
A Novel Strategy for Identifying NSCLC MicroRNA Biomarkers and Their Mechanism Analysis Based on a Brand-New CeRNA-Hub-FFL Network


Abstract
:1. Introduction
2. Results
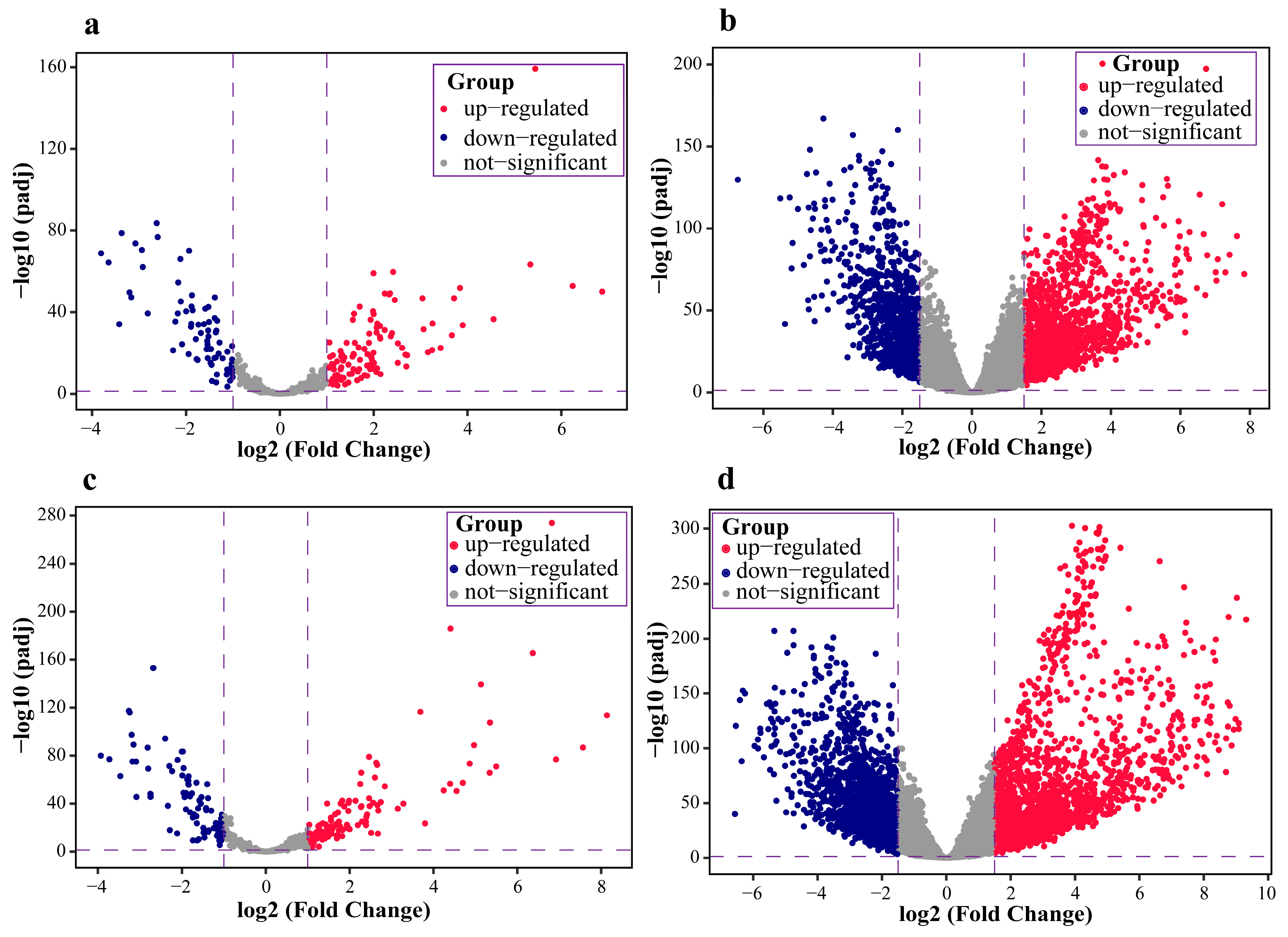
2.1. Data Pre-Processing and Differential Expression Analysis Results
2.2. Screening and Validation of the Lung Cancer Biomarkers
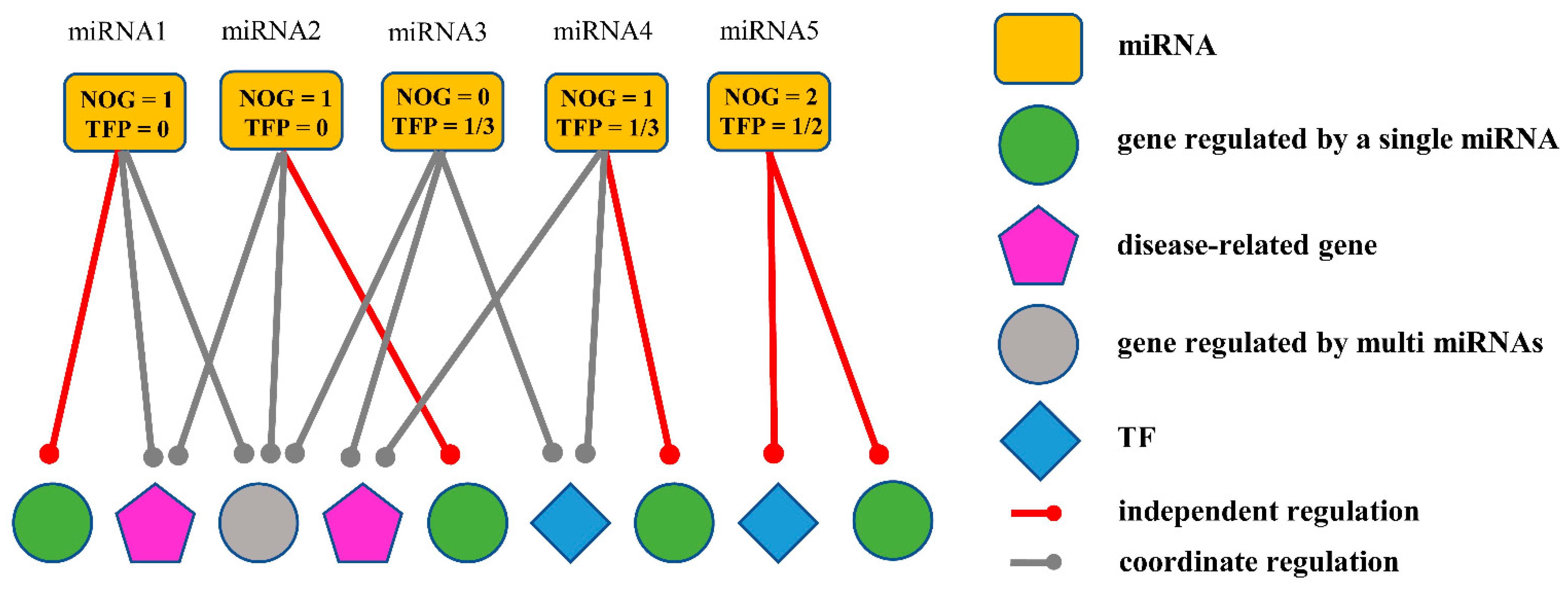
2.2.1. Obtaining miRNA Biomarkers Based on the Independent Regulatory Model
2.2.2. Identifying Candidate miRNA Biomarkers Based on Biological Significance of Target Genes
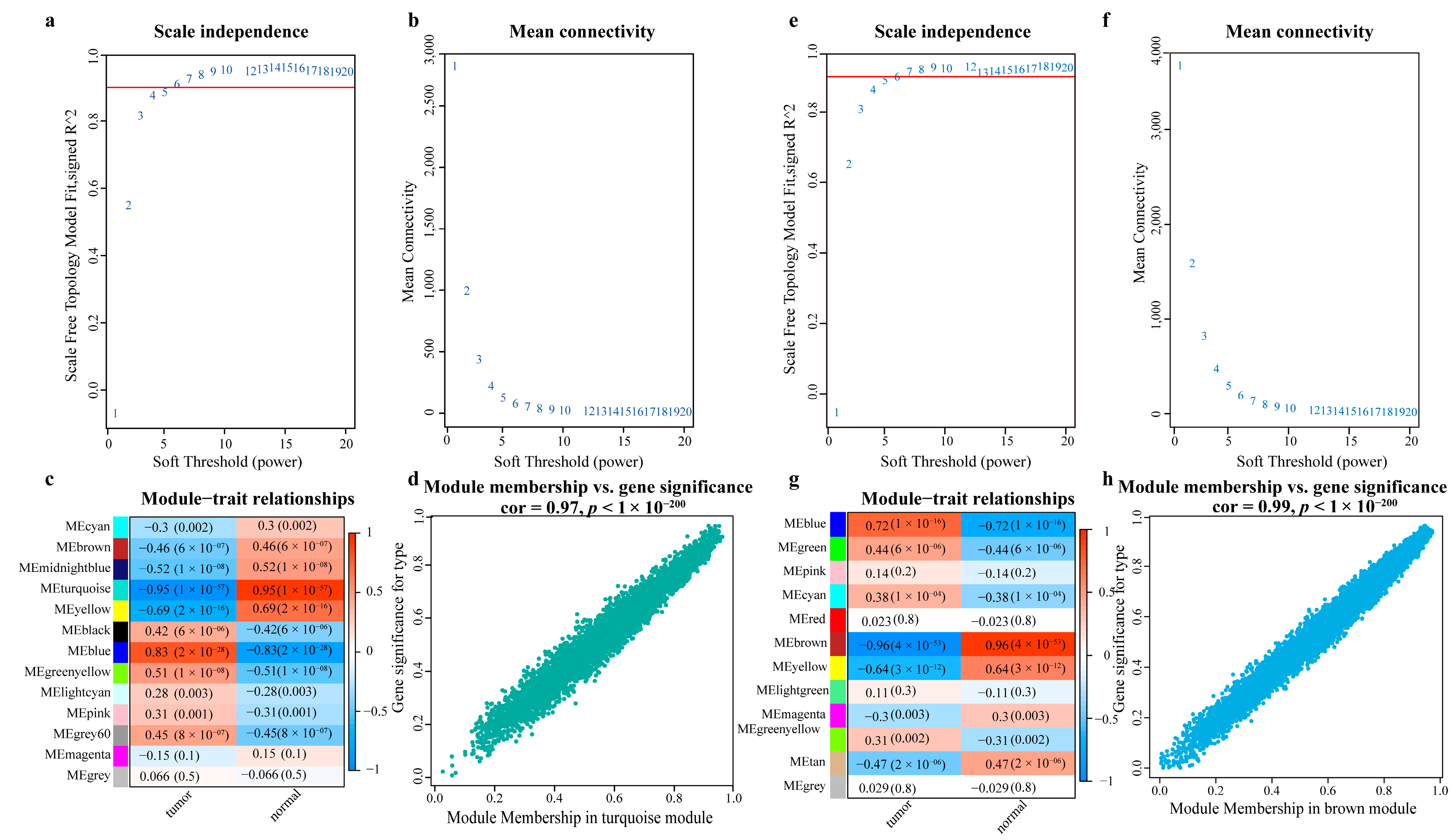
- Obtaining the most relevant gene sets for LC using WGCNA
- Screening LC-related genes based on databases of oncogenes and tumor suppressor genes
- Identifying miRNA biomarkers based on biological significance of genes
2.2.3. The Final miRNA Biomarkers for LC
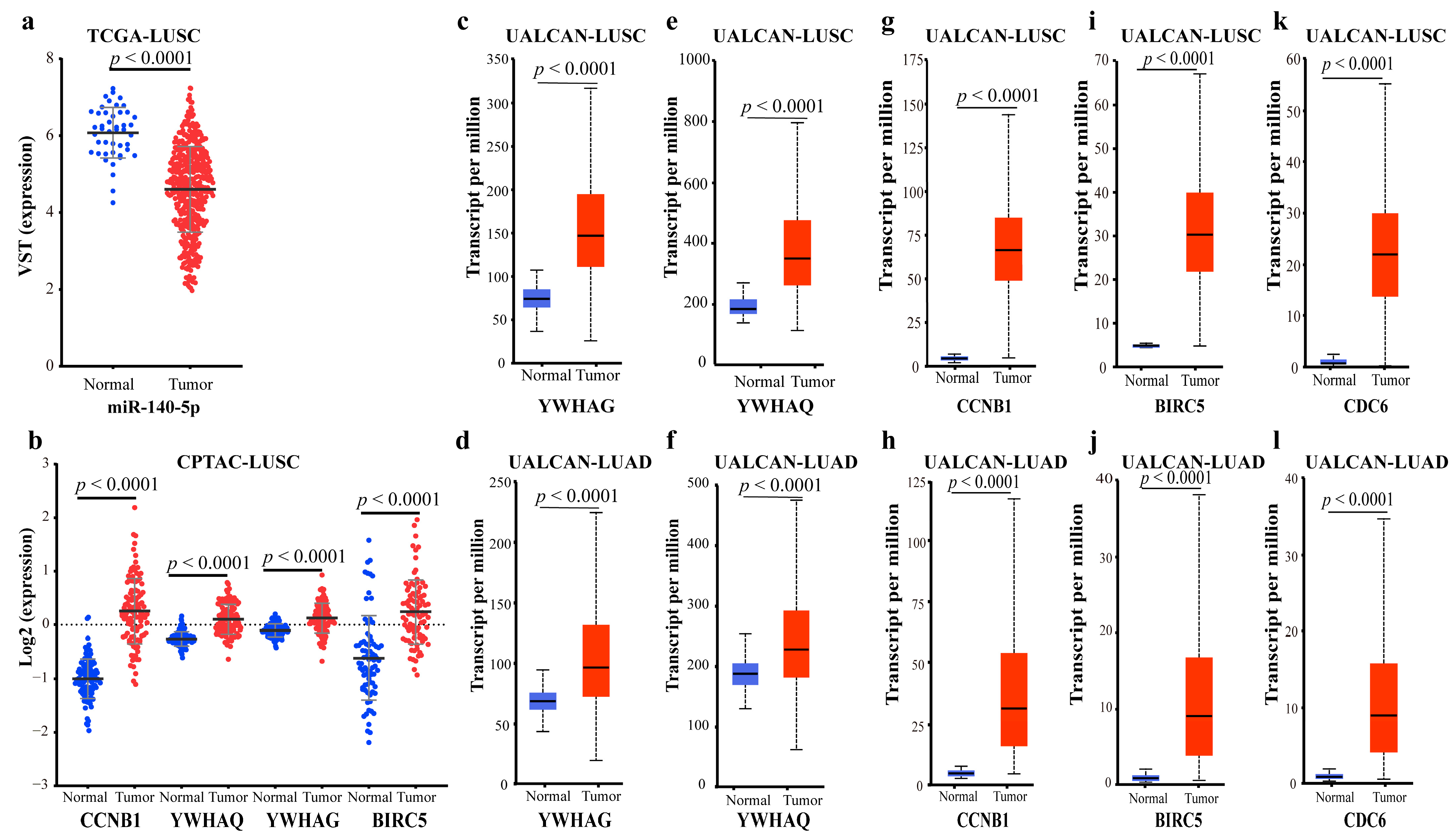
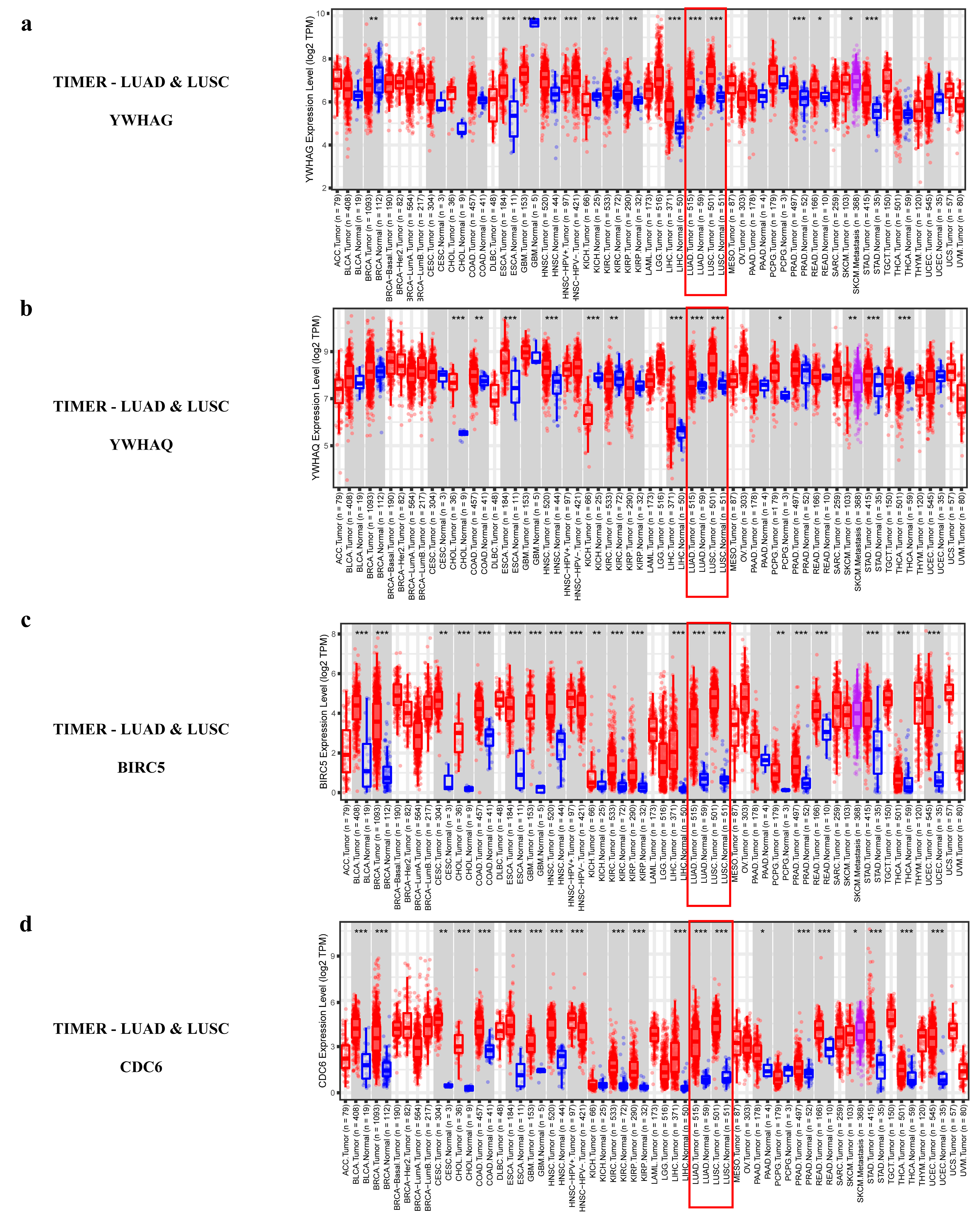
2.2.4. Validating the Reliability and Rationality of miRNA Biomarkers
- Validation using literature
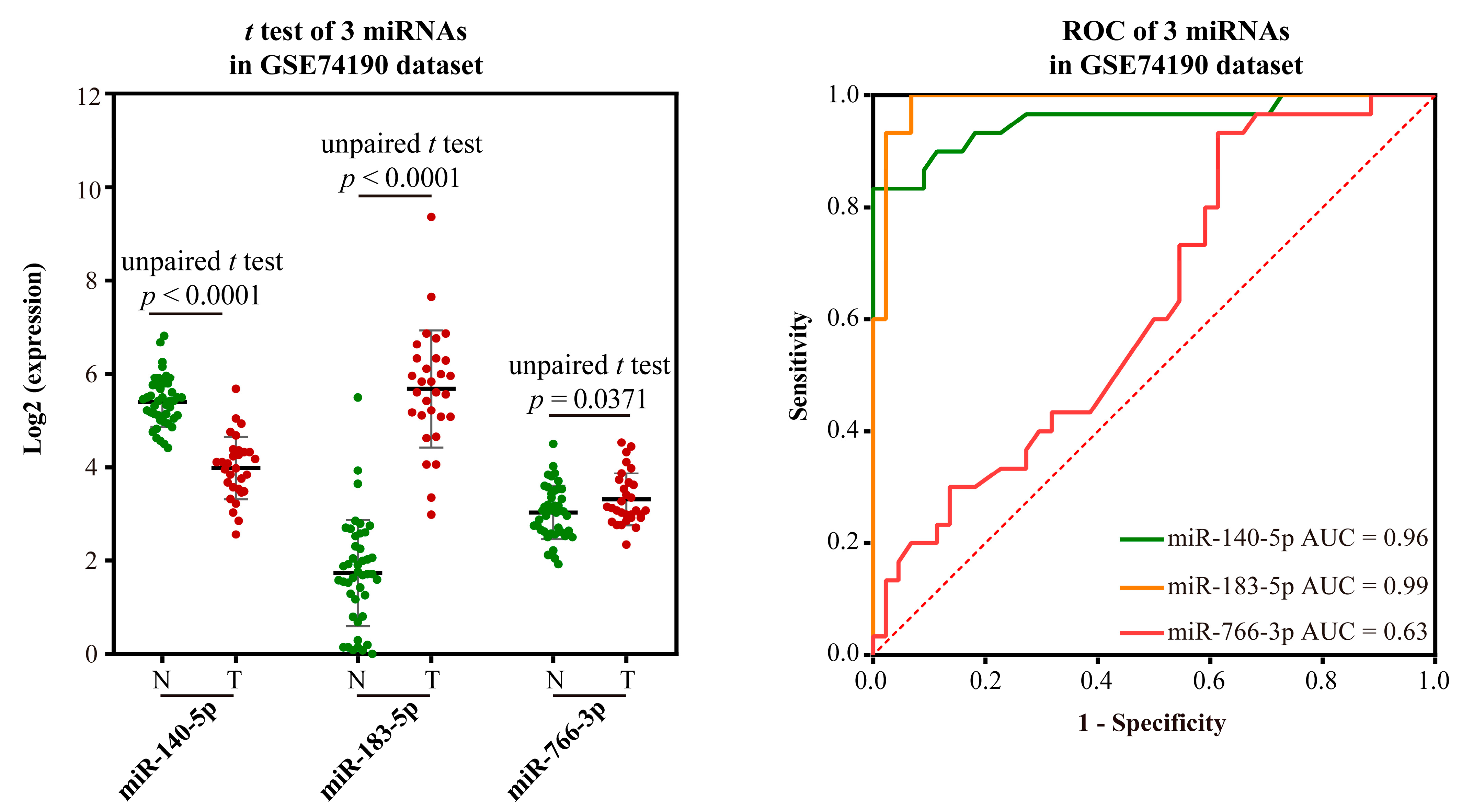
- Validation using external datasets
2.3. Construction of ceRNA-hub-FFL Network Based on miRNA Biomarkers
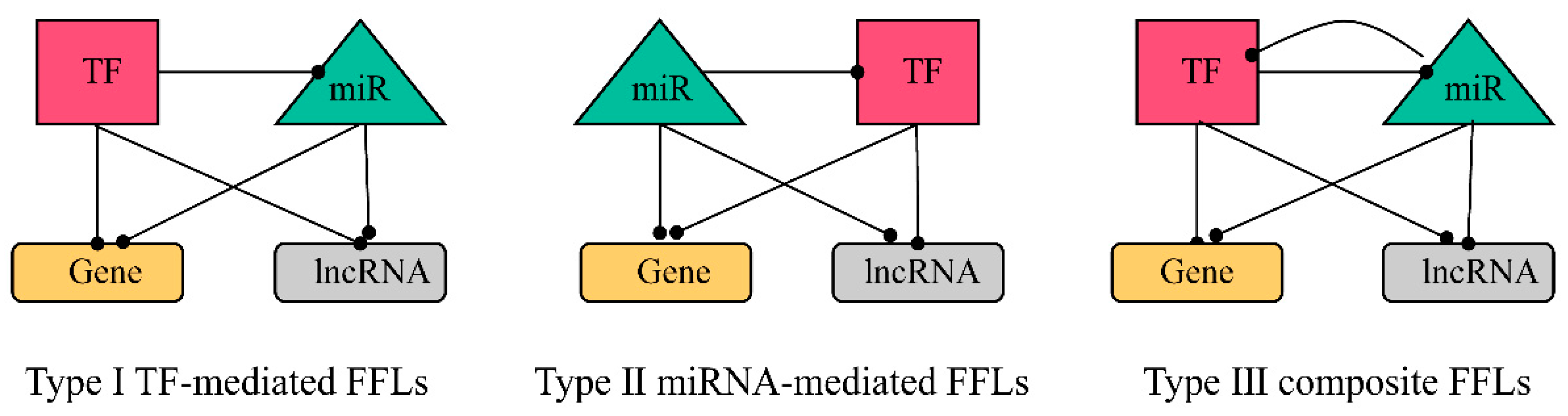
2.3.1. Construction of FFL Network
2.3.2. Obtaining hub-FFL Network and Further Extracting ceRNA-hub-FFL Network
2.4. Analysis of Potential Molecular Mechanisms of Lung Cancer Based on ceRNA-hub-FFL Network
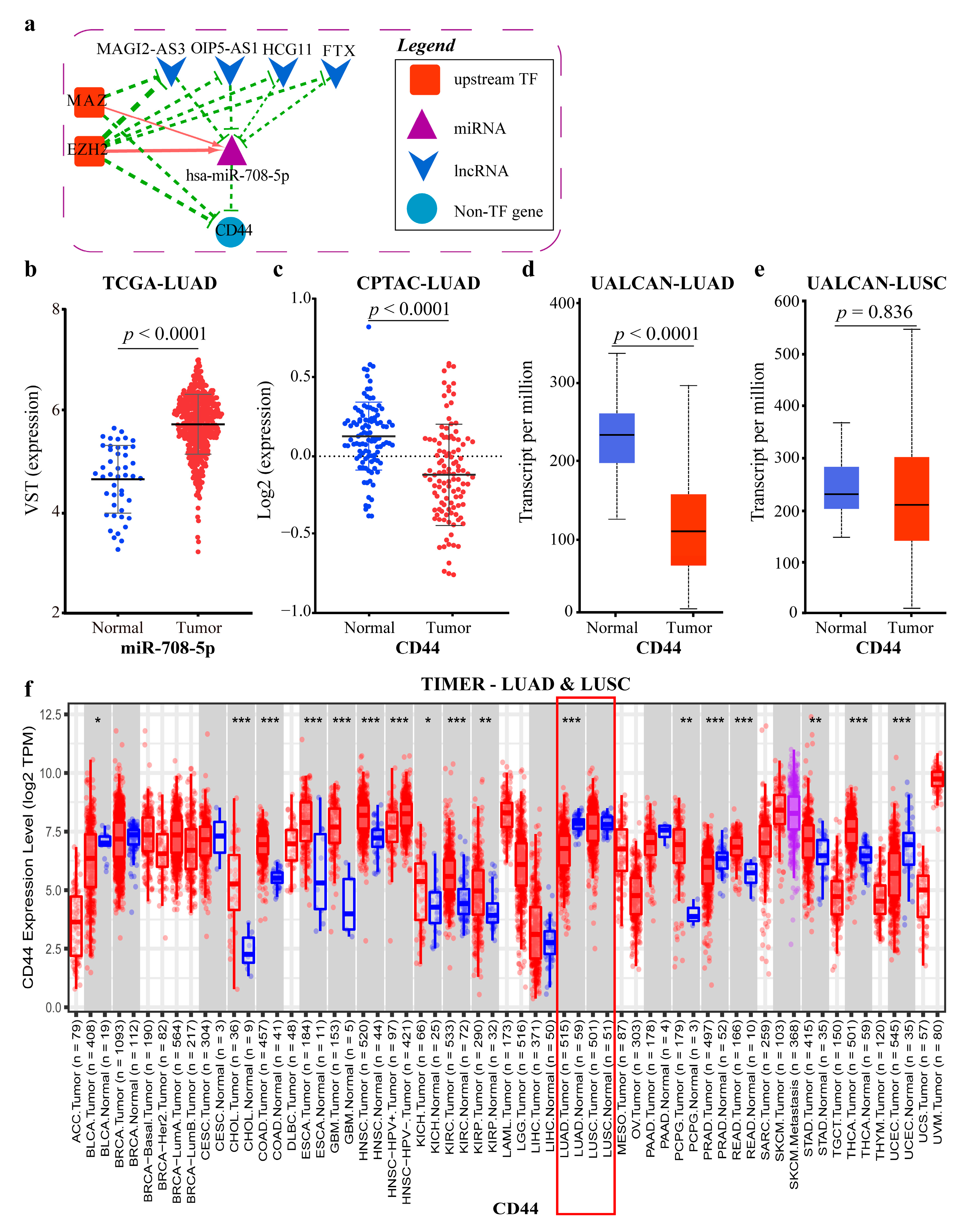
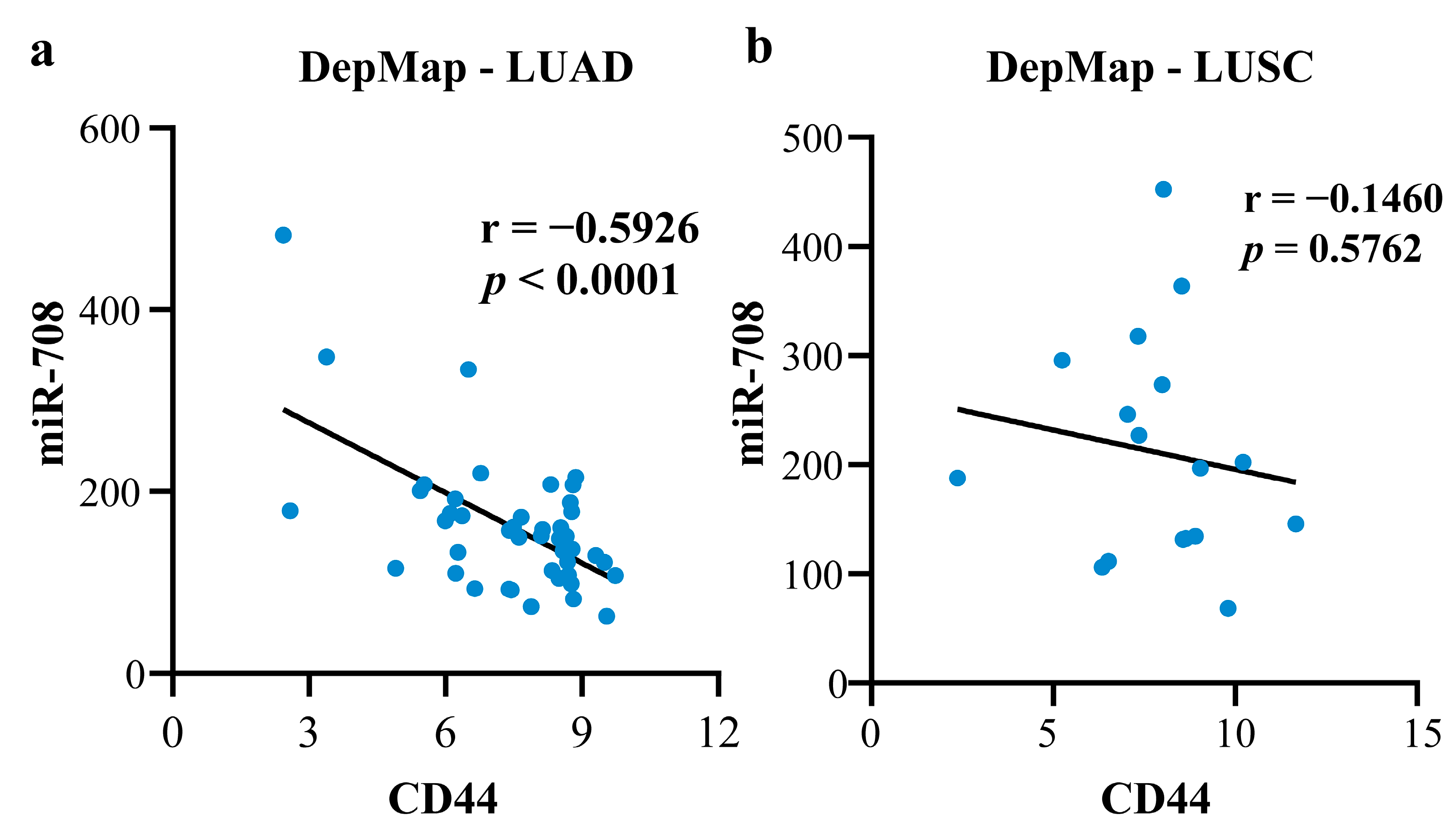
2.4.1. Mechanism Revealed by miR-708-5p Related ceRNA-hub-FFL Regulatory Subnetwork
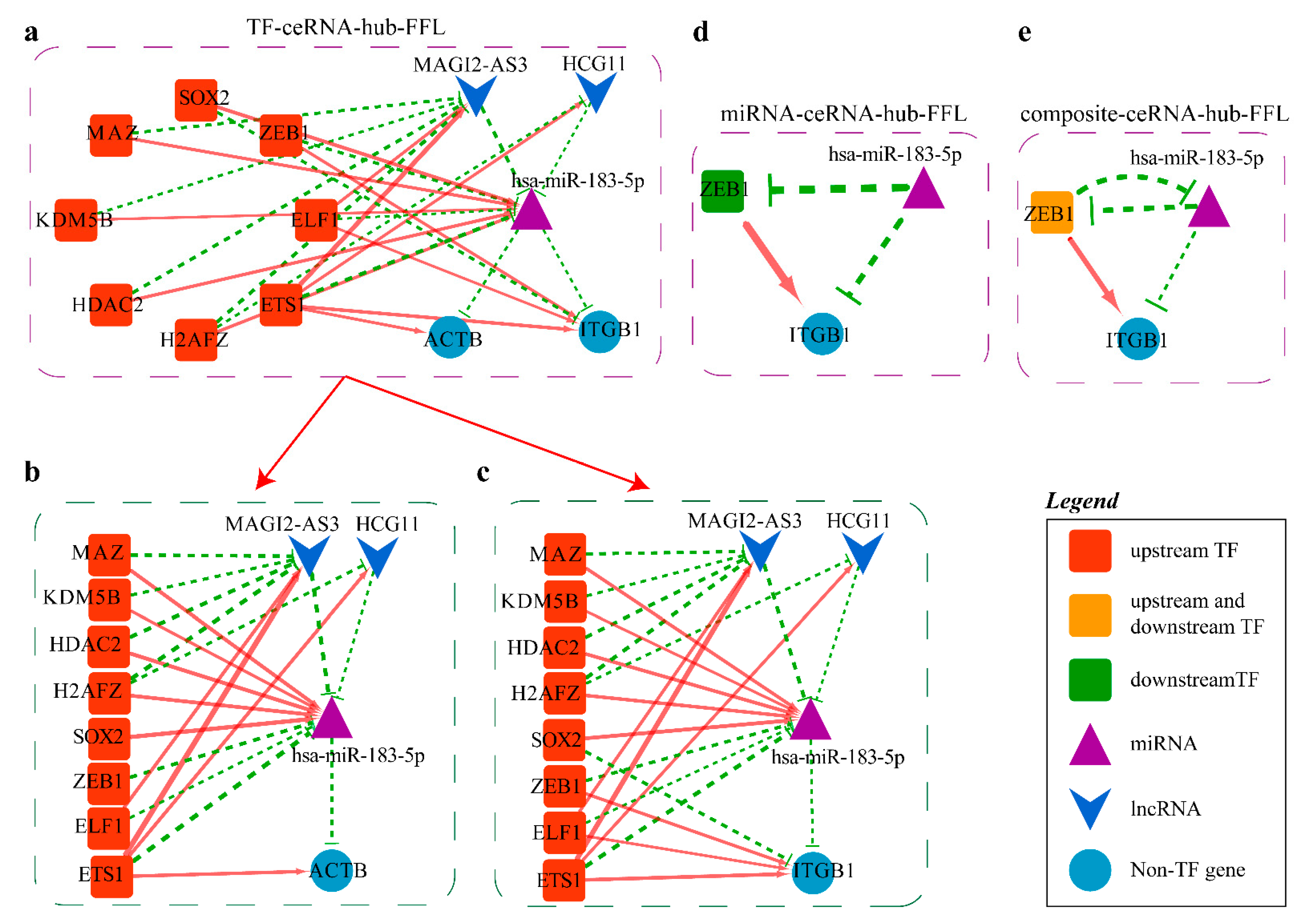
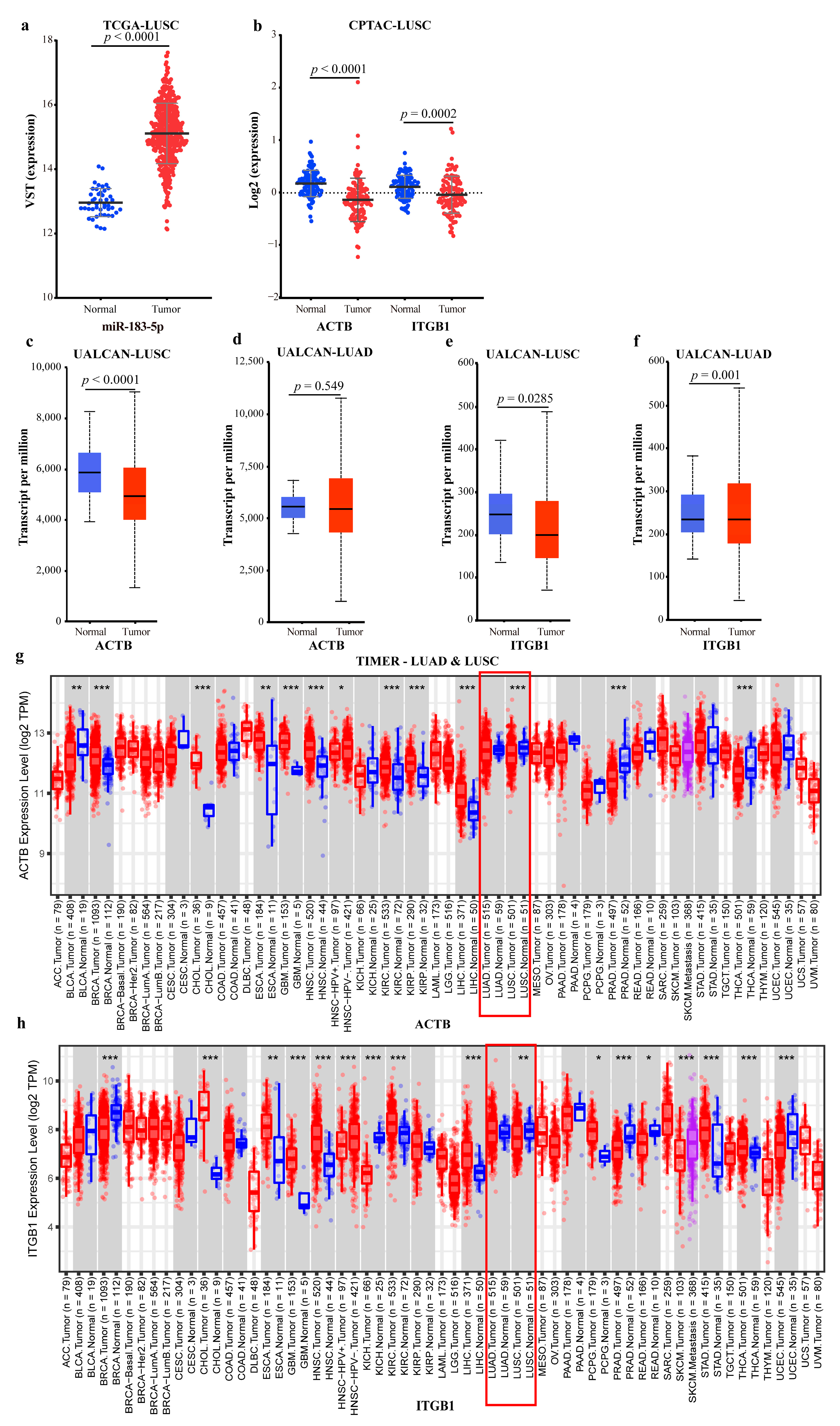
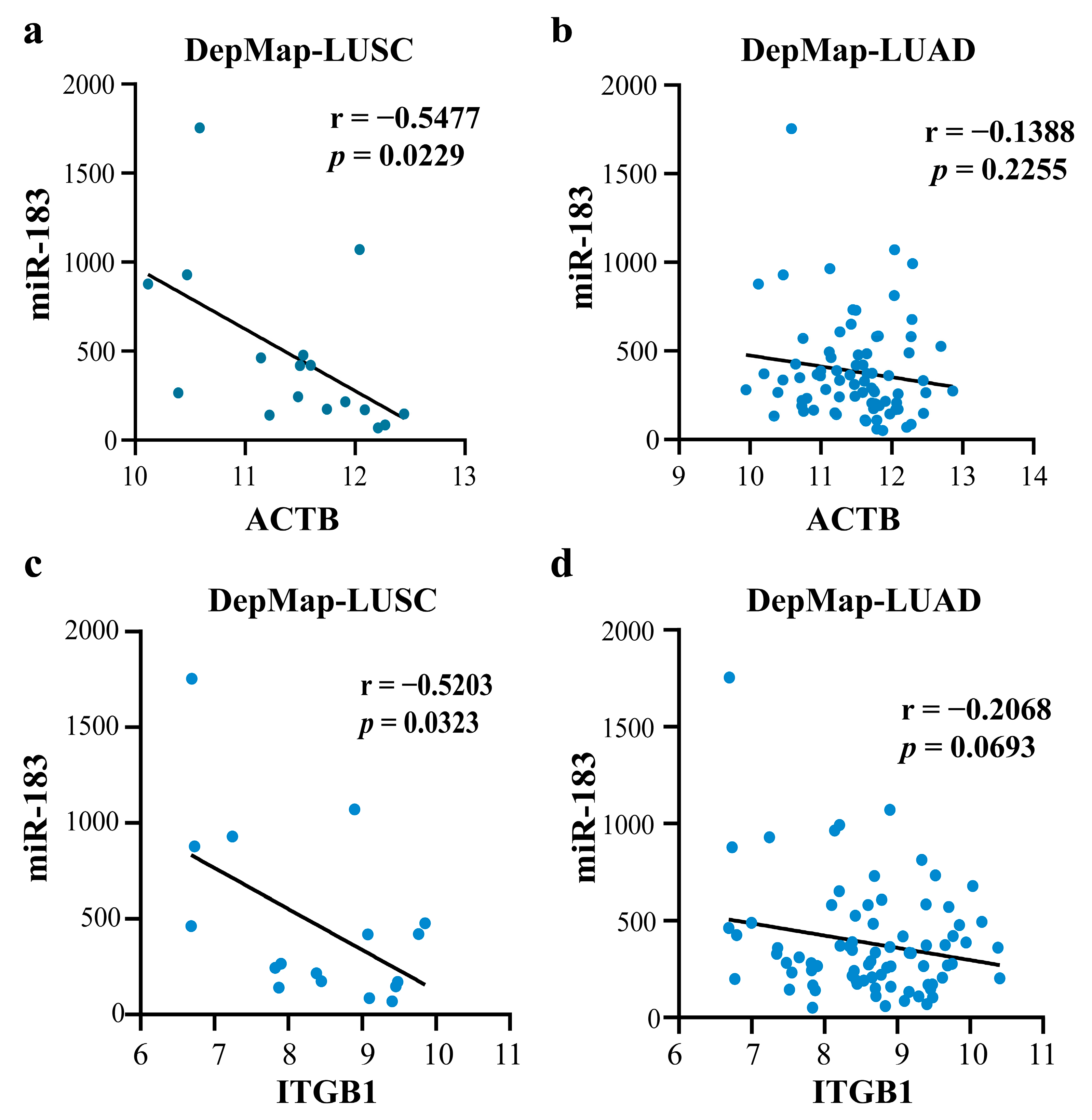
2.4.2. Mechanism Revealed by miR-183-5p Related ceRNA-hub-FFL Regulatory Subnetworks
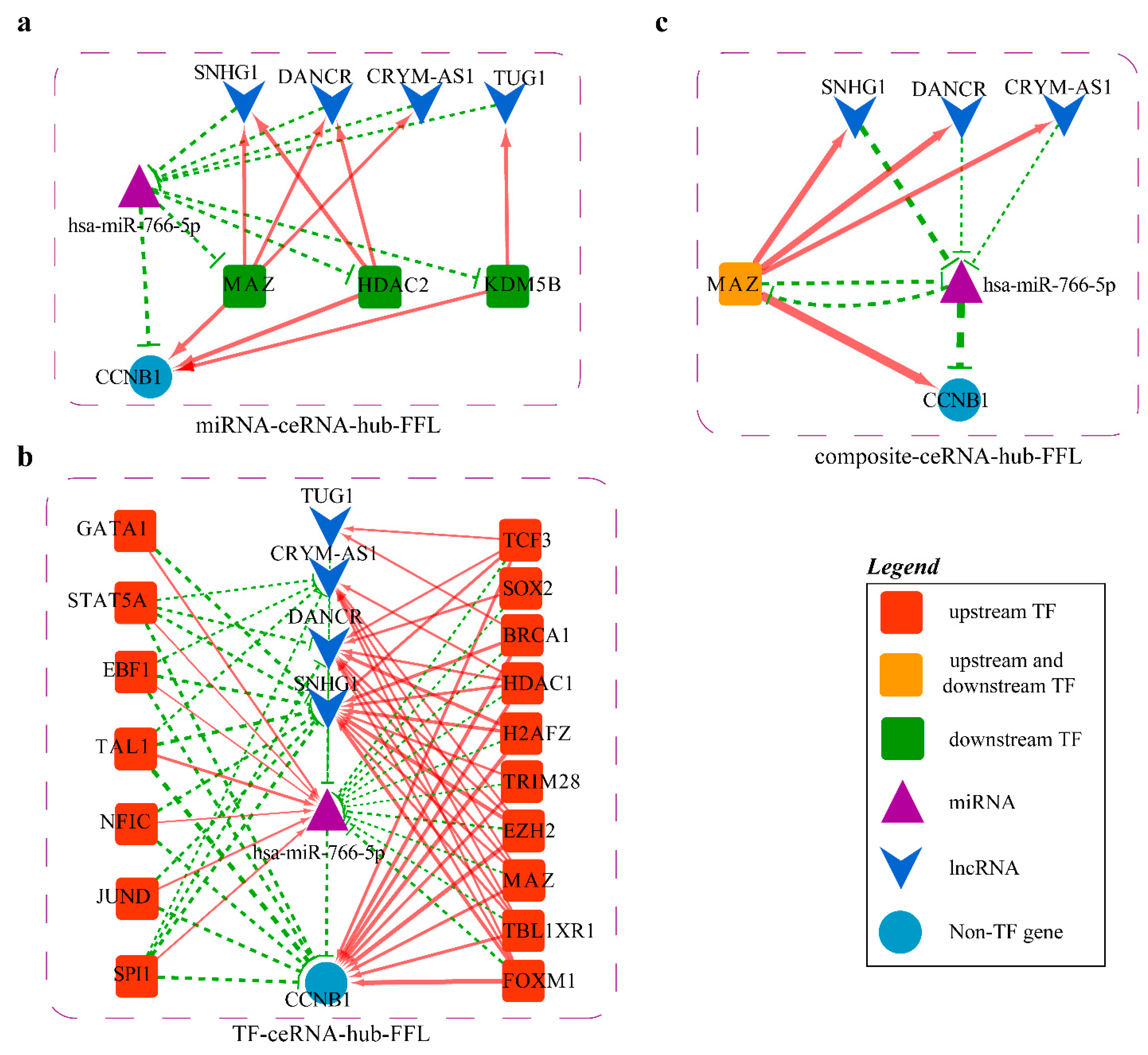
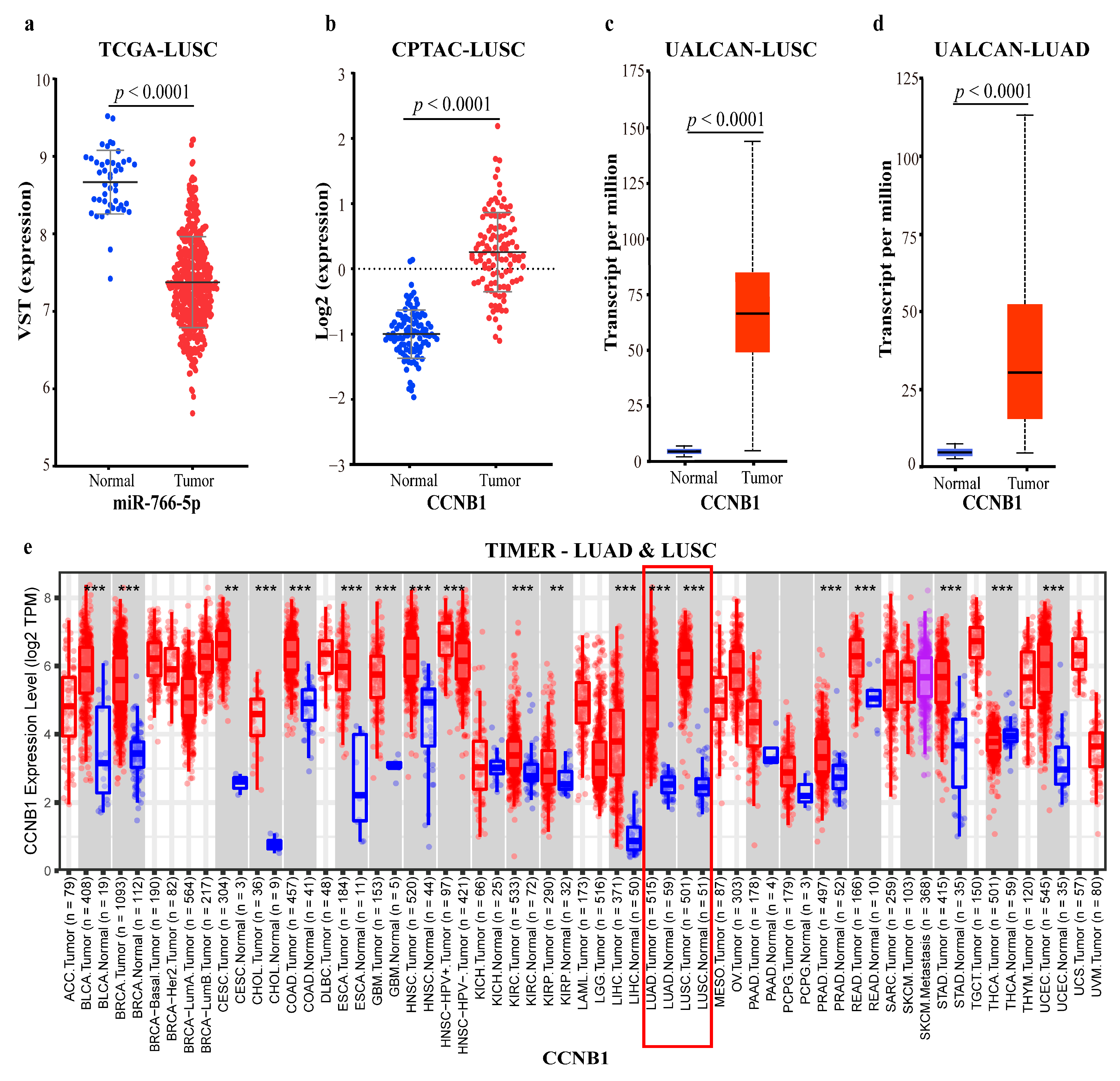
2.4.3. Mechanism Revealed by miR-766-5p Related ceRNA-hub-FFL Regulatory Subnetworks
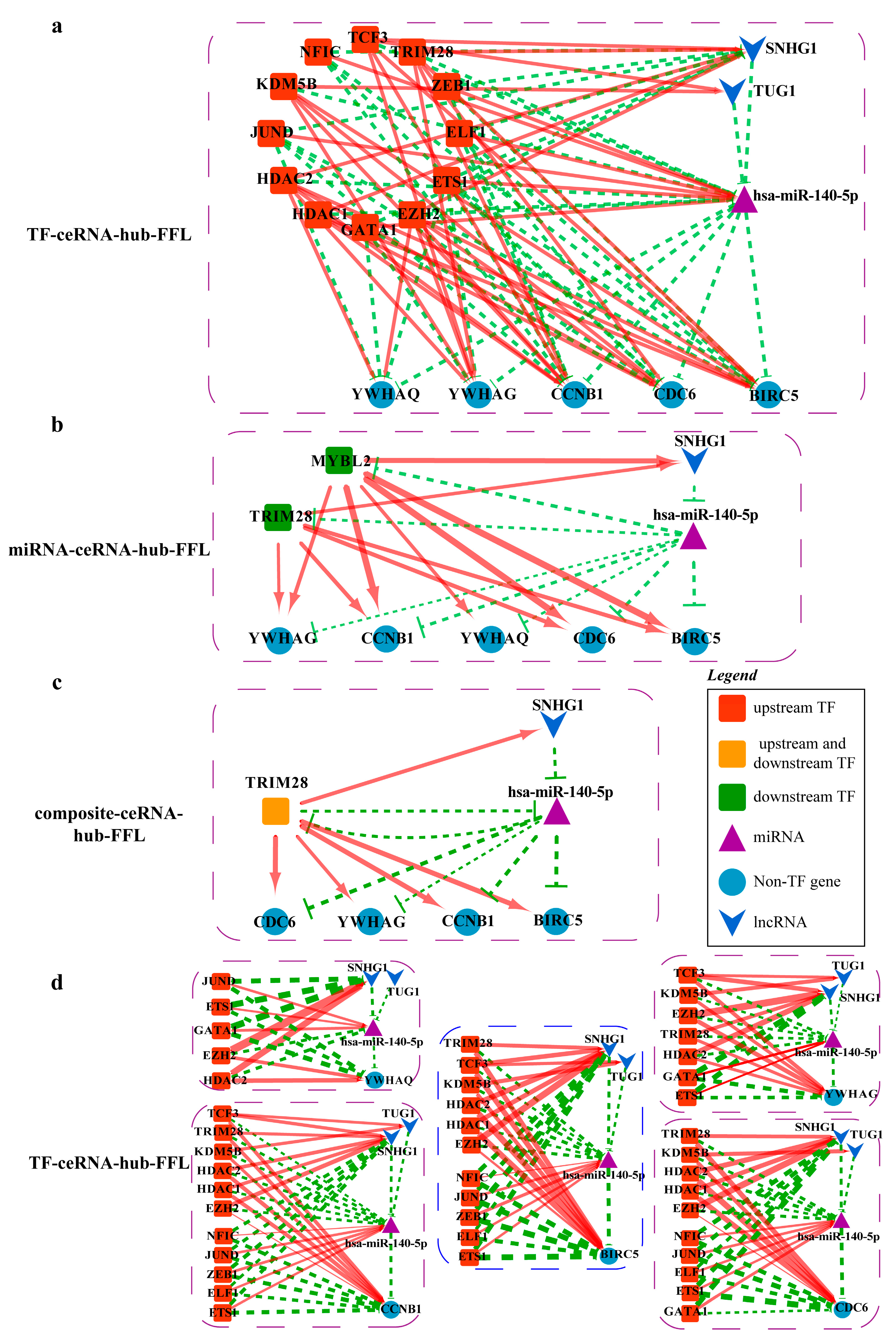
2.4.4. Mechanisms Revealed by miR-140-5p Related ceRNA-hub-FFL Regulatory Subnetworks
3. Discussion
4. Methods and Materials
4.1. Data Download and Pre-Processing
4.2. Differential Expression Analysis of mRNAs and miRNAs
4.3. Screening and Validation of miRNA as Biomarkers
4.3.1. Identification of miRNA Biomarkers Based on the Independent Regulation Model
- Construction of human miRNA-mRNA regulatory network
- Obtaining candidate biomarkers based on miRNA independent regulation model
4.3.2. Screening miRNA Biomarkers Based on the Biological Significance of Target Genes
- Obtaining the gene set most relevant to LC using WGCNA
- Screening LC-related genes based on databases of oncogenes and tumor suppressor genes
- Construction of the strong relationship pairs between LC-related genes and miRNAs
4.3.3. The Final miRNA Biomarkers for LC
4.3.4. Validating the Reliability and Rationality of Biomarkers
- Validation based on the existing literature
- Validation based on external datasets
4.4. Construction of 4-Node ceRNA-hub-FFL Network Based on miRNA Biomarkers
4.4.1. Identifying the Interaction Pairs between TFs, miRNAs, Genes, and lncRNAs and Calculating the Direction of Interactions
