.; Jia, Y.L.; Chen, L.; Zeng, Q.; Zhou, J.N.; Fu, C.J.; Chen, H.X.; Yuan, H.F.; Li, Z.W.; Shi, L.; et al. Hepatocellular carcinoma-associated mesenchymal stem cells promote hepatocarcinoma progression: Role of the S100A4-miR155-SOCS1-MMP9 axis. Hepatology 2013, 57, 2274–2286. [Google Scholar] [CrossRef] [PubMed]
- De Ponti, A.; Wiechert, L.; Schneller, D.; Pusterla, T.; Longerich, T.; Hogg, N.; Vogel, A.; Schirmacher, P.; Hess, J.; Angel, P. A pro-tumorigenic function of S100A8/A9 in carcinogen-induced hepatocellular carcinoma. Cancer Lett. 2015, 369, 396–404. [Google Scholar] [CrossRef]
- Németh, J.; Stein, I.; Haag, D.; Riehl, A.; Longerich, T.; Horwitz, E.; Breuhahn, K.; Gebhardt, C.; Schirmacher, P.; Hahn, M.; et al. S100A8 and S100A9 are novel nuclear factor kappa B target genes during malignant progression of murine and human liver carcinogenesis. Hepatology 2009, 50, 1251–1262. [Google Scholar] [CrossRef]
- Hua, X.; Zhang, H.; Jia, J.; Chen, S.; Sun, Y.; Zhu, X. Roles of S100 family members in drug resistance in tumors: Status and prospects. Biomed. Pharmacother. 2020, 127, 110156. [Google Scholar] [CrossRef]
- Bolander, A.; Agnarsdóttir, M.; Wagenius, G.; Strömberg, S.; Pontén, F.; Ekman, S.; Brattström, D.; Larsson, A.; Einarsson, R.; Ullenhag, G.; et al. Serological and immunohistochemical analysis of S100 and new derivatives as markers for prognosis in patients with malignant melanoma. Melanoma Res. 2008, 18, 412–419. [Google Scholar] [CrossRef]
- Marenholz, I.; Heizmann, C.W.; Fritz, G. S100 proteins in mouse and man: From evolution to function and pathology (including an update of the nomenclature). Biochem. Biophys. Res. Commun. 2004, 322, 1111–1122. [Google Scholar] [CrossRef]
- Bresnick, A.R. S100 proteins as therapeutic targets. Biophys. Rev. 2018, 10, 1617–1629. [Google Scholar] [CrossRef]
- Kiss, B.; Ecsédi, P.; Simon, M.; Nyitray, L. Isolation and Characterization of S100 Protein-Protein Complexes. Methods Mol. Biol. 2019, 1929, 325–338. [Google Scholar] [CrossRef]
- Oslejsková, L.; Grigorian, M.; Hulejová, H.; Vencovsky, J.; Pavelka, K.; Klingelhöfer, J.; Gay, S.; Neidhart, M.; Brabcová, H.; Suchy, D.; et al. Metastasis-inducing S100A4 protein is associated with the disease activity of rheumatoid arthritis. Rheumatology 2009, 48, 1590–1594. [Google Scholar] [CrossRef] [PubMed]
- Donato, R. Intracellular and extracellular roles of S100 proteins. Microsc. Res. Tech. 2003, 60, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, J.A.; May, R.; Tanousis, K.; McNeill, E.; Mathies, M.; Gebhardt, C.; Henderson, R.; Robinson, M.J.; Hogg, N. Myeloid cell function in MRP-14 (S100A9) null mice. Mol. Cell. Biol. 2003, 23, 2564–2576. [Google Scholar] [CrossRef]
- Heizmann, C.W. The multifunctional S100 protein family. Methods Mol. Biol. 2002, 172, 69–80. [Google Scholar] [CrossRef]
- Saiki, Y.; Horii, A. Multiple functions of S100A10, an important cancer promoter. Pathol. Int. 2019, 69, 629–636. [Google Scholar] [CrossRef]
- Réty, S.; Sopkova, J.; Renouard, M.; Osterloh, D.; Gerke, V.; Tabaries, S.; Russo-Marie, F.; Lewit-Bentley, A. The crystal structure of a complex of p11 with the annexin II N-terminal peptide. Nat. Struct. Biol. 1999, 6, 89–95. [Google Scholar] [CrossRef]
- Damo, S.M.; Kehl-Fie, T.E.; Sugitani, N.; Holt, M.E.; Rathi, S.; Murphy, W.J.; Zhang, Y.; Betz, C.; Hench, L.; Fritz, G.; et al. Molecular basis for manganese sequestration by calprotectin and roles in the innate immune response to invading bacterial pathogens. Proc. Natl. Acad. Sci. USA 2013, 110, 3841–3846. [Google Scholar] [CrossRef] [PubMed]
- Moroz, O.V.; Antson, A.A.; Grist, S.J.; Maitland, N.J.; Dodson, G.G.; Wilson, K.S.; Lukanidin, E.; Bronstein, I.B. Structure of the human S100A12-copper complex: Implications for host-parasite defence. Acta Crystallogr. D Biol. Crystallogr. 2003, 59, 859–867. [Google Scholar] [CrossRef]
- Schäfer, B.W.; Heizmann, C.W. The S100 family of EF-hand calcium-binding proteins: Functions and pathology. Trends Biochem. Sci. 1996, 21, 134–140. [Google Scholar] [CrossRef]
- He, H.; Li, J.; Weng, S.; Li, M.; Yu, Y. S100A11: Diverse function and pathology corresponding to different target proteins. Cell Biochem. Biophys. 2009, 55, 117–126. [Google Scholar] [CrossRef]
- Hsu, K.; Champaiboon, C.; Guenther, B.D.; Sorenson, B.S.; Khammanivong, A.; Ross, K.F.; Geczy, C.L.; Herzberg, M.C. Anti-infective Protective Properties of S100 Calgranulins. Antiinflamm. Antiallergy Agents Med. Chem. 2009, 8, 290–305. [Google Scholar] [CrossRef] [PubMed]
- Chottekalapanda, R.U.; Kalik, S.; Gresack, J.; Ayala, A.; Gao, M.; Wang, W.; Meller, S.; Aly, A.; Schaefer, A.; Greengard, P. AP-1 controls the p11-dependent antidepressant response. Mol. Psychiatry 2020, 25, 1364–1381. [Google Scholar] [CrossRef] [PubMed]
