
Deep Learning Approaches for Detection of Breast Adenocarcinoma Causing Carcinogenic Mutations



Abstract
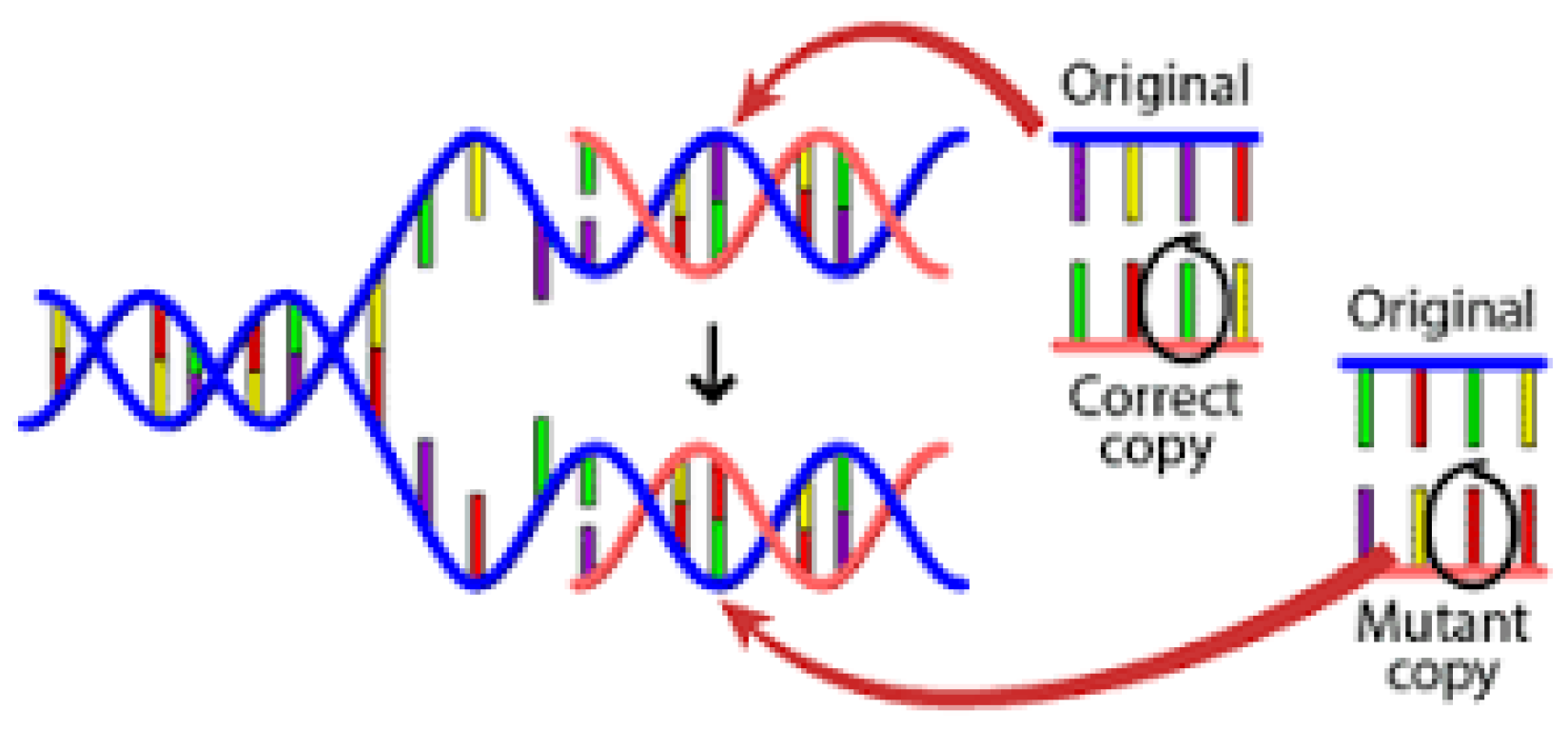