4.4.2. Construction of FFL Network
4.4.3. Obtaining hub-FFL Network
4.4.4. Extraction of ceRNA-hub-FFL Network
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Goldstraw, P.; Chansky, K.; Crowley, J.; Rami-Porta, R.; Asamura, H.; Eberhardt, W.E.; Nicholson, A.G.; Groome, P.; Mitchell, A.; Bolejack, V.; et al. The IASLC Lung Cancer Staging Project: Proposals for Revision of the TNM Stage Groupings in the Forthcoming (Eighth) Edition of the TNM Classification for Lung Cancer. J. Thorac. Oncol. 2016, 11, 39–51. [Google Scholar] [CrossRef]
- Ettinger, D.S.; Akerley, W.; Bepler, G.; Blum, M.G.; Chang, A.; Cheney, R.T.; Chirieac, L.R.; D’Amico, T.A.; Demmy, T.L.; Ganti, A.K.P.; et al. Non–Small Cell Lung Cancer. J. Natl. Compr. Cancer Netw. 2010, 8, 740–801. [Google Scholar] [CrossRef] [PubMed]
- Xi, K.; Wang, W.; Wen, Y.; Chen, Y.; Zhang, X.; Wu, Y.; Zhang, R.; Wang, G.; Huang, Z.; Zhang, L. Combining Plasma miRNAs and Computed Tomography Features to Differentiate the Nature of Pulmonary Nodules. Front. Oncol. 2019, 9, 975. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef]
- Jung, M.; Schaefer, A.; Steiner, I.; Kempkensteffen, C.; Stephan, C.; Erbersdobler, A.; Jung, K. Robust MicroRNA Stability in Degraded RNA Preparations from Human Tissue and Cell Samples. Clin. Chem. 2010, 56, 998–1006. [Google Scholar] [CrossRef]
- Huang, X.; Xiao, S.; Zhu, X.; Yu, Y.; Cao, M.; Zhang, X.; Li, S.; Zhu, W.; Wu, F.; Zheng, X.; et al. miR-196b-5p-mediated downregulation of FAS promotes NSCLC progression by activating IL6-STAT3 signaling. Cell Death Dis. 2020, 11, 758. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsson, A.; Dabrosin, C. Tissue specific expression of extracellular microRNA in human breast cancers and normal human breast tissue in vivo. Oncotarget 2015, 6, 22959–22969. [Google Scholar] [CrossRef]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.-L.; Chen, H.-Y.; Chang, G.-C.; Chen, C.-Y.; Chen, H.-W.; Singh, S.; Cheng, C.-L.; Yu, C.-J.; Lee, Y.-C.; Chen, H.-S.; et al. MicroRNA Signature Predicts Survival and Relapse in Lung Cancer. Cancer Cell 2008, 13, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Saikia, J.; Sharawat, S.K.; Malik, P.S.; Kumar, S.; Mohan, A. Analysis of miR-375-3p, miR-197-3p, and miR-15a-5p Expression and Their Clinical Relevance as Biomarkers in Lung Cancer. Technol. Cancer Res. Treat. 2022, 21, 15330338221080981. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, C.-X.; Lv, J.-Y.; Li, Y.-S.; Xiao, Y.; Shao, T.-T.; Huo, X.; Li, X.; Zou, Y.; Han, Q.-L.; et al. Prioritizing Candidate Disease miRNAs by Topological Features in the miRNA Target–Dysregulated Network: Case Study of Prostate Cancer. Mol. Cancer Ther. 2011, 10, 1857–1866. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Cao, F.; Liu, F.; Liu, S.; Meng, L.; Gu, L.; Zhao, H.; Sang, M.; Shan, B. Identification of potential circular RNA biomarkers in lung adenocarcinoma: A bioinformatics analysis and retrospective clinical study. Oncol. Lett. 2022, 23, 144. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, W.; Sun, Z.; Shen, L.; Shen, B. MiRNA-BD: An evidence-based bioinformatics model and software tool for microRNA biomarker discovery. RNA Biol. 2018, 15, 1093–1105. [Google Scholar] [CrossRef]