- Tsoporis, J.N.; Izhar, S.; Parker, T.G. Expression of S100A6 in cardiac myocytes limits apoptosis induced by tumor necrosis factor-alpha. J. Biol. Chem. 2008, 283, 30174–30183. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, N.; Kido, J.; Kido, R.; Wada, C.; Kataoka, M.; Shinohara, Y.; Nagata, T. Regulation of calprotectin expression by interleukin-1alpha and transforming growth factor-beta in human gingival keratinocytes. J. Periodontal. Res. 2007, 42, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sobolewski, C.; Abegg, D.; Berthou, F.; Dolicka, D.; Calo, N.; Sempoux, C.; Fournier, M.; Maeder, C.; Ay, A.S.; Clavien, P.A.; et al. S100A11/ANXA2 belongs to a tumour suppressor/oncogene network deregulated early with steatosis and involved in inflammation and hepatocellular carcinoma development. Gut 2020, 69, 1841–1854. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Xie, H.; Long, X.; Zhou, M.; Xu, Z.; Shi, B.; Jiang, H.; Li, Z. EGFRvIII mediates hepatocellular carcinoma cell invasion by promoting S100 calcium binding protein A11 expression. PLoS ONE 2013, 8, e83332. [Google Scholar] [CrossRef]
- Wang, Q.; Williamson, M.; Bott, S.; Brookman-Amissah, N.; Freeman, A.; Nariculam, J.; Hubank, M.J.; Ahmed, A.; Masters, J.R. Hypomethylation of WNT5A, CRIP1 and S100P in prostate cancer. Oncogene 2007, 26, 6560–6565. [Google Scholar] [CrossRef]
- Wang, X.H.; Zhang, L.H.; Zhong, X.Y.; Xing, X.F.; Liu, Y.Q.; Niu, Z.J.; Peng, Y.; Du, H.; Zhang, G.G.; Hu, Y.; et al. S100A6 overexpression is associated with poor prognosis and is epigenetically up-regulated in gastric cancer. Am. J. Pathol. 2010, 177, 586–597. [Google Scholar] [CrossRef]
- Gjorgjieva, M.; Sobolewski, C.; Dolicka, D.; Correia de Sousa, M.; Foti, M. miRNAs and NAFLD: From pathophysiology to therapy. Gut 2019, 68, 2065–2079. [Google Scholar] [CrossRef]
- Choe, N.; Kwon, D.H.; Shin, S.; Kim, Y.S.; Kim, Y.K.; Kim, J.; Ahn, Y.; Eom, G.H.; Kook, H. The microRNA miR-124 inhibits vascular smooth muscle cell proliferation by targeting S100 calcium-binding protein A4 (S100A4). FEBS Lett. 2017, 591, 1041–1052. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Zhao, Q.; Du, B.; Chen, H.Y.; Zhou, D.Z. MicroRNA-187 Inhibits Growth and Metastasis of Osteosarcoma by Downregulating S100A4. Cancer Investig. 2018, 36, 1–9. [Google Scholar] [CrossRef]
- Shan, X.; Miao, Y.; Fan, R.; Qian, H.; Chen, P.; Liu, H.; Yan, X.; Li, J.; Zhou, F. MiR-590-5P inhibits growth of HepG2 cells via decrease of S100A10 expression and Inhibition of the Wnt pathway. Int. J. Mol. Sci. 2013, 14, 8556–8569. [Google Scholar] [CrossRef]
- Zhao, J.T.; Chi, B.J.; Sun, Y.; Chi, N.N.; Zhang, X.M.; Sun, J.B.; Chen, Y.; Xia, Y. LINC00174 is an oncogenic lncRNA of hepatocellular carcinoma and regulates miR-320/S100A10 axis. Cell Biochem. Funct. 2020, 38, 859–869. [Google Scholar] [CrossRef]
- Kligman, D.; Hilt, D.C. The S100 protein family. Trends Biochem. Sci. 1988, 13, 437–443. [Google Scholar] [CrossRef]
- Morera-Fumero, A.L.; Abreu-Gonzalez, P.; Henry-Benitez, M.; Yelmo-Cruz, S.; Diaz-Mesa, E. Summer/winter changes in serum S100B protein concentration as a source of research variance. J. Psychiatr. Res. 2013, 47, 791–795. [Google Scholar] [CrossRef]
- Nogueira, M.I.; Abbas, S.Y.; Campos, L.G.; Allemandi, W.; Lawson, P.; Takada, S.H.; Azmitia, E.C. S100beta protein expression: Gender- and age-related daily changes. Neurochem. Res. 2009, 34, 1355–1362. [Google Scholar] [CrossRef]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef]
- Rohde, D.; Ritterhoff, J.; Voelkers, M.; Katus, H.A.; Parker, T.G.; Most, P. S100A1: A multifaceted therapeutic target in cardiovascular disease. J. Cardiovasc. Transl. Res. 2010, 3, 525–537. [Google Scholar] [CrossRef]
- Passey, R.J.; Williams, E.; Lichanska, A.M.; Wells, C.; Hu, S.; Geczy, C.L.; Little, M.H.; Hume, D.A. A null mutation in the inflammation-associated S100 protein S100A8 causes early resorption of the mouse embryo. J. Immunol. 1999, 163, 2209–2216. [Google Scholar]
- Kan, J.; Zhao, C.; Lu, S.; Shen, G.; Yang, J.; Tong, P.; Xi, L.; Zhang, R.; Liang, X.; Su, D.; et al. S100A16, a novel lipogenesis promoting factor in livers of mice and hepatocytes in vitro. J. Cell. Physiol. 2019, 234, 21395–21406. [Google Scholar] [CrossRef]
- Mofid, A.; Newman, N.S.; Lee, P.J.; Abbasi, C.; Matkar, P.N.; Rudenko, D.; Kuliszewski, M.A.; Chen, H.H.; Afrasiabi, K.; Tsoporis, J.N.; et al. Cardiac Overexpression of S100A6 Attenuates Cardiomyocyte Apoptosis and Reduces Infarct Size After Myocardial Ischemia-Reperfusion. J. Am. Heart Assoc. 2017, 6, e004738. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.F.; Ma, J.X.; Wang, Y.L.; Ma, X.L. Calcyclin (S100A6) Attenuates Inflammatory Response and Mediates Apoptosis of Chondrocytes in Osteoarthritis via the PI3K/AKT Pathway. Orthop. Surg. 2021, 13, 1094–1101. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, T.; Miao, H.; Liang, B. The Calcium Binding Protein S100A11 and Its Roles in Diseases. Front Cell Dev. Biol. 2021, 9, 693262. [Google Scholar] [CrossRef]
- Fernandez-Fernandez, M.R.; Rutherford, T.J.; Fersht, A.R. Members of the S100 family bind p53 in two distinct ways. Protein. Sci. 2008, 17, 1663–1670. [Google Scholar] [CrossRef]
- Meng, M.; Sang, L.; Wang, X. S100 Calcium Binding Protein A11 (S100A11) Promotes The Proliferation, Migration And Invasion Of Cervical Cancer Cells, And Activates Wnt/β-Catenin Signaling. Onco. Targets Ther. 2019, 12, 8675–8685. [Google Scholar] [CrossRef]
- Zhang, M.X.; Gan, W.; Jing, C.Y.; Zheng, S.S.; Yi, Y.; Zhang, J.; Xu, X.; Lin, J.J.; Zhang, B.H.; Qiu, S.J. S100A11 promotes cell proliferation via P38/MAPK signaling pathway in intrahepatic cholangiocarcinoma. Mol. Carcinog. 2019, 58, 19–30. [Google Scholar] [CrossRef]
- Liu, Y.; Buck, D.C.; Neve, K.A. Novel interaction of the dopamine D2 receptor and the Ca2+ binding protein S100B: Role in D2 receptor function. Mol. Pharmacol. 2008, 74, 371–378. [Google Scholar] [CrossRef]