:1. Introduction
2. Results
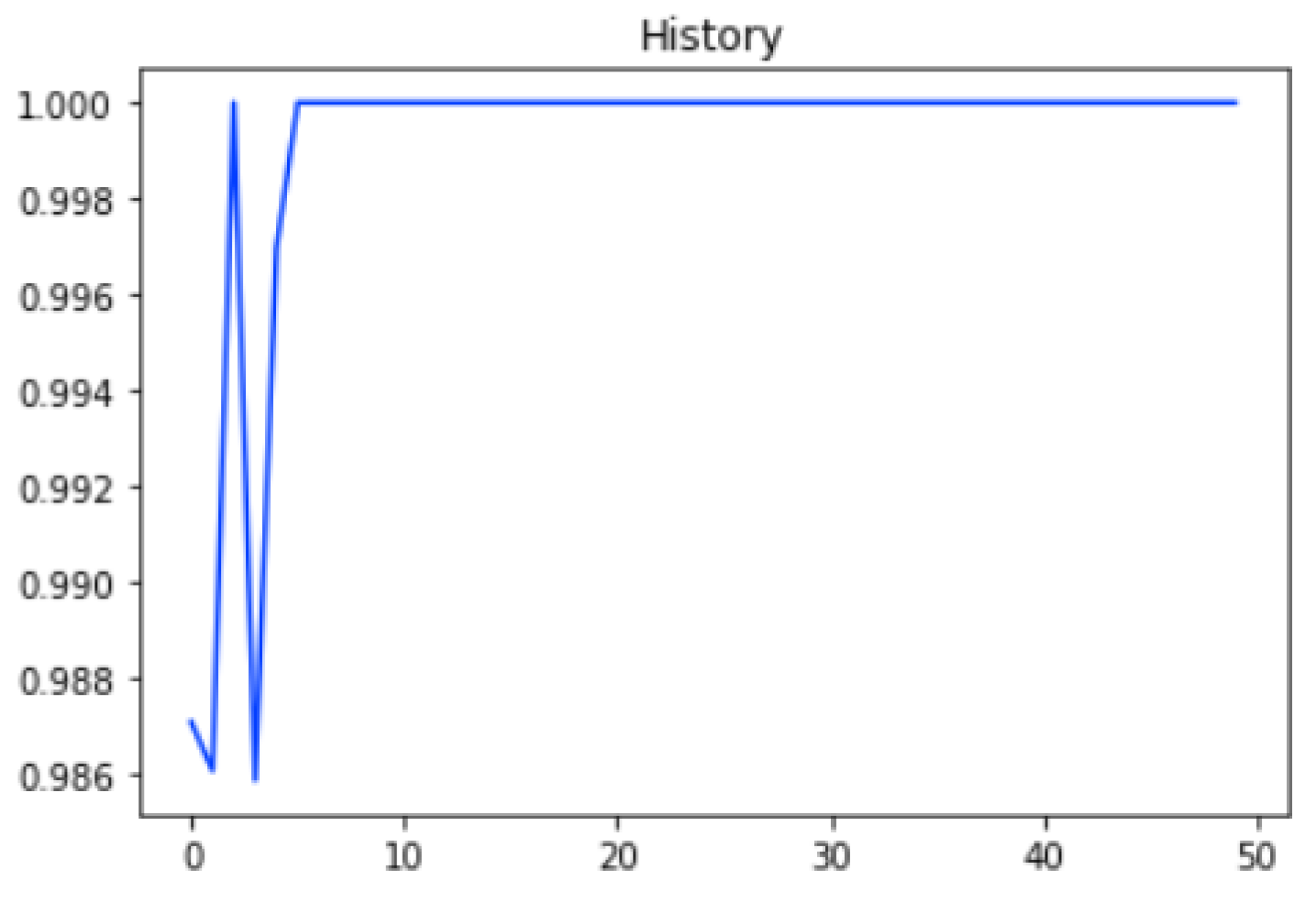
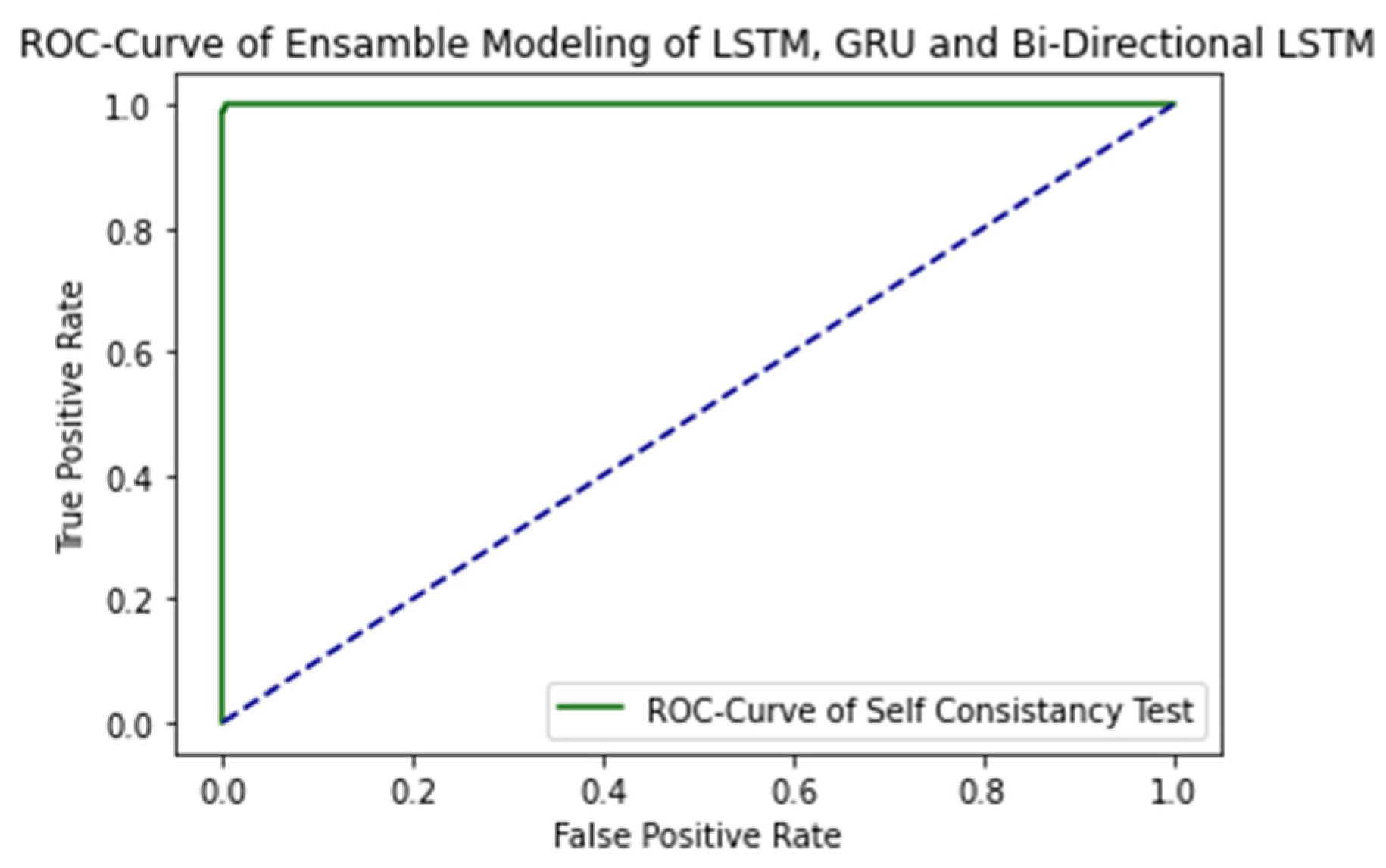
2.1. Self-Consistency Testing