- Nie, R.; Niu, W.; Tang, T.; Zhang, J.; Zhang, X. Integrating microRNA expression, miRNA-mRNA regulation network and signal pathway: A novel strategy for lung cancer biomarker discovery. PeerJ 2021, 9, e12369. [Google Scholar] [CrossRef]
- Zhang, W.; Zang, J.; Jing, X.; Sun, Z.; Yan, W.; Yang, D.; Guo, F.; Shen, B. Identification of candidate miRNA biomarkers from miRNA regulatory network with application to prostate cancer. J. Transl. Med. 2014, 12, 66. [Google Scholar] [CrossRef]
- Zhu, Y.; Peng, Q.; Lin, Y.; Zou, L.; Shen, P.; Chen, F.; Min, M.; Shen, L.; Chen, J.; Shen, B. Identification of biomarker microRNAs for predicting the response of colorectal cancer to neoadjuvant chemoradiotherapy based on microRNA regulatory network. Oncotarget 2017, 8, 2233–2248. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Lin, Y.; Yan, W.; Sun, Z.; Jiang, Z.; Shen, B.; Jiang, X.; Shi, J. Novel Biomarker MicroRNAs for Subtyping of Acute Coronary Syndrome: A Bioinformatics Approach. BioMed Res. Int. 2016, 2016, 4618323. [Google Scholar] [CrossRef]
- Ding, D.; Zhang, J.; Luo, Z.; Wu, H.; Lin, Z.; Liang, W.; Xue, X. Analysis of the lncRNA–miRNA–mRNA Network Reveals a Potential Regulatory Mechanism of EGFR-TKI Resistance in NSCLC. Front. Genet. 2022, 13, 851391. [Google Scholar] [CrossRef]
- Hajheidari, M.; Huang, S.-S.C. Elucidating the biology of transcription factor–DNA interaction for accurate identification of cis-regulatory elements. Curr. Opin. Plant Biol. 2022, 68, 102232. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Jia, P.; Mallik, S.; Fei, R.; Yoshioka, H.; Suzuki, A.; Iwata, J.; Zhao, Z. Critical microRNAs and regulatory motifs in cleft palate identified by a conserved miRNA–TF–gene network approach in humans and mice. Brief. Bioinform. 2020, 21, 1465–1478. [Google Scholar] [CrossRef] [PubMed]
- Bracken, C.P.; Scott, H.S.; Goodall, G.J. A network-biology perspective of microRNA function and dysfunction in cancer. Nat. Rev. Genet. 2016, 17, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Mitra, R.; Lin, C.-C.; Wang, Q.; Cheng, F.; Zhao, Z. Systematic dissection of dysregulated transcription factor–miRNA feed-forward loops across tumor types. Brief. Bioinform. 2016, 17, 996–1008. [Google Scholar] [CrossRef]
- Somel, M.; Guo, S.; Fu, N.; Yan, Z.; Hu, H.Y.; Xu, Y.; Yuan, Y.; Ning, Z.; Hu, Y.; Menzel, C.; et al. MicroRNA, mRNA, and protein expression link development and aging in human and macaque brain. Genome Res. 2010, 20, 1207–1218. [Google Scholar] [CrossRef]
- Kong, L.-M.; Liao, C.-G.; Zhang, Y.; Xu, J.; Li, Y.; Huang, W.; Zhang, Y.; Bian, H.; Chen, Z.-N. A Regulatory Loop Involving miR-22, Sp1, and c-Myc Modulates CD147 Expression in Breast Cancer Invasion and Metastasis. Cancer Res. 2014, 74, 3764–3778. [Google Scholar] [CrossRef]
- Bhan, A.; Soleimani, M.; Mandal, S.S. Long Noncoding RNA and Cancer: A New Paradigm. Cancer Res. 2017, 77, 3965–3981. [Google Scholar] [CrossRef]
- Uchida, S.; Dimmeler, S. Long Noncoding RNAs in Cardiovascular Diseases. Circ. Res. 2015, 116, 737–750. [Google Scholar] [CrossRef]
- Goodall, G.J.; Wickramasinghe, V.O. RNA in cancer. Nat. Cancer 2021, 21, 22–36. [Google Scholar] [CrossRef]