- Fang, D.; Zhang, C.; Xu, P.; Liu, Y.; Mo, X.; Sun, Q.; Abdelatty, A.; Hu, C.; Xu, H.; Zhou, G.; et al. S100A16 promotes metastasis and progression of pancreatic cancer through FGF19-mediated AKT and ERK1/2 pathways. Cell Biol. Toxicol. 2021, 37, 555–571. [Google Scholar] [CrossRef]
- Foertsch, F.; Teichmann, N.; Kob, R.; Hentschel, J.; Laubscher, U.; Melle, C. S100A11 is involved in the regulation of the stability of cell cycle regulator p21(CIP1/WAF1) in human keratinocyte HaCaT cells. FEBS J. 2013, 280, 3840–3853. [Google Scholar] [CrossRef]
- Hiroshima, Y.; Hsu, K.; Tedla, N.; Wong, S.W.; Chow, S.; Kawaguchi, N.; Geczy, C.L. S100A8/A9 and S100A9 reduce acute lung injury. Immunol. Cell Biol. 2017, 95, 461–472. [Google Scholar] [CrossRef]
- Doi, S.; Fujioka, N.; Ohtsuka, S.; Kondo, R.; Yamamoto, M.; Denda, M.; Magari, M.; Kanayama, N.; Hatano, N.; Morishita, R.; et al. Regulation of the tubulin polymerization-promoting protein by Ca(2+)/S100 proteins. Cell Calcium 2021, 96, 102404. [Google Scholar] [CrossRef] [PubMed]
- Ismail, T.M.; Fernig, D.G.; Rudland, P.S.; Terry, C.J.; Wang, G.; Barraclough, R. The basic C-terminal amino acids of calcium-binding protein S100A4 promote metastasis. Carcinogenesis 2008, 29, 2259–2266. [Google Scholar] [CrossRef] [PubMed]
- Leśniak, W.; Słomnicki, Ł.P.; Filipek, A. S100A6—New facts and features. Biochem. Biophys. Res. Commun. 2009, 390, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Vogl, T.; Ludwig, S.; Goebeler, M.; Strey, A.; Thorey, I.S.; Reichelt, R.; Foell, D.; Gerke, V.; Manitz, M.P.; Nacken, W.; et al. MRP8 and MRP14 control microtubule reorganization during transendothelial migration of phagocytes. Blood 2004, 104, 4260–4268. [Google Scholar] [CrossRef]
- Elenjord, R.; Ljones, H.; Sundkvist, E.; Loennechen, T.; Winberg, J.O. Dysregulation of matrix metalloproteinases and their tissue inhibitors by S100A4. Connect. Tissue Res. 2008, 49, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Tao, Z.; Zhou, Q.; Zhao, D.; Gu, L.; Zhu, S.; Chen, J. Effects of S100 calcium-binding protein A8 (S100A8) and S100 calcium-binding protein A9 (S100A9) on matrix metalloproteinase (MMP) expression in nasopharyngeal carcinoma CNE-2 cells. Transl. Cancer Res. 2021, 10, 1874–1884. [Google Scholar] [CrossRef]
- O’Connell, P.A.; Surette, A.P.; Liwski, R.S.; Svenningsson, P.; Waisman, D.M. S100A10 regulates plasminogen-dependent macrophage invasion. Blood 2010, 116, 1136–1146. [Google Scholar] [CrossRef]
- Sapkota, D.; Bruland, O.; Costea, D.E.; Haugen, H.; Vasstrand, E.N.; Ibrahim, S.O. S100A14 regulates the invasive potential of oral squamous cell carcinoma derived cell-lines in vitro by modulating expression of matrix metalloproteinases, MMP1 and MMP9. Eur. J. Cancer 2011, 47, 600–610. [Google Scholar] [CrossRef]
- Donato, R.; Sorci, G.; Giambanco, I. S100A6 protein: Functional roles. Cell Mol. Life Sci. 2017, 74, 2749–2760. [Google Scholar] [CrossRef]
- Jin, Q.; Chen, H.; Luo, A.; Ding, F.; Liu, Z. S100A14 stimulates cell proliferation and induces cell apoptosis at different concentrations via receptor for advanced glycation end products (RAGE). PLoS ONE 2011, 6, e19375. [Google Scholar] [CrossRef]
- Hofmann Bowman, M.; Wilk, J.; Heydemann, A.; Kim, G.; Rehman, J.; Lodato, J.A.; Raman, J.; McNally, E.M. S100A12 mediates aortic wall remodeling and aortic aneurysm. Circ. Res. 2010, 106, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surette, A.P.; Madureira, P.A.; Phipps, K.D.; Miller, V.A.; Svenningsson, P.; Waisman, D.M. Regulation of fibrinolysis by S100A10 in vivo. Blood 2011, 118, 3172–3181. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, J.K.; Lauritzen, S.P.; Scheffer, L.; Sakaguchi, M.; Bunkenborg, J.; Simon, S.M.; Kallunki, T.; Jäättelä, M.; Nylandsted, J. S100A11 is required for efficient plasma membrane repair and survival of invasive cancer cells. Nat. Commun. 2014, 5, 3795. [Google Scholar] [CrossRef] [PubMed]
- Foertsch, F.; Melle, C. Analysis of S100A11 in DNA Damage Repair. Methods Mol. Biol. 2019, 1929, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100A8/A9 in Inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef] [PubMed]
- Prudovsky, I.; Tarantini, F.; Landriscina, M.; Neivandt, D.; Soldi, R.; Kirov, A.; Small, D.; Kathir, K.M.; Rajalingam, D.; Kumar, T.K. Secretion without Golgi. J. Cell Biochem. 2008, 103, 1327–1343. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E. HMGB1, IL-1α, IL-33 and S100 proteins: Dual-function alarmins. Cell. Mol. Immunol. 2017, 14, 43–64. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Drury, S.; Fu, C.; Qu, W.; Taguchi, A.; Lu, Y.; Avila, C.; Kambham, N.; Bierhaus, A.; Nawroth, P.; et al. RAGE mediates a novel proinflammatory axis: A central cell surface receptor for S100/calgranulin polypeptides. Cell 1999, 97, 889–901. [Google Scholar] [CrossRef]
- Tondera, C.; Laube, M.; Pietzsch, J. Insights into binding of S100 proteins to scavenger receptors: Class B scavenger receptor CD36 binds S100A12 with high affinity. Amino. Acids. 2017, 49, 183–191. [Google Scholar] [CrossRef]
- Teng, F.; Jiang, J.; Zhang, J.; Yuan, Y.; Li, K.; Zhou, B.; Zhou, X.; Liu, W.; Zhang, P.; Liu, D.; et al. The S100 calcium-binding protein A11 promotes hepatic steatosis through RAGE-mediated AKT-mTOR signaling. Metabolism 2021, 117, 154725. [Google Scholar] [CrossRef]