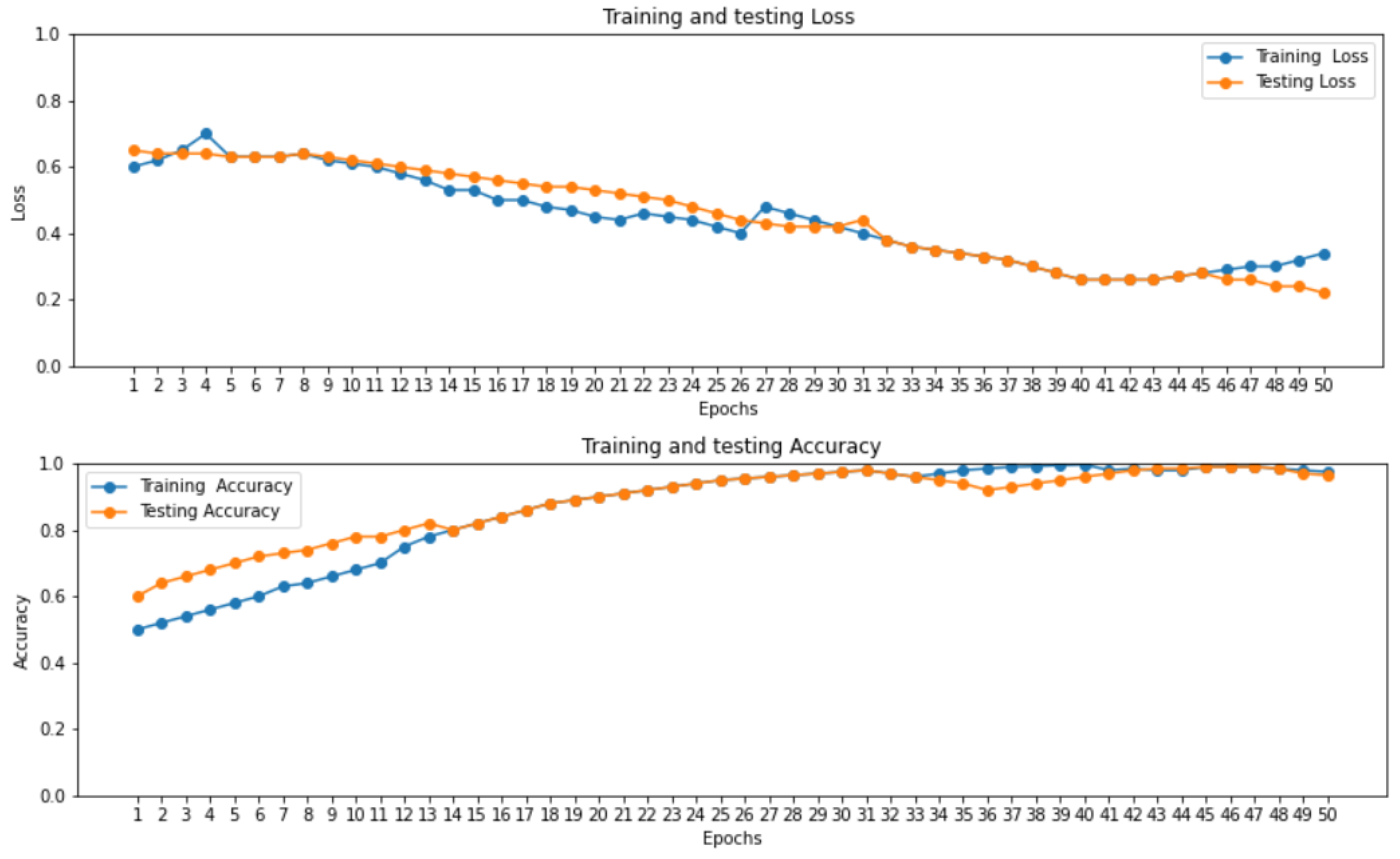
2.2. Independent Set Testing
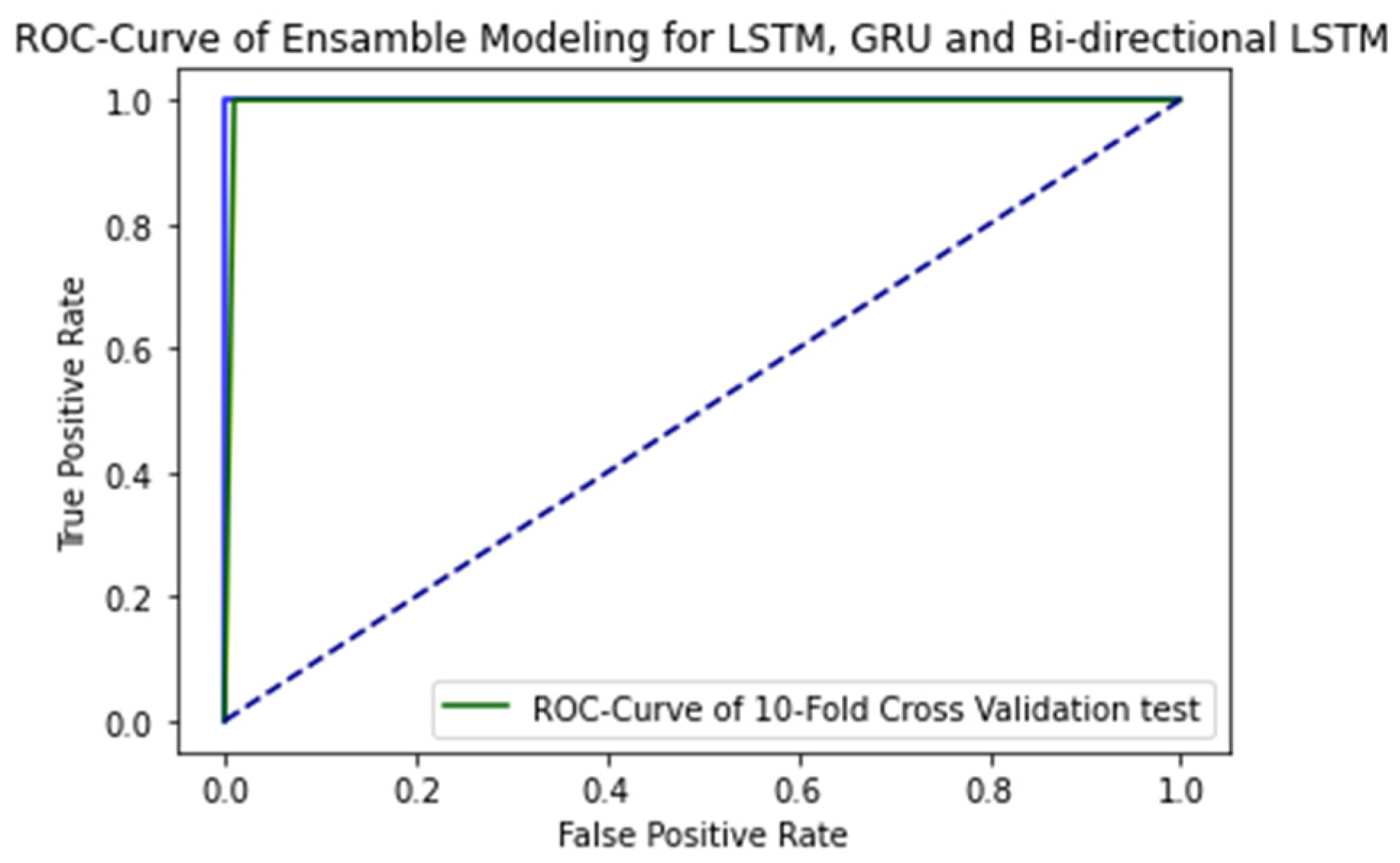
2.3. 10-Fold Cross-Validation Test