- Kargi, H.; Kuyucuoğlu, M.; Alakavuklar, M.; Akpmar, O.; Erk, S. CD44 expression in metastatic and non-metastatic non-small cell lung cancers. Cancer Lett. 1997, 119, 27–30. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef]
- Saini, S.; Majid, S.; Shahryari, V.; Arora, S.; Yamamura, S.; Chang, I.; Zaman, M.S.; Deng, G.; Tanaka, Y.; Dahiya, R. miRNA-708 Control of CD44+ Prostate Cancer–Initiating Cells. Cancer Res. 2012, 72, 3618–3630. [Google Scholar] [CrossRef]
- Yin, J.; Zhang, H.; Wu, X.; Zhang, Y.; Li, J.; Shen, J.; Zhao, Y.; Xiao, Z.; Lu, L.; Huang, C.; et al. CD44 inhibition attenuates EGFR signaling and enhances cisplatin sensitivity in human EGFR wild-type non-small-cell lung cancer cells. Int. J. Mol. Med. 2020, 45, 1783–1792. [Google Scholar] [CrossRef]
- Dominguez, R.; Holmes, K.C. Actin Structure and Function. Annu. Rev. Biophys. 2011, 40, 169–186. [Google Scholar] [CrossRef]
- Tang, M.; Hu, Z.; Rao, C.; Chen, J.; Yuan, S.; Zhang, J.; Mao, C.; Yan, J.; Xia, Y.; Zhang, M.; et al. Burkholderia pseudomallei interferes with host lipid metabolism via NR1D2-mediated PNPLA2/ATGL suppression to block autophagy-dependent inhibition of infection. Autophagy 2021, 17, 1918–1933. [Google Scholar] [CrossRef]
- Latham, S.L.; Ehmke, N.; Reinke, P.Y.A.; Taft, M.H.; Eicke, D.; Reindl, T.; Stenzel, W.; Lyons, M.J.; Friez, M.J.; Lee, J.A.; et al. Variants in exons 5 and 6 of ACTB cause syndromic thrombocytopenia. Nat. Commun. 2018, 9, 4250. [Google Scholar] [CrossRef]
- Rubie, C.; Kempf, K.; Hans, J.; Su, T.; Tilton, B.; Georg, T.; Brittner, B.; Ludwig, B.; Schilling, M. Housekeeping gene variability in normal and cancerous colorectal, pancreatic, esophageal, gastric and hepatic tissues. Mol. Cell. Probes 2005, 19, 101–109. [Google Scholar] [CrossRef]
- Gu, Y.; Tang, S.; Wang, Z.; Cai, L.; Lian, H.; Shen, Y.; Zhou, Y. A pan-cancer analysis of the prognostic and immunological role of β-actin (ACTB) in human cancers. Bioengineered 2021, 12, 6166–6185. [Google Scholar] [CrossRef]
- Kuwada, S.K.; Kuang, J.; Li, X. Integrin α5/β1 Expression Mediates HER-2 Down-regulation in Colon Cancer Cells. J. Biol. Chem. 2005, 280, 19027–19035. [Google Scholar] [CrossRef] [Green Version]
- Lanzafame, S.; Emmanuele, C.; Torrisi, A. Correlation of α2ß1 Integrin Expression with Histological Type and Hormonal Receptor Status in Breast Carcinomas. Pathol. Res. Pactr. 1996, 192, 1031–1038. [Google Scholar] [CrossRef]
- Liu, Z.; Feng, J.-G.; Tuersun, A.; Liu, T.; Liu, H.; Liu, Q.; Zheng, S.-T.; Huang, C.-G.; Lv, G.-D.; Sheyhidin, I.; et al. Proteomic identification of differentially-expressed proteins in esophageal cancer in three ethnic groups in Xinjiang. Mol. Biol. Rep. 2011, 38, 3261–3269. [Google Scholar] [CrossRef]
- Saviozzi, S.; Cordero, F.; Iacono, M.L.; Novello, S.; Scagliotti, G.V.; Calogero, R.; Silvia, S.; Francesca, C.; Marco, L.I.; Silvia, N.; et al. Selection of suitable reference genes for accurate normalization of gene expression profile studies in non-small cell lung cancer. BMC Cancer 2006, 6, 200. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, X.; Zhang, Y.; Wang, Z. Curcumin suppresses the malignancy of non-small cell lung cancer by modulating the circ-PRKCA/miR-384/ITGB1 pathway. Biomed. Pharmacother. 2021, 138, 111439. [Google Scholar] [CrossRef]