- Mukai, K.; Miyagi, T.; Nishio, K.; Yokoyama, Y.; Yoshioka, T.; Saito, Y.; Tanaka, S.; Shigekawa, M.; Nawa, T.; Hikita, H.; et al. S100A8 Production in CXCR2-Expressing CD11b+Gr-1high Cells Aggravates Hepatitis in Mice Fed a High-Fat and High-Cholesterol Diet. J. Immunol. 2016, 196, 395–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhang, Z.; Li, C.; Zhu, T.; Gao, J.; Zhou, H.; Zheng, Y.; Chang, Q.; Wang, M.; Wu, J.; et al. S100A11 Promotes Liver Steatosis via FOXO1-Mediated Autophagy and Lipogenesis. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 697–724. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, R.; Xin, J.; Sun, Y.; Li, J.; Wei, D.; Zhao, A.Z. Identification of S100A16 as a novel adipogenesis promoting factor in 3T3-L1 cells. Endocrinology 2011, 152, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, R.; Zhu, W.; Xue, Y.; Zhang, Y.; Huang, Q.; Liu, M.; Liu, Y. S100A16 inhibits osteogenesis but stimulates adipogenesis. Mol. Biol. Rep. 2013, 40, 3465–3473. [Google Scholar] [CrossRef]
- Hou, S.; Jiao, Y.; Yuan, Q.; Zhai, J.; Tian, T.; Sun, K.; Chen, Z.; Wu, Z.; Zhang, J. S100A4 protects mice from high-fat diet-induced obesity and inflammation. Lab. Investig. 2018, 98, 1025–1038. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Y.; Ming, Y.; Song, Y.; Zhang, J.; Chen, X.; Zeng, M.; Mao, Y. S100A9: A Potential Biomarker for the Progression of Non-Alcoholic Fatty Liver Disease and the Diagnosis of Non-Alcoholic Steatohepatitis. PLoS ONE 2015, 10, e0127352. [Google Scholar] [CrossRef]
- Anguita-Ruiz, A.; Mendez-Gutierrez, A.; Ruperez, A.I.; Leis, R.; Bueno, G.; Gil-Campos, M.; Tofe, I.; Gomez-Llorente, C.; Moreno, L.A.; Gil, Á.; et al. The protein S100A4 as a novel marker of insulin resistance in prepubertal and pubertal children with obesity. Metabolism 2020, 105, 154187. [Google Scholar] [CrossRef]
- Arner, P.; Petrus, P.; Esteve, D.; Boulomié, A.; Näslund, E.; Thorell, A.; Gao, H.; Dahlman, I.; Rydén, M. Screening of potential adipokines identifies S100A4 as a marker of pernicious adipose tissue and insulin resistance. Int. J. Obes 2018, 42, 2047–2056. [Google Scholar] [CrossRef]
- Mortensen, O.H.; Nielsen, A.R.; Erikstrup, C.; Plomgaard, P.; Fischer, C.P.; Krogh-Madsen, R.; Lindegaard, B.; Petersen, A.M.; Taudorf, S.; Pedersen, B.K. Calprotectin--a novel marker of obesity. PLoS ONE 2009, 4, e7419. [Google Scholar] [CrossRef]
- Ortega, F.J.; Sabater, M.; Moreno-Navarrete, J.M.; Pueyo, N.; Botas, P.; Delgado, E.; Ricart, W.; Frühbeck, G.; Fernández-Real, J.M. Serum and urinary concentrations of calprotectin as markers of insulin resistance and type 2 diabetes. Eur. J. Endocrinol. 2012, 167, 569–578. [Google Scholar] [CrossRef]
- Herder, C.; Maalmi, H.; Strassburger, K.; Zaharia, O.P.; Ratter, J.M.; Karusheva, Y.; Elhadad, M.A.; Bódis, K.; Bongaerts, B.W.C.; Rathmann, W.; et al. Differences in Biomarkers of Inflammation Between Novel Subgroups of Recent-Onset Diabetes. Diabetes 2021, 70, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Song, F.; Hurtado del Pozo, C.; Rosario, R.; Zou, Y.S.; Ananthakrishnan, R.; Xu, X.; Patel, P.R.; Benoit, V.M.; Yan, S.F.; Li, H.; et al. RAGE regulates the metabolic and inflammatory response to high-fat feeding in mice. Diabetes 2014, 63, 1948–1965. [Google Scholar] [CrossRef]
- Wan, J.; Wu, X.; Chen, H.; Xia, X.; Song, X.; Chen, S.; Lu, X.; Jin, J.; Su, Q.; Cai, D.; et al. Aging-induced aberrant RAGE/PPARα axis promotes hepatic steatosis via dysfunctional mitochondrial β oxidation. Aging Cell 2020, 19, e13238. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Ma, Q.; Ding, P.; Li, J.; Chen, L.L.; Ao, K.J.; Tian, Y.Y. S100A4 Gene is Crucial for Methionine-Choline-Deficient Diet-Induced Non-Alcoholic Fatty Liver Disease in Mice. Yonsei Med. J. 2018, 59, 1064–1071. [Google Scholar] [CrossRef]
- Averill, M.M.; Barnhart, S.; Becker, L.; Li, X.; Heinecke, J.W.; Leboeuf, R.C.; Hamerman, J.A.; Sorg, C.; Kerkhoff, C.; Bornfeldt, K.E. S100A9 differentially modifies phenotypic states of neutrophils, macrophages, and dendritic cells: Implications for atherosclerosis and adipose tissue inflammation. Circulation 2011, 123, 1216–1226. [Google Scholar] [CrossRef] [PubMed]
- Manitz, M.P.; Horst, B.; Seeliger, S.; Strey, A.; Skryabin, B.V.; Gunzer, M.; Frings, W.; Schönlau, F.; Roth, J.; Sorg, C.; et al. Loss of S100A9 (MRP14) results in reduced interleukin-8-induced CD11b surface expression, a polarized microfilament system, and diminished responsiveness to chemoattractants in vitro. Mol. Cell. Biol. 2003, 23, 1034–1043. [Google Scholar] [CrossRef]
- Huang, Y.K.; Chou, R.H.; Yu, C. Tranilast Blocks the Interaction between the Protein S100A11 and Receptor for Advanced Glycation End Products (RAGE) V Domain and Inhibits Cell Proliferation. J. Biol. Chem. 2016, 291, 14300–14310. [Google Scholar] [CrossRef] [PubMed]
- Uno, M.; Kurita, S.; Misu, H.; Ando, H.; Ota, T.; Matsuzawa-Nagata, N.; Kita, Y.; Nabemoto, S.; Akahori, H.; Zen, Y.; et al. Tranilast, an antifibrogenic agent, ameliorates a dietary rat model of nonalcoholic steatohepatitis. Hepatology 2008, 48, 109–118. [Google Scholar] [CrossRef]
- Jiao, J.; González, Á.; Stevenson, H.L.; Gagea, M.; Sugimoto, H.; Kalluri, R.; Beretta, L. Depletion of S100A4(+) stromal cells does not prevent HCC development but reduces the stem cell-like phenotype of the tumors. Exp. Mol. Med. 2018, 50, e422. [Google Scholar] [CrossRef]