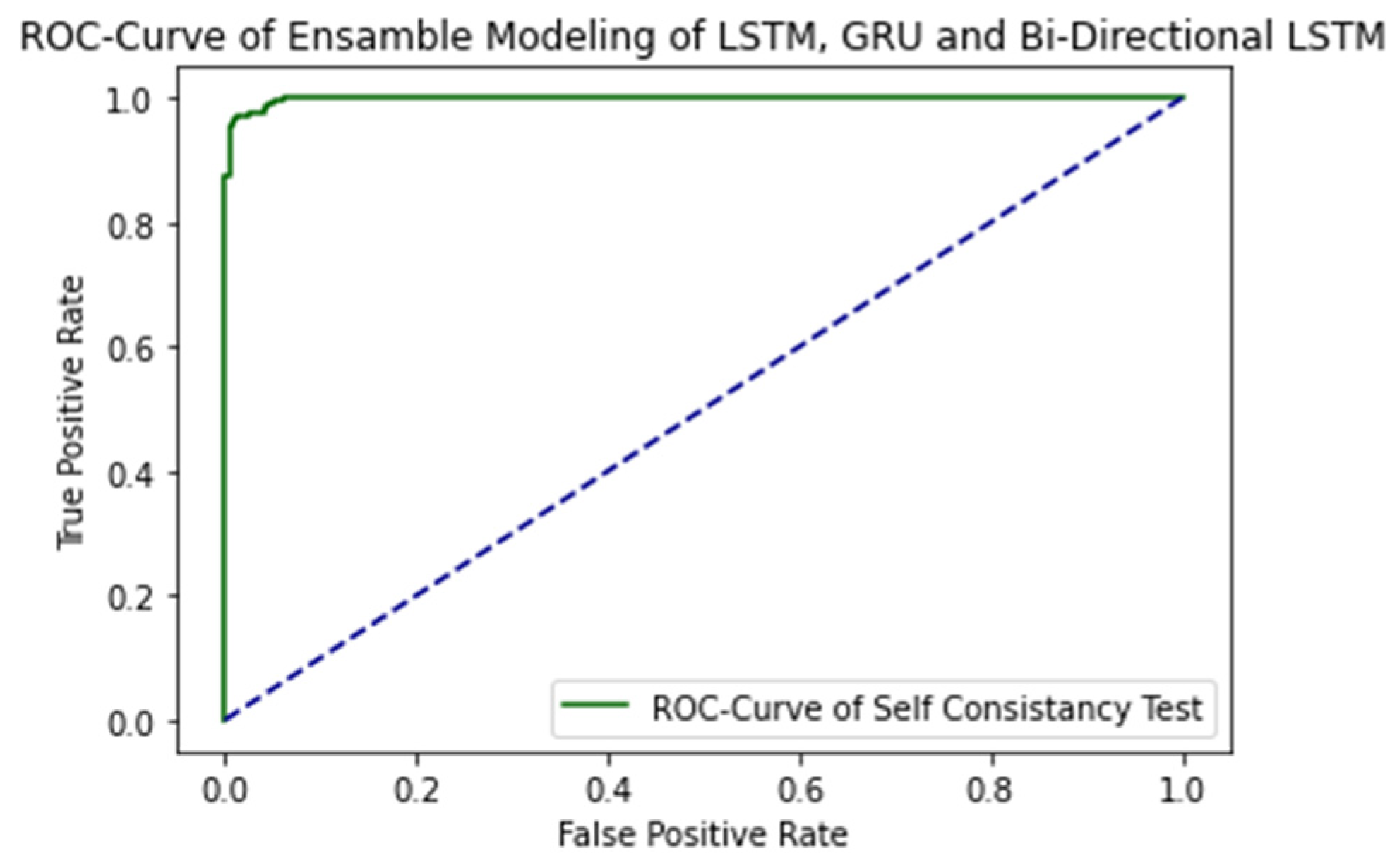
2.4. Results Comparison
3. Analysis and Discussion
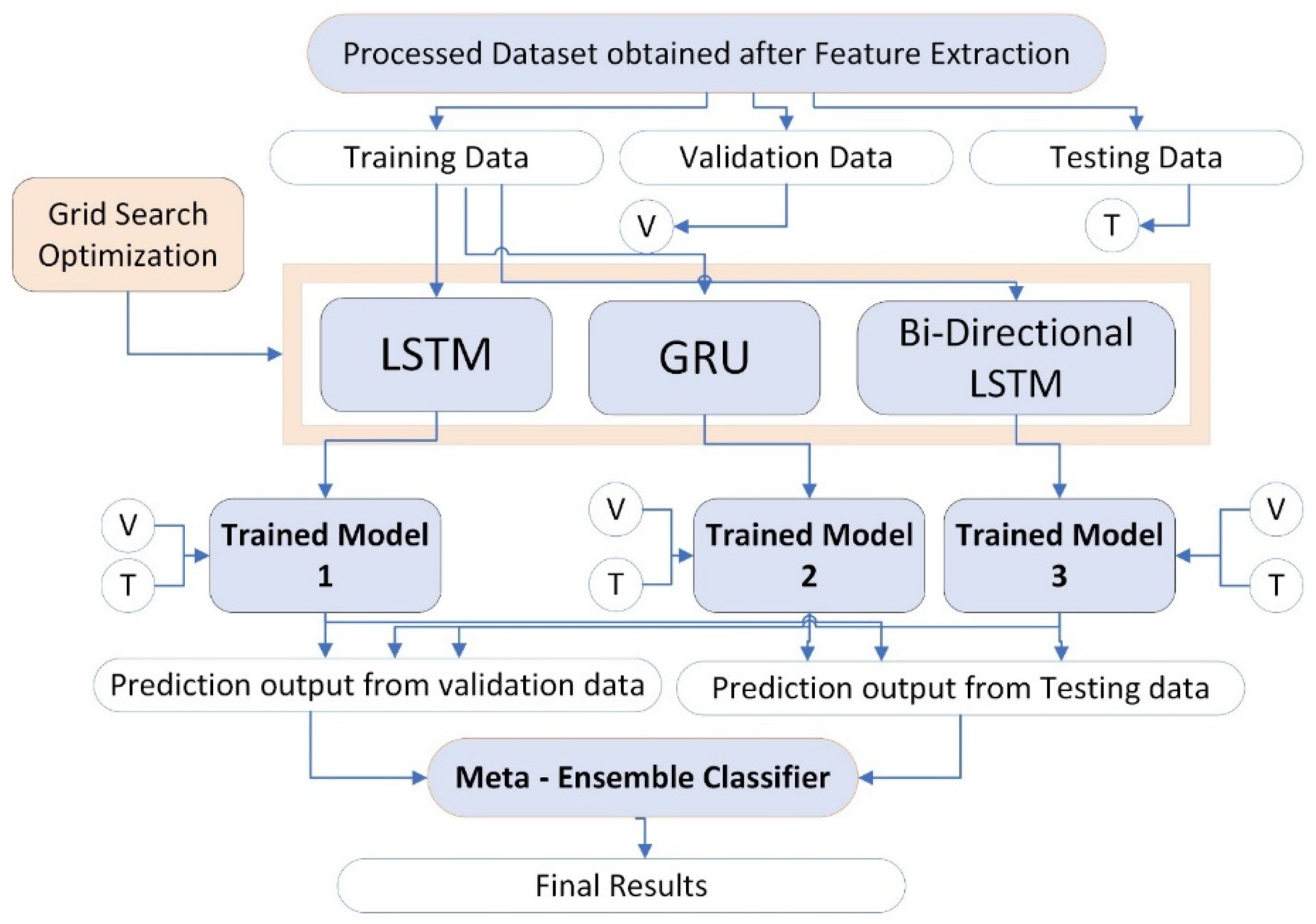
4. Materials and Methods