- Zhao, M.; Tong, C.; Hao, Z.; Zhao, R.; Wang, L. MicroRNA -374b mediates the initiation of non-small cell lung cancer by regulating ITGB1 and p53 expressions. Thorac. Cancer 2020, 11, 1670–1678. [Google Scholar] [CrossRef]
- Deng, H.; Huang, Y.; Wang, L.; Chen, M. High Expression of UBB, RAC1, and ITGB1 Predicts Worse Prognosis among Nonsmoking Patients with Lung Adenocarcinoma through Bioinformatics Analysis. BioMed Res. Int. 2020, 2020, 2071593. [Google Scholar] [CrossRef]
- Sadek, K.H.; Cagampang, F.R.; Bruce, K.D.; Macklon, N.; Cheong, Y. Variation in stability of housekeeping genes in healthy and adhesion-related mesothelium. Fertil. Steril. 2012, 98, 1023–1027. [Google Scholar] [CrossRef]
- Oh, H.-N.; Kwak, A.-W.; Lee, M.-H.; Kim, E.; Yoon, G.; Cho, S.-S.; Liu, K.; Chae, J.-I.; Shim, J.-H. Targeted inhibition of c-MET by podophyllotoxin promotes caspase-dependent apoptosis and suppresses cell growth in gefitinib-resistant non-small cell lung cancer cells. Phytomedicine 2021, 80, 153355. [Google Scholar] [CrossRef]
- Park, C.; Cha, H.-J.; Lee, H.; Hwang-Bo, H.; Ji, S.Y.; Kim, M.Y.; Hong, S.H.; Jeong, J.-W.; Han, M.H.; Choi, S.H.; et al. Induction of G2/M Cell Cycle Arrest and Apoptosis by Genistein in Human Bladder Cancer T24 Cells through Inhibition of the ROS-Dependent PI3k/Akt Signal Transduction Pathway. Antioxidants 2019, 8, 327. [Google Scholar] [CrossRef]
- Zha, J.; Harada, H.; Yang, E.; Jockel, J.; Korsmeyer, S.J. Serine Phosphorylation of Death Agonist BAD in Response to Survival Factor Results in Binding to 14-3-3 Not BCL-XL. Cell 1996, 87, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Aitken, A. 14-3-3 proteins: A historic overview. Semin. Cancer Biol. 2006, 16, 162–172. [Google Scholar] [CrossRef]
- Com, E.; Clavreul, A.; Lagarrigue, M.; Michalak, S.; Menei, P.; Pineau, C. Quantitative proteomic Isotope-Coded Protein Label (ICPL) analysis reveals alteration of several functional processes in the glioblastoma. J. Proteom. 2012, 75, 3898–3913. [Google Scholar] [CrossRef]
- Kim, J.-O.; Kim, S.-R.; Lim, K.-H.; Kim, J.-H.; Ajjappala, B.; Lee, H.-J.; Choi, J.-I.; Baek, K.-H. Deubiquitinating enzyme USP37 regulating oncogenic function of 14-3-3γ. Oncotarget 2015, 6, 36551–36576. [Google Scholar] [CrossRef]
- Wang, P.; Deng, Y.; Fu, X. MiR-509-5p suppresses the proliferation, migration, and invasion of non-small cell lung cancer by targeting YWHAG. Biochem. Biophys. Res. Commun. 2017, 482, 935–941. [Google Scholar] [CrossRef]
- Liu, J.; Lu, Y.; Huang, W.; He, Z. Comprehensive Analysis of Inhibitor of Apoptosis Protein Expression and Prognostic Significance in Non–Small Cell Lung Cancer. Front. Genet. 2021, 12, 764270. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Y.; Jia, Y.; Zhang, Q.; Zhu, P.; Ma, X. α5-nAChR and survivin: Two potential biological targets in lung adenocarcinoma. J. Cell. Physiol. 2021, 236, 1787–1797. [Google Scholar] [CrossRef]
- Mazur, J.; Roy, K.; Kanwar, J.R. Recent advances in nanomedicine and survivin targeting in brain cancers. Nanomedicine 2018, 13, 105–137. [Google Scholar] [CrossRef]