- Peyrou, M.; Bourgoin, L.; Poher, A.L.; Altirriba, J.; Maeder, C.; Caillon, A.; Fournier, M.; Montet, X.; Rohner-Jeanrenaud, F.; Foti, M. Hepatic PTEN deficiency improves muscle insulin sensitivity and decreases adiposity in mice. J. Hepatol. 2015, 62, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Li, J.; Zhang, J.; Dai, C.; Liu, X.; Wang, J.; Gao, Z.; Guo, H.; Wang, R.; Lu, S.; et al. S100A4 promotes liver fibrosis via activation of hepatic stellate cells. J. Hepatol. 2015, 62, 156–164. [Google Scholar] [CrossRef]
- Li, Y.; Bao, J.; Bian, Y.; Erben, U.; Wang, P.; Song, K.; Liu, S.; Li, Z.; Gao, Z.; Qin, Z. S100A4(+) Macrophages Are Necessary for Pulmonary Fibrosis by Activating Lung Fibroblasts. Front. Immunol. 2018, 9, 1776. [Google Scholar] [CrossRef]
- Xia, H.; Gilbertsen, A.; Herrera, J.; Racila, E.; Smith, K.; Peterson, M.; Griffin, T.; Benyumov, A.; Yang, L.; Bitterman, P.B.; et al. Calcium-binding protein S100A4 confers mesenchymal progenitor cell fibrogenicity in idiopathic pulmonary fibrosis. J. Clin. Investig. 2017, 127, 2586–2597. [Google Scholar] [CrossRef]
- Qian, L.; Hong, J.; Zhang, Y.; Zhu, M.; Wang, X.; Zhang, Y.; Chu, M.; Yao, J.; Xu, D. Downregulation of S100A4 Alleviates Cardiac Fibrosis via Wnt/β -Catenin Pathway in Mice. Cell. Physiol. Biochem. 2018, 46, 2551–2560. [Google Scholar] [CrossRef]
- Österreicher, C.H.; Penz-Österreicher, M.; Grivennikov, S.I.; Guma, M.; Koltsova, E.K.; Datz, C.; Sasik, R.; Hardiman, G.; Karin, M.; Brenner, D.A. Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc. Natl. Acad. Sci. USA 2011, 108, 308–313. [Google Scholar] [CrossRef]
- Dahlmann, M.; Okhrimenko, A.; Marcinkowski, P.; Osterland, M.; Herrmann, P.; Smith, J.; Heizmann, C.W.; Schlag, P.M.; Stein, U. RAGE mediates S100A4-induced cell motility via MAPK/ERK and hypoxia signaling and is a prognostic biomarker for human colorectal cancer metastasis. Oncotarget 2014, 5, 3220–3233. [Google Scholar] [CrossRef]
- Xia, P.; He, H.; Kristine, M.S.; Guan, W.; Gao, J.; Wang, Z.; Hu, J.; Han, L.; Li, J.; Han, W.; et al. Therapeutic effects of recombinant human S100A6 and soluble receptor for advanced glycation end products(sRAGE) on CCl(4)-induced liver fibrosis in mice. Eur. J. Pharmacol. 2018, 833, 86–93. [Google Scholar] [CrossRef]
- Krenkel, O.; Hundertmark, J.; Abdallah, A.T.; Kohlhepp, M.; Puengel, T.; Roth, T.; Branco, D.P.P.; Mossanen, J.C.; Luedde, T.; Trautwein, C.; et al. Myeloid cells in liver and bone marrow acquire a functionally distinct inflammatory phenotype during obesity-related steatohepatitis. Gut 2020, 69, 551–563. [Google Scholar] [CrossRef]
- Louka, M.L.; Ramzy, M.M. Involvement of fibroblast-specific protein 1 (S100A4) and matrix metalloproteinase-13 (MMP-13) in CCl4-induced reversible liver fibrosis. Gene 2016, 579, 29–33. [Google Scholar] [CrossRef]
- Dong, Y.; Zheng, Q.; Wang, Z.; Lin, X.; You, Y.; Wu, S.; Wang, Y.; Hu, C.; Xie, X.; Chen, J.; et al. Higher matrix stiffness as an independent initiator triggers epithelial-mesenchymal transition and facilitates HCC metastasis. J. Hematol. Oncol. 2019, 12, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristensen, D.B.; Kawada, N.; Imamura, K.; Miyamoto, Y.; Tateno, C.; Seki, S.; Kuroki, T.; Yoshizato, K. Proteome analysis of rat hepatic stellate cells. Hepatology 2000, 32, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.S.; Zhang, R.; Ge, Y.; Wang, D.; Hu, Y.; Qin, X.; Kan, J.; Liu, Y. S100a16 deficiency prevents hepatic stellate cells activation and liver fibrosis via inhibiting CXCR4 expression. Metabolism 2022, 135, 155271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yao, R.; Chen, J.; Zou, Q.; Zeng, L. S100 family members: Potential therapeutic target in patients with hepatocellular carcinoma: A STROBE study. Medicine 2021, 100, e24135. [Google Scholar] [CrossRef]
- Kim, B.; Jung, S.; Kim, H.; Kwon, J.O.; Song, M.K.; Kim, M.K.; Kim, H.J.; Kim, H.H. The role of S100A4 for bone metastasis in prostate cancer cells. BMC Cancer 2021, 21, 137. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zheng, J.; Huang, Y.; Song, L.; Yin, Y.; Ou, D.; He, S.; Chen, X.; Ouyang, X. Impact of S100A4 Expression on Clinicopathological Characteristics and Prognosis in Pancreatic Cancer: A Meta-Analysis. Dis. Markers 2016, 2016, 8137378. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Liu, S.; Xin, X.; Cai, L.; Shi, R.; Chi, S.; Feng, D.; Wang, H. S100A4 promotes endometrial cancer progress through epithelial-mesenchymal transition regulation. Oncol. Rep. 2016, 35, 3419–3426. [Google Scholar] [CrossRef]
- Zhai, X.; Zhu, H.; Wang, W.; Zhang, S.; Zhang, Y.; Mao, G. Abnormal expression of EMT-related proteins, S100A4, vimentin and E-cadherin, is correlated with clinicopathological features and prognosis in HCC. Med. Oncol. 2014, 31, 970. [Google Scholar] [CrossRef]
- Boye, K.; Maelandsmo, G.M. S100A4 and metastasis: A small actor playing many roles. Am. J. Pathol. 2010, 176, 528–535. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Song, K.; Liu, S.; Zhang, H.; Wang, F.; Ni, C.; Zhai, W.; Liang, J.; Qin, Z.; et al. S100A4 promotes hepatocellular carcinogenesis by intensifying fibrosis-associated cancer cell stemness. Oncoimmunology 2020, 9, 1725355. [Google Scholar] [CrossRef]