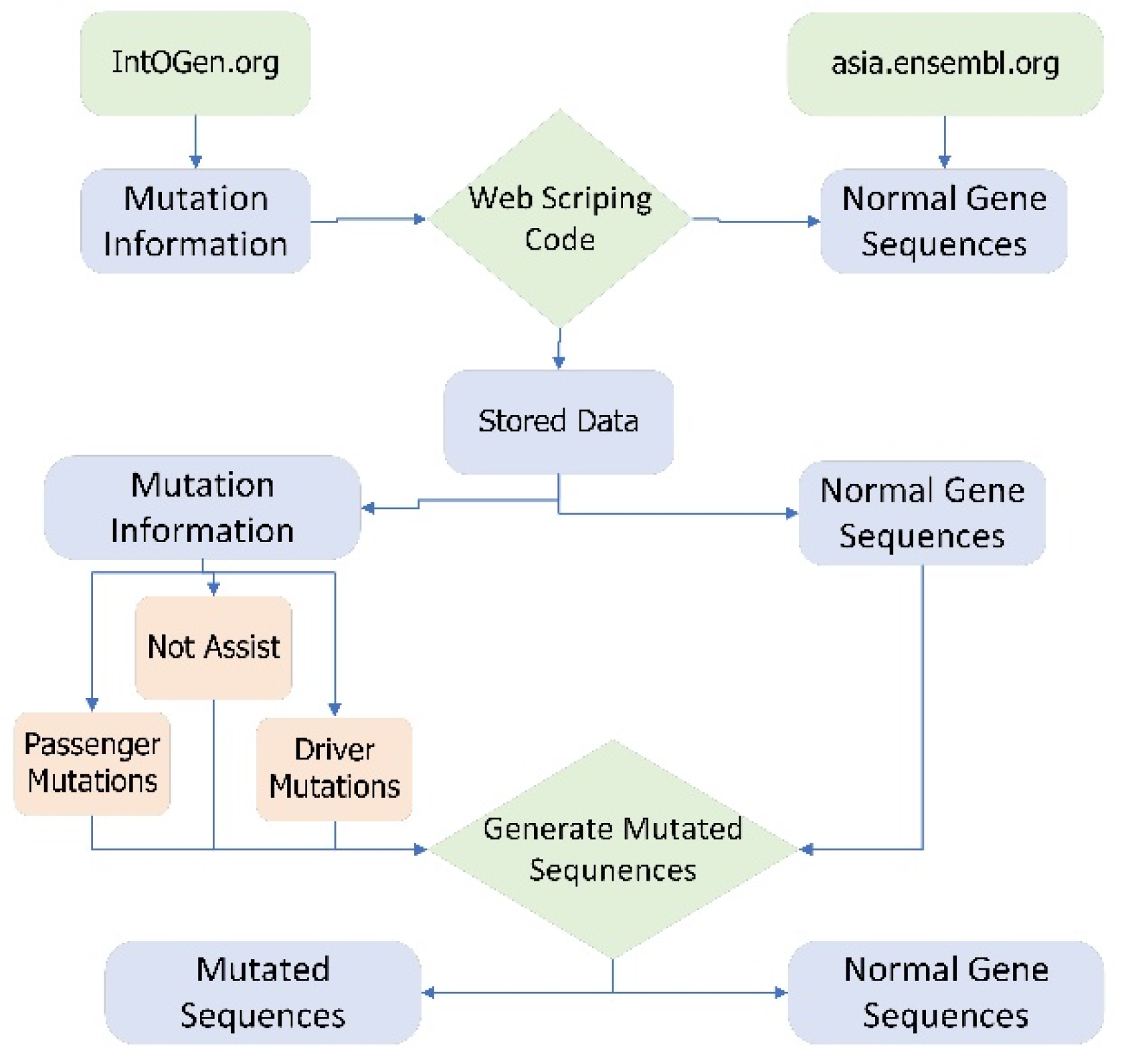
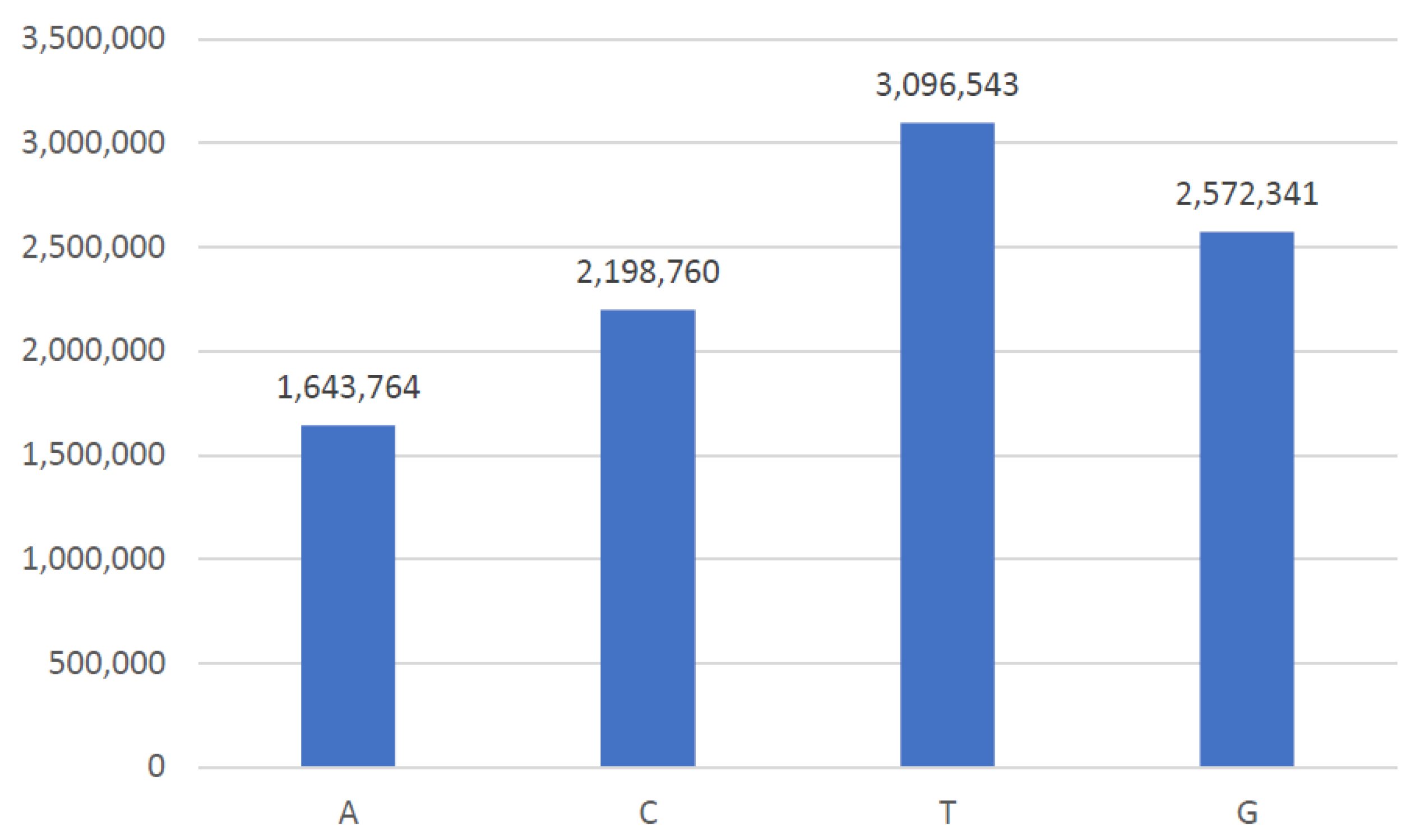
4.1. Data Acquisition Framework
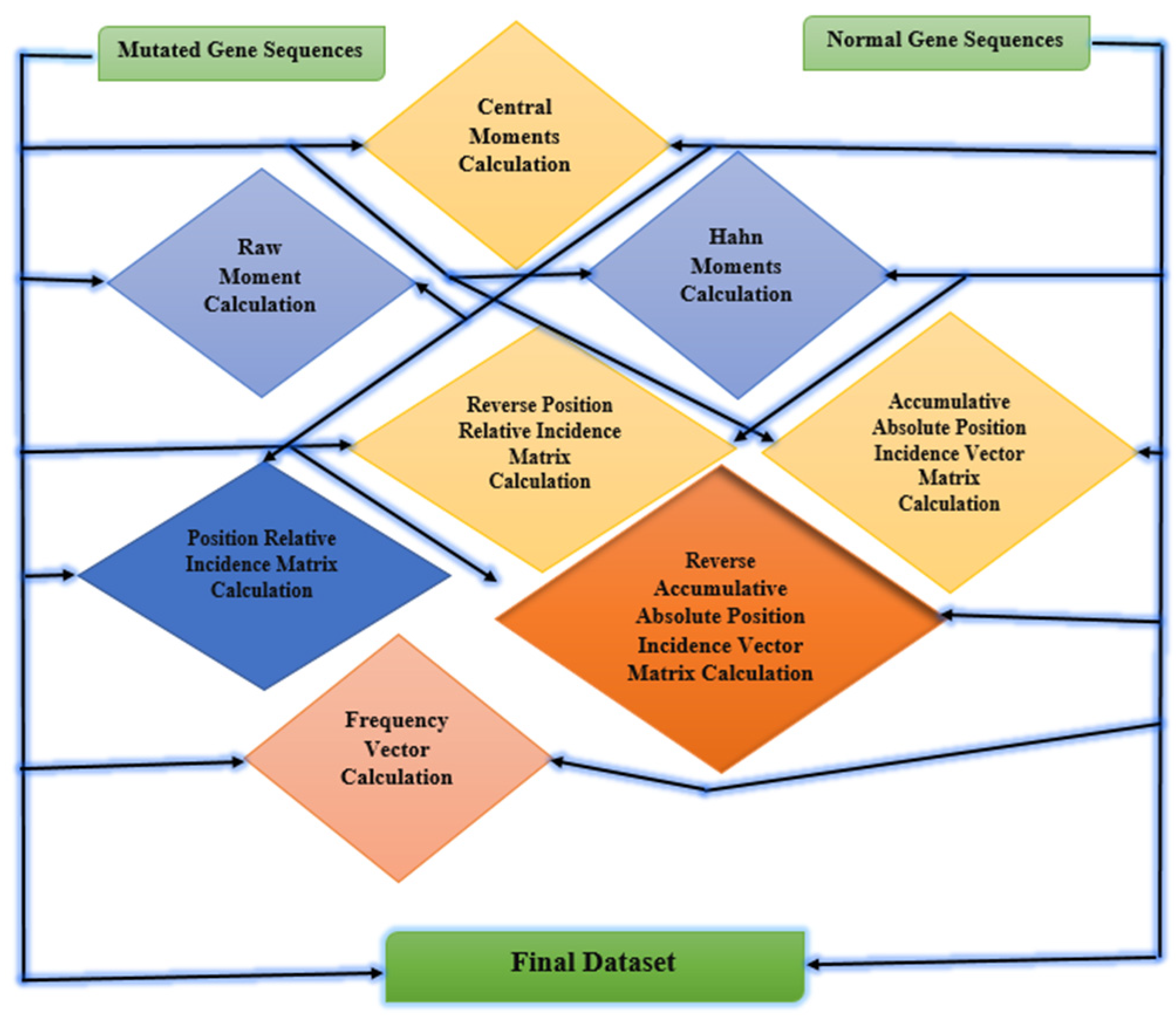
4.2. Feature Extraction
4.2.1. Hahn Moment Calculation
4.2.2. Raw Moment Calculation
4.2.3. Central Moment Calculation
4.2.4. Position Relative Incidence Matrix (PRIM)
4.2.5. Reverse Position Relative Incidence Matrix (RPRIM)
4.2.6. Accumulative Absolute Position Incidence Vector (AAPIV)
4.2.7. Reverse Accumulative Absolute Position Incidence Vector (RAAPIV)
4.2.8. Frequency Vector Calculation
4.3. Algorithm for Predictive Modeling