- Nitschkowski, D.; Marwitz, S.; Kotanidou, S.A.; Reck, M.; Kugler, C.; Rabe, K.F.; Ammerpohl, O.; Goldmann, T. Live and let die: Epigenetic modifications of Survivin and Regucalcin in non-small cell lung cancer tissues contribute to malignancy. Clin. Epigenetics 2019, 11, 157. [Google Scholar] [CrossRef]
- Faghihloo, E.; Sadeghizadeh, M.; Shahmahmoodi, S.; Mokhtari-Azad, T. Cdc6 expression is induced by HPV16 E6 and E7 oncogenes and represses E-cadherin expression. Cancer Gene Ther. 2016. online ahead of print. [Google Scholar] [CrossRef]
- Borlado, L.R.; Méndez, J. CDC6: From DNA replication to cell cycle checkpoints and oncogenesis. Carcinogenesis 2008, 29, 237–243. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Lian, T.; Fan, W.; Zhang, G.; Chen, Z.; Gou, X.; Jha, R.K. Long-Noncoding RNA CASC9 Promotes Progression of Non-Small Cell Lung Cancer by Promoting the Expression of CDC6 Through Binding to HuR. Cancer Manag. Res. 2020, 12, 9033–9043. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Mitra, R.; Maulik, U.; Zhang, M.Q. Development of the human cancer microRNA network. Silence 2010, 1, 6–14. [Google Scholar] [CrossRef]
- Lin, Y.; Qian, F.; Shen, L.; Chen, F.; Chen, J.; Shen, B. Computer-aided biomarker discovery for precision medicine: Data resources, models and applications. Brief. Bioinform. 2019, 20, 952–975. [Google Scholar] [CrossRef]
- Santo, G.D.; Frasca, M.; Bertoli, G.; Castiglioni, I.; Cava, C. Identification of key miRNAs in prostate cancer progression based on miRNA-mRNA network construction. Comput. Struct. Biotechnol. J. 2022, 20, 864–873. [Google Scholar] [CrossRef]
- Zhang, Q.; Kang, L.; Lingkai, K.; Li, Z.; Wen, S.; Fu, X. Bioinformatics Analysis Predicts hsa_circ_0026337/miR-197-3p as a Potential Oncogenic ceRNA Network for Non-small Cell Lung Cancers. Anti-Cancer Agents Med. Chem. 2022, 22, 874–886. [Google Scholar] [CrossRef]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A Long Noncoding RNA Controls Muscle Differentiation by Functioning as a Competing Endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar] [CrossRef]
- Hobert, O. Gene Regulation by Transcription Factors and MicroRNAs. Science 2008, 319, 1785–1786. [Google Scholar] [CrossRef]
- Crudele, F.; Bianchi, N.; Reali, E.; Galasso, M.; Agnoletto, C.; Volinia, S. The network of non-coding RNAs and their molecular targets in breast cancer. Mol. Cancer 2020, 19, 61. [Google Scholar] [CrossRef]
- Schulte-Sasse, R.; Budach, S.; Hnisz, D.; Marsico, A. Integration of multiomics data with graph convolutional networks to identify new cancer genes and their associated molecular mechanisms. Nat. Mach. Intell. 2021, 3, 513–526. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Liu, W. Roles of circRNAs in the Tumorigenesis and Metastasis of HCC: A Mini Review. Cancer Manag. Res. 2022, 14, 1847–1856. [Google Scholar] [CrossRef]
- Xia, X.; Li, X.; Li, F.; Wu, X.; Zhang, M.; Zhou, H.; Huang, N.; Yang, X.; Xiao, F.; Liu, D.; et al. A novel tumor suppressor protein encoded by circular AKT3 RNA inhibits glioblastoma tumorigenicity by competing with active phosphoinositide-dependent Kinase-1. Mol. Cancer 2019, 18, 131. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Wang, Y.; Hao, Y.; Juan, L.; Teng, M.; Zhang, X.; Li, M.; Wang, G.; Liu, Y. miR2Disease: A manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009, 37, D98–D104. [Google Scholar] [CrossRef]