- Cui, J.F.; Liu, Y.K.; Zhang, L.J.; Shen, H.L.; Song, H.Y.; Dai, Z.; Yu, Y.L.; Zhang, Y.; Sun, R.X.; Chen, J.; et al. Identification of metastasis candidate proteins among HCC cell lines by comparative proteome and biological function analysis of S100A4 in metastasis in vitro. Proteomics 2006, 6, 5953–5961. [Google Scholar] [CrossRef]
- Sun, H.; Wang, C.; Hu, B.; Gao, X.; Zou, T.; Luo, Q.; Chen, M.; Fu, Y.; Sheng, Y.; Zhang, K.; et al. Exosomal S100A4 derived from highly metastatic hepatocellular carcinoma cells promotes metastasis by activating STAT3. Signal Transduct. Target. Ther. 2021, 6, 187. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, D.L.; Jiao, X.L.; Dong, Q. S100A4 regulates migration and invasion in hepatocellular carcinoma HepG2 cells via NF-κB-dependent MMP-9 signal. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 2372–2382. [Google Scholar]
- Arai, K.; Yamada, T.; Nozawa, R. Immunohistochemical investigation of migration inhibitory factor-related protein (MRP)-14 expression in hepatocellular carcinoma. Med. Oncol. 2000, 17, 183–188. [Google Scholar] [CrossRef]
- Oura, K.; Morishita, A.; Tani, J.; Masaki, T. Tumor Immune Microenvironment and Immunosuppressive Therapy in Hepatocellular Carcinoma: A Review. Int. J. Mol. Sci. 2021, 22, 5801. [Google Scholar] [CrossRef]
- Wei, R.; Zhu, W.W.; Yu, G.Y.; Wang, X.; Gao, C.; Zhou, X.; Lin, Z.F.; Shao, W.Q.; Wang, S.H.; Lu, M.; et al. S100 calcium-binding protein A9 from tumor-associated macrophage enhances cancer stem cell-like properties of hepatocellular carcinoma. Int. J. Cancer 2021, 148, 1233–1244. [Google Scholar] [CrossRef]
- Wu, R.; Duan, L.; Cui, F.; Cao, J.; Xiang, Y.; Tang, Y.; Zhou, L. S100A9 promotes human hepatocellular carcinoma cell growth and invasion through RAGE-mediated ERK1/2 and p38 MAPK pathways. Exp. Cell Res. 2015, 334, 228–238. [Google Scholar] [CrossRef]
- Wu, R.; Duan, L.; Ye, L.; Wang, H.; Yang, X.; Zhang, Y.; Chen, X.; Zhang, Y.; Weng, Y.; Luo, J.; et al. S100A9 promotes the proliferation and invasion of HepG2 hepatocellular carcinoma cells via the activation of the MAPK signaling pathway. Int. J. Oncol. 2013, 42, 1001–1010. [Google Scholar] [CrossRef]
- De Ponti, A.; Wiechert, L.; Stojanovic, A.; Longerich, T.; Marhenke, S.; Hogg, N.; Vogel, A.; Cerwenka, A.; Schirmacher, P.; Hess, J.; et al. Chronic liver inflammation and hepatocellular carcinogenesis are independent of S100A9. Int. J. Cancer 2015, 136, 2458–2463. [Google Scholar] [CrossRef]
- Zhou, X.; Shi, M.; Cao, J.; Yuan, T.; Yu, G.; Chen, Y.; Fang, W.; Li, H. S100 Calcium Binding Protein A10, A Novel Oncogene, Promotes the Proliferation, Invasion, and Migration of Hepatocellular Carcinoma. Front. Genet. 2021, 12, 695036. [Google Scholar] [CrossRef]
- Bharadwaj, A.G.; Dahn, M.L.; Liu, R.Z.; Colp, P.; Thomas, L.N.; Holloway, R.W.; Marignani, P.A.; Too, C.K.; Barnes, P.J.; Godbout, R.; et al. S100A10 Has a Critical Regulatory Function in Mammary Tumor Growth and Metastasis: Insights Using MMTV-PyMT Oncomice and Clinical Patient Sample Analysis. Cancers 2020, 12, 3673. [Google Scholar] [CrossRef]
- Yanagi, H.; Watanabe, T.; Nishimura, T.; Hayashi, T.; Kono, S.; Tsuchida, H.; Hirata, M.; Kijima, Y.; Takao, S.; Okada, S.; et al. Upregulation of S100A10 in metastasized breast cancer stem cells. Cancer Sci. 2020, 111, 4359–4370. [Google Scholar] [CrossRef]
- Phipps, K.D.; Surette, A.P.; O’Connell, P.A.; Waisman, D.M. Plasminogen receptor S100A10 is essential for the migration of tumor-promoting macrophages into tumor sites. Cancer Res. 2011, 71, 6676–6683. [Google Scholar] [CrossRef]
- Brenner, A.K.; Bruserud, Ø. S100 Proteins in Acute Myeloid Leukemia. Neoplasia 2018, 20, 1175–1186. [Google Scholar] [CrossRef]
- Reyes, M.; González, L.; Ibeas, K.; Cereijo, R.; Taxerås, S.D.; Pellitero, S.; Martínez, E.; Tarascó, J.; Moreno, P.; Malagón, P.; et al. White adipose tissue-infiltrated CD11b+ myeloid cells are a source of S100A4, a new potential marker of hepatic damage. Eur. J. Endocrinol. 2021, 184, 533–541. [Google Scholar] [CrossRef]
- Meng, J.; Gu, F.; Fang, H.; Qu, B. Elevated Serum S100A9 Indicated Poor Prognosis in Hepatocellular Carcinoma after Curative Resection. J. Cancer 2019, 10, 408–415. [Google Scholar] [CrossRef]
- Qi, L.N.; Ma, L.; Wu, F.X.; Chen, Y.Y.; Xing, W.T.; Jiang, Z.J.; Zhong, J.H.; Chen, Z.S.; Gong, W.F.; Ye, J.Z.; et al. S100P as a novel biomarker of microvascular invasion and portal vein tumor thrombus in hepatocellular carcinoma. Hepatol. Int. 2021, 15, 114–126. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, G.; Zhang, P.; Zhang, J.; Li, X.; Gan, D.; Cao, X.; Han, M.; Du, H.; Ye, Y. The threshold of alpha-fetoprotein (AFP) for the diagnosis of hepatocellular carcinoma: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0228857. [Google Scholar] [CrossRef]
- Sumida, Y.; Yoneda, M. Current and future pharmacological therapies for NAFLD/NASH. J. Gastroenterol. 2018, 53, 362–376. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Cheng, A.L.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Lim, H.Y.; Kudo, M.; Breder, V.; Merle, P.; et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J. Hepatol. 2022, 76, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Zhong, C.; Li, Y.; Yang, J.; Jin, S.; Chen, G.; Li, D.; Fan, X.; Lin, H. Immunotherapy for Hepatocellular Carcinoma: Current Limits and Prospects. Front. Oncol. 2021, 11, 589680. [Google Scholar] [CrossRef] [PubMed]