- Xiao, F.; Zuo, Z.; Cai, G.; Kang, S.; Gao, X.; Li, T. miRecords: An integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009, 37, D105–D110. [Google Scholar] [CrossRef]
- Huang, H.-Y.; Lin, Y.-C.; Cui, S.; Huang, Y.; Tang, Y.; Xu, J.; Bao, J.; Li, Y.; Wen, J.; Zuo, H.; et al. miRTarBase update 2022: An informative resource for experimentally validated miRNA–target interactions. Nucleic Acids Res. 2021, 50, D222–D230. [Google Scholar] [CrossRef]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA–gene interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. miRWalk: An online resource for prediction of microRNA binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Jiao, X.; Sherman, B.T.; Huang, D.W.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. DAVID-WS: A stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012, 28, 1805–1806. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Kim, P.; Mitra, R.; Zhao, J.; Zhao, Z. TSGene 2.0: An updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res. 2016, 44, D1023–D1031. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, J.; Zhao, M. ONGene: A literature-based database for human oncogenes. J. Genet. Genom. 2017, 44, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Kulakovskiy, I.V.; Vorontsov, I.E.; Yevshin, I.S.; Sharipov, R.N.; Fedorova, A.D.; Rumynskiy, E.I.; Medvedeva, Y.A.; Magana-Mora, A.; Bajic, V.B.; Papatsenko, D.A.; et al. HOCOMOCO: Towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2017, 46, D252–D259. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Miao, Y.-R.; Jia, L.-H.; Yu, Q.-Y.; Zhang, Q.; Guo, A.-Y. AnimalTFDB 3.0: A comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res. 2019, 47, D33–D38. [Google Scholar] [CrossRef]
- Han, H.; Cho, J.-W.; Lee, S.-Y.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef]
- The ENCODE Project Consortium. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Matys, V.; Kel-Margoulis, O.V.; Fricke, E.; Liebich, I.; Land, S.; Barre-Dirrie, A.; Reuter, I.; Chekmenev, D.; Krull, M.; Hornischer, K.; et al. TRANSFAC(R) and its module TRANSCompel(R): Transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006, 34, D108–D110. [Google Scholar] [CrossRef]
- Keenan, A.B.; Torre, D.; Lachmann, A.; Leong, A.K.; Wojciechowicz, M.L.; Utti, V.; Jagodnik, K.M.; Kropiwnicki, E.; Wang, Z.; Ma’Ayan, A. ChEA3: Transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 2019, 47, W212–W224. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.; Cui, Q.; Wang, J.; Zhou, Y. TransmiR v2.0: An updated transcription factor-microRNA regulation database. Nucleic Acids Res. 2019, 47, D253–D258. [Google Scholar] [CrossRef]
- Bovolenta, L.A.; Acencio, M.L.; Lemke, N. HTRIdb: An open-access database for experimentally verified human transcriptional regulation interactions. BMC Genom. 2012, 13, 405. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-H.; Liu, S.; Zhou, H.; Qu, L.-H.; Yang, J.-H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [PubMed]
- Karagkouni, D.; Paraskevopoulou, M.D.; Tastsoglou, S.; Skoufos, G.; Karavangeli, A.; Pierros, V.; Zacharopoulou, E.; Hatzigeorgiou, A.G. DIANA-LncBase v3: Indexing experimentally supported miRNA targets on non-coding transcripts. Nucleic Acids Res. 2020, 48, D101–D110. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Bhattacharyya, M. PuTmiR: A database for extracting neighboring transcription factors of human microRNAs. BMC Bioinform. 2010, 11, 190. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Lin, Y.; Chen, J.; Shen, B. Decoding competing endogenous RNA networks for cancer biomarker discovery. Brief. Bioinform. 2020, 21, 441–457. [Google Scholar] [CrossRef] [PubMed]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA ID | NOG Value | TFP Value | Important Gene | NOG Gene | AUC |
|---|---|---|---|---|---|
| miR-101-3p | 8 | 0.177 | EMP1, KLF6 | OCIAD2, BRIP1, NPC1L1, KL, FOXF1, AJAP1, NKX6-1, MAOB | 0.838 |
| miR-708-5p | 6 | 0.1628 | SLIT2 | JAM2, TMEM88, MAG, KANK4, DNAH10, FAM107A | 0.894 |
| miRNA ID | NOG Value | TFP Value | Important Gene | NOG Gene | AUC |
|---|---|---|---|---|---|
| miR-101-3p | 12 | 0.1572 | FHL1, KLF2 | SNX31, CNTN4, PHACTR3, RNASE4, RNASE1, KL, FOXF1, C17orf104, KRT10, CACNA1D, ADD2, MAOB | 0.792 |
| miR-140-5p | 7 | 0.2195 | BIRC5, CCNB1 | TEX19, RASL11B, GRIN1, SRD5A1, FAM162A, ADA, CADPS2 | 0.874 |
| miR-183-5p | 7 | 0.16 | FOS, CAV1, KLF6 | E2F8, RAI2, HIST1H2AI, GBP4, DNAH3, FAM83A, FMN1 | 0.976 |
| miR-766-5p | 10 | 0.1667 | CCNB1 | EFR3B, NPM3, CALCOCO1, CYP27C1, ASIC1, IL16, PCDHA3, GPBAR1, PPFIA4, KCTD1 | 0.957 |
| miR-766-3p | 16 | 0.1587 | DLC1 | SP8, TRIM45, TRIM15, TTBK1, TTC25, IL1RL2, RCCD1, TEKT1, HIST2H2AB, ZNF670, CENPH, TRPV3, CGNL1, VAMP5, KIRREL2, TNNT1 | 0.973 |
| miRNA ID | LUAD | LUSC | NSCLC | Subtype Study | Total |
|---|---|---|---|---|---|
| miR-708-5p | 1 | 1 | 6 | 1 | 9 |
| miR-766-3p | 1 | - | 1 | - | 2 |
| miR-766-5p | 1 | - | - | - | 1 |
| miR-183-5p | 12 | 2 | 19 | - | 33 |
| miR-140-5p | 4 | - | 19 | 1 | 24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Nie, R.; Liu, M.; Zhang, X. A Novel Strategy for Identifying NSCLC MicroRNA Biomarkers and Their Mechanism Analysis Based on a Brand-New CeRNA-Hub-FFL Network. Int. J. Mol. Sci. 2022, 23, 11303. https://doi.org/10.3390/ijms231911303
Zhang J, Nie R, Liu M, Zhang X. A Novel Strategy for Identifying NSCLC MicroRNA Biomarkers and Their Mechanism Analysis Based on a Brand-New CeRNA-Hub-FFL Network. International Journal of Molecular Sciences. 2022; 23(19):11303. https://doi.org/10.3390/ijms231911303
Chicago/Turabian StyleZhang, Jin, Renqing Nie, Mengxi Liu, and Xiaoyi Zhang. 2022. "A Novel Strategy for Identifying NSCLC MicroRNA Biomarkers and Their Mechanism Analysis Based on a Brand-New CeRNA-Hub-FFL Network" International Journal of Molecular Sciences 23, no. 19: 11303. https://doi.org/10.3390/ijms231911303
APA StyleZhang, J., Nie, R., Liu, M., & Zhang, X. (2022). A Novel Strategy for Identifying NSCLC MicroRNA Biomarkers and Their Mechanism Analysis Based on a Brand-New CeRNA-Hub-FFL Network. International Journal of Molecular Sciences, 23(19), 11303. https://doi.org/10.3390/ijms231911303