- Shishibori, T.; Oyama, Y.; Matsushita, O.; Yamashita, K.; Furuichi, H.; Okabe, A.; Maeta, H.; Hata, Y.; Kobayashi, R. Three distinct anti-allergic drugs, amlexanox, cromolyn and tranilast, bind to S100A12 and S100A13 of the S100 protein family. Biochem. J. 1999, 338 Pt 3, 583–589. [Google Scholar] [CrossRef]
- Cho, C.C.; Chou, R.H.; Yu, C. Amlexanox Blocks the Interaction between S100A4 and Epidermal Growth Factor and Inhibits Cell Proliferation. PLoS ONE 2016, 11, e0161663. [Google Scholar] [CrossRef]
- Okada, M.; Tokumitsu, H.; Kubota, Y.; Kobayashi, R. Interaction of S100 proteins with the antiallergic drugs, olopatadine, amlexanox, and cromolyn: Identification of putative drug binding sites on S100A1 protein. Biochem. Biophys. Res. Commun. 2002, 292, 1023–1030. [Google Scholar] [CrossRef]
- Kishimoto, K.; Kaneko, S.; Ohmori, K.; Tamura, T.; Hasegawa, K. Olopatadine suppresses the migration of THP-1 monocytes induced by S100A12 protein. Mediators Inflamm. 2006, 2006, 42726. [Google Scholar] [CrossRef]
- Zhang, X.; Wei, L.; Wang, J.; Qin, Z.; Wang, J.; Lu, Y.; Zheng, X.; Peng, Q.; Ye, Q.; Ai, F.; et al. Suppression Colitis and Colitis-Associated Colon Cancer by Anti-S100a9 Antibody in Mice. Front. Immunol. 2017, 8, 1774. [Google Scholar] [CrossRef]
- Kinoshita, R.; Sato, H.; Yamauchi, A.; Takahashi, Y.; Inoue, Y.; Sumardika, I.W.; Chen, Y.; Tomonobu, N.; Araki, K.; Shien, K.; et al. Newly developed anti-S100A8/A9 monoclonal antibody efficiently prevents lung tropic cancer metastasis. Int. J. Cancer 2019, 145, 569–575. [Google Scholar] [CrossRef]
- Hernández, J.L.; Padilla, L.; Dakhel, S.; Coll, T.; Hervas, R.; Adan, J.; Masa, M.; Mitjans, F.; Martinez, J.M.; Coma, S.; et al. Therapeutic targeting of tumor growth and angiogenesis with a novel anti-S100A4 monoclonal antibody. PLoS ONE 2013, 8, e72480. [Google Scholar] [CrossRef]
- Dakhel, S.; Padilla, L.; Adan, J.; Masa, M.; Martinez, J.M.; Roque, L.; Coll, T.; Hervas, R.; Calvis, C.; Messeguer, R.; et al. S100P antibody-mediated therapy as a new promising strategy for the treatment of pancreatic cancer. Oncogenesis 2014, 3, e92. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
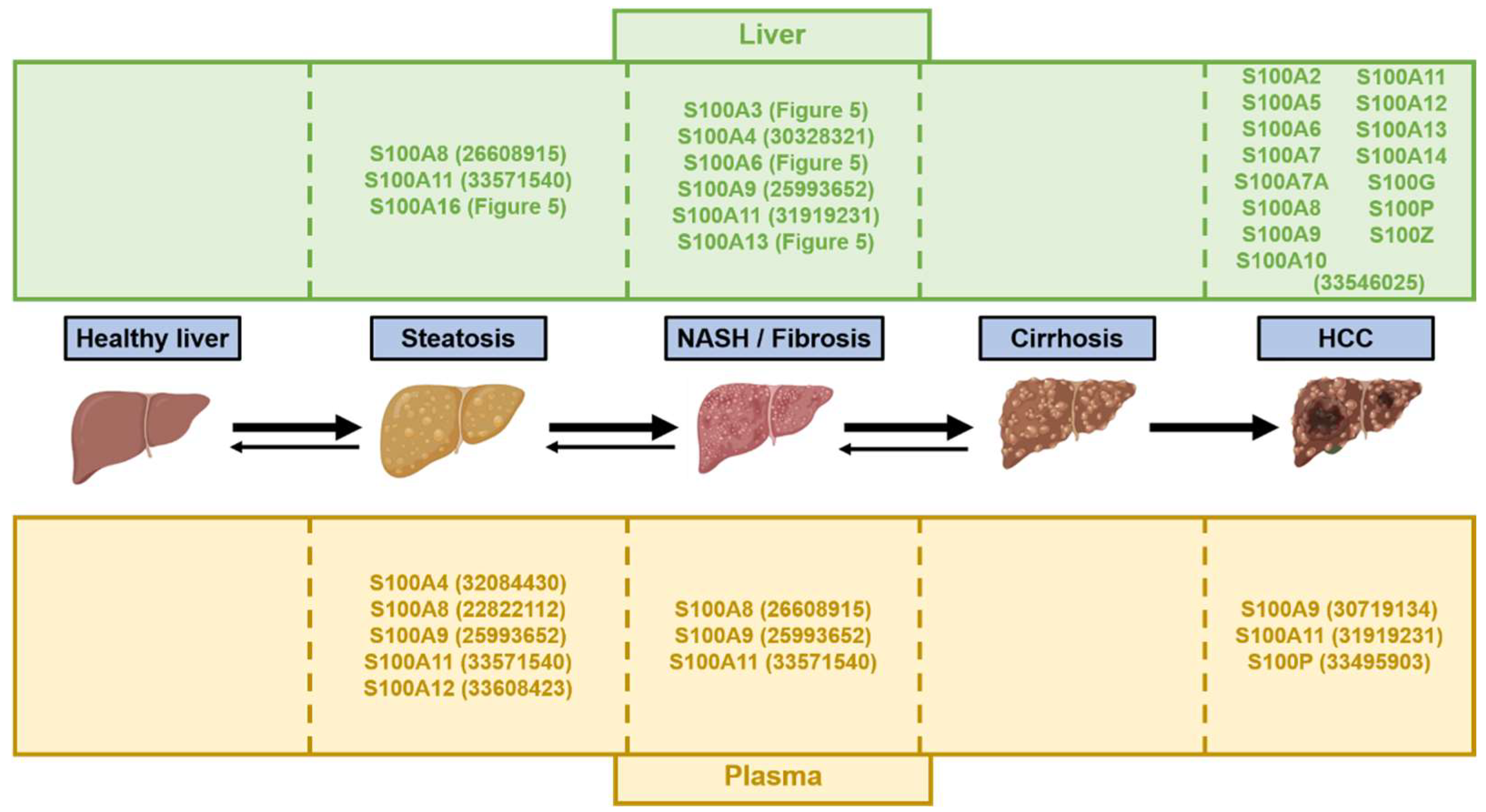{kind=link}
| Protein Name | Amino Acids (Molecular Weight) | Chromosomal Gene Location | EF-Hand Domain 1 (Affinity of the Ca2+ Binding Site) | EF-Hand Domain 2 (Affinity of the Ca2+ Binding Site) | Oligomerization Status |
|---|---|---|---|---|---|
| S100A1 | 94 aa (10.5 kDa) | 1q21.3 | EF-hand domain 1 (low Ca2+ affinity) | EF-hand domain 2 (high Ca2+ affinity) | Homodimer (21296671) Heterodimer with S100B (30719832) or S100P (30719832) |
| S100A2 | 98 aa (11.1 kDa) | 1q21.3 | EF-hand domain 1 (low Ca2+ affinity) | EF-hand domain 2 (high Ca2+ affinity) | Homodimer (10951287) |
| S100A3 | 101 aa (11.7 kDa) | 1q21.3 | EF-hand domain 1 (low Ca2+ affinity) | EF-hand domain 2 (high Ca2+ affinity, bind also Zn2+) | Homodimer and homotetramer (18083705) |
| S100A4 | 101 aa (11.7 kDa) | 1q21.3 | EF-hand domain 1 (low Ca2+ affinity) | EF-hand domain 2 (high Ca2+ affinity) | Homodimer and Multimeric (19828600) |
| S100A5 | 92 aa (10.7 kDa) | 1q21.3 | EF-hand domain 1 (low Ca2+ affinity) | EF-hand domain 2 (high Ca2+ affinity) | Homodimer (19536568) |
| S100A6 | 90 aa (10.2 kDa) | 1q21.3 | EF-hand domain 1 (Ca2+ affinity Nd) | EF-hand domain 2 (Ca2+ affinity Nd) | Homodimer (11937060) |
| S100A7 | 101 aa (11.4 kDa) | 1q21.3 | EF-hand domain 1 (bind Zn2+) | EF-hand domain 2 (high Ca2+ affinity) | Homodimer (28976190) |
| S100A8 | 93 aa (10.8 kDa) | 1q21.3 | EF-hand domain 1 (low Ca2+ affinity, bind also Zn2+) | EF-hand domain 2 (high Ca2+ affinity) | Homodimer Heterodimer or Heterotetramer with S100A9 (17553524) |
| S100A9 | 114 aa (13.2 kDa) | 1q21.3 | EF-hand domain 1 (low Ca2+ affinity, bind also Zn2+) | EF-hand domain 2 (high Ca2+ affinity, bind also Zn2+) | Homodimer Heterodimer or Heterotetramer with S100A8 (17553524) |
| S100A10 | 97 aa (11.2 kDa) | 1q21.3 | Related EF-hand domain (No Ca2+ binding) | Related EF-hand domain (No Ca2+ binding) | Heterotetramer with ANXA2 (9886297) |
| S100A11 | 105 aa (11.7 kDa) | 1q21.3 | EF-hand domain 1 (low Ca2+ affinity) | EF-hand domain 2 (high Ca2+ affinity) | Homodimer (16503655) Heterodimer with S100B (30719832) |
| S100A12 | 92 aa (10.6 kDa) | 1q21.3 | EF-hand domain 1 (low Ca2+ affinity, bind also Zn2+ and Cu2+) | EF-hand domain 2 (high Ca2+ affinity, bind also Zn2+ and Cu2+) | Homodimer (18443896) Homooligomer (tetramer or hexamer) (19386136) |
| S100A13 | 98 aa (11.5 kDa) | 1q21.3 | EF-hand domain 1 (Ca2+ affinity Nd) | No EF-hand domain (Ca2+ affinity Nd) | Homodimer (16122705) |
| S100A14 | 104 aa (11.6 kDa) | 1q21.3 | None | EF-hand domain (No Ca2+ binding) | Homodimer (23197251) |
| S100A16 | 103 aa (11.8 kDa) | 1q21.3 | Degenerated EF-hand domain 1 (No Ca2+ binding) | EF-hand domain 2 (high Ca2+ affinity) | Homodimer (21046186) |
| S100A7A | 101 aa (11.3 kDa) | 1q21.3 | EF-hand domain 1 (No Ca2+ binding, bind also Zn2+) | EF-hand domain 2 (high Ca2+ affinity, bind also Zn2+) | Nd |
| S100A7L2 | 101 aa (11.3 kDa) | 1q21.3 | EF-hand domain 1 (No Ca2+ binding) | EF-hand domain 2 (high Ca2+ affinity, bind also Zn2+) | Nd |
| S100B | 92 aa (10.7 kDa) | 21q22.3 | EF-hand domain 1 (low Ca2+ affinity) | EF-hand domain 2 (high Ca2+ affinity) | Homodimer (32027773) Heterodimer with S100A1 (30719832), S100A11 (30719832), S100A6 (9925766) |
| S100G | 79 aa (9 kDa) | Xp22.2 | EF-hand domain 1 (low Ca2+ affinity) | EF-hand domain 2 (high Ca2+ affinity) | Monomer (30710283) |
| S100P | 95 aa (10.4 kDa) | 4p16.1 | EF-hand domain 1 (low Ca2+ affinity) | EF-hand domain 2 (high Ca2+ affinity) | Homodimer (12808036) Heterodimer with S100A1 (30719832) |
| S100Z | 99 aa (11.6 kDa) | 5q13.3 | EF-hand domain 1 (low Ca2+ affinity) | EF-hand domain 2 (high Ca2+ affinity) | Homodimer (11747429) |
| Protein | MicroRNA | Validated | miRTarbase ID |
|---|---|---|---|
| S100A4 | hsa-miR-6745 | no | |
| S100A6 | hsa-miR-141-3p | yes | MIRT731072 |
| S100A8 | hsa-miR-125b-5p | yes | MIRT045918 |
| hsa-miR-24-3p | yes | MIRT052953 | |
| hsa-miR-98-5p | yes | MIRT027768 | |
| S100A9 | hsa-miR-1204 | yes | MIRT710086 |
| hsa-miR-132-5p | yes | MIRT710087 | |
| hsa-miR-196a-5p | yes | MIRT000220 | |
| hsa-miR-4252 | yes | MIRT4911293/MIRT710084 | |
| hsa-miR-4679 | yes | MIRT710085/MIRT4911292 | |
| hsa-miR-4701-5p | yes | MIRT710083/MIRT4911294 | |
| hsa-miR-588 | yes | MIRT710082/MIRT4911295 | |
| hsa-miR-660-3p | no | ||
| hsa-miR-663b | no | ||
| hsa-miR-766-5p | no | ||
| S100A10 | hsa-miR-100-5p | yes | MIRT048454 |
| hsa-miR-3122 | no | ||
| hsa-miR-3151-5p | no | ||
| hsa-miR-4270 | no | ||
| hsa-miR-4298 | no | ||
| hsa-miR-486-3p | no | ||
| hsa-miR-6847-3p | no | ||
| S100A11 | hsa-miR-1-3p | yes | MIRT023889 |
| hsa-miR-1207-5p | yes | - | |
| hsa-miR-1293 | no | ||
| hsa-miR-142-3p | yes | MIRT500051 | |
| hsa-miR-155-5p | yes | MIRT020889 | |
| hsa-miR-2861 | no | ||
| hsa-miR-3591-5p | yes | MIRT500050 | |
| hsa-miR-3609 | yes | MIRT460529 | |
| hsa-miR-3665 | yes | ||
| hsa-miR-3934-3p | yes | MIRT500053 | |
| hsa-miR-4307 | yes | MIRT460527 | |
| hsa-miR-4736 | yes | - | |
| hsa-miR-4741 | no | ||
| hsa-miR-548ah-5p | yes | MIRT460528 | |
| hsa-miR-548az-5p | yes | MIRT460531 | |
| hsa-miR-548t-5p | yes | MIRT460530 | |
| hsa-miR-556-3p | yes | MIRT460526 | |
| hsa-miR-6076 | yes | MIRT500055 | |
| hsa-miR-6134 | yes | MIRT500056 | |
| hsa-miR-648 | yes | MIRT500058 | |
| hsa-miR-6516-3p | yes | MIRT460525 | |
| hsa-miR-6797-3p | yes | MIRT500054 | |
| hsa-miR-7854-3p | yes | MIRT500057 | |
| hsa-miR-876-3p | yes | MIRT500052 | |
| S100A12 | hsa-miR-146a-5p | yes | MIRT437615 MIRT437621 |
| hsa-miR-4505 | no | ||
| hsa-miR-4710 | no | ||
| hsa-miR-5787 | no | ||
| hsa-miR-6858-5p | no | ||
| S100A16 | hsa-miR-1-3p | yes | MIRT024074 |
| hsa-miR-1207-5p | yes | - | |
| hsa-miR-1247-3p | yes | - | |
| hsa-miR-1249-5p | yes | - | |
| hsa-miR-1293 | yes | - | |
| hsa-miR-1912-3p | no | ||
| hsa-miR-193b-3p | yes | MIRT016530 | |
| hsa-miR-24-3p | yes | - | |
| hsa-miR-2467-5p | yes | - | |
| hsa-miR-3116 | no | ||
| hsa-miR-3184-5p | no | ||
| hsa-miR-363-5p | yes | - | |
| hsa-miR-3929 | yes | - | |
| hsa-miR-423-5p | no | ||
| hsa-miR-4478 | yes | - | |
| hsa-miR-4481 | no | ||
| hsa-miR-4510 | yes | - | |
| hsa-miR-4514 | yes | - | |
| hsa-miR-4537 | yes | - | |
| hsa-miR-4689 | yes | - | |
| hsa-miR-4692 | yes | - | |
| hsa-miR-4695-5p | yes | - | |
| hsa-miR-4736 | yes | - | |
| hsa-miR-4746-3p | yes | - | |
| hsa-miR-4784 | yes | - | |
| hsa-miR-498-5p | no | ||
| hsa-miR-541-3p | yes | - | |
| hsa-miR-6085 | no | ||
| hsa-miR-6127 | yes | - | |
| hsa-miR-6129 | yes | - | |
| hsa-miR-6130 | yes | - | |
| hsa-miR-6515-5p | no | ||
| hsa-miR-665 | yes | - | |
| hsa-miR-6715b-5p | yes | - | |
| hsa-miR-6721-5p | yes | - | |
| hsa-miR-6745 | yes | - | |
| hsa-miR-6756-5p | yes | - | |
| hsa-miR-6760-5p | no | ||
| hsa-miR-6766-5p | yes | - | |
| hsa-miR-6774-5p | yes | - | |
| hsa-miR-6775-3p | yes | - | |
| hsa-miR-6776-5p | yes | - | |
| hsa-miR-6791-5p | yes | - | |
| hsa-miR-6808-5p | yes | - | |
| hsa-miR-6813-5p | no | ||
| hsa-miR-6827-5p | yes | - | |
| hsa-miR-6847-5p | no | ||
| hsa-miR-6858-5p | yes | - | |
| hsa-miR-6884-5p | yes | - | |
| hsa-miR-6893-5p | yes | - | |
| hsa-miR-7150 | yes | - | |
| hsa-miR-7157-5p | no | ||
| hsa-miR-7160-5p | yes | - | |
| hsa-miR-765 | yes | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delangre, E.; Oppliger, E.; Berkcan, S.; Gjorgjieva, M.; Correia de Sousa, M.; Foti, M. S100 Proteins in Fatty Liver Disease and Hepatocellular Carcinoma. Int. J. Mol. Sci. 2022, 23, 11030. https://doi.org/10.3390/ijms231911030
Delangre E, Oppliger E, Berkcan S, Gjorgjieva M, Correia de Sousa M, Foti M. S100 Proteins in Fatty Liver Disease and Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2022; 23(19):11030. https://doi.org/10.3390/ijms231911030
Chicago/Turabian StyleDelangre, Etienne, Ezia Oppliger, Serkan Berkcan, Monika Gjorgjieva, Marta Correia de Sousa, and Michelangelo Foti. 2022. "S100 Proteins in Fatty Liver Disease and Hepatocellular Carcinoma" International Journal of Molecular Sciences 23, no. 19: 11030. https://doi.org/10.3390/ijms231911030
APA StyleDelangre, E., Oppliger, E., Berkcan, S., Gjorgjieva, M., Correia de Sousa, M., & Foti, M. (2022). S100 Proteins in Fatty Liver Disease and Hepatocellular Carcinoma. International Journal of Molecular Sciences, 23(19), 11030. https://doi.org/10.3390/ijms231911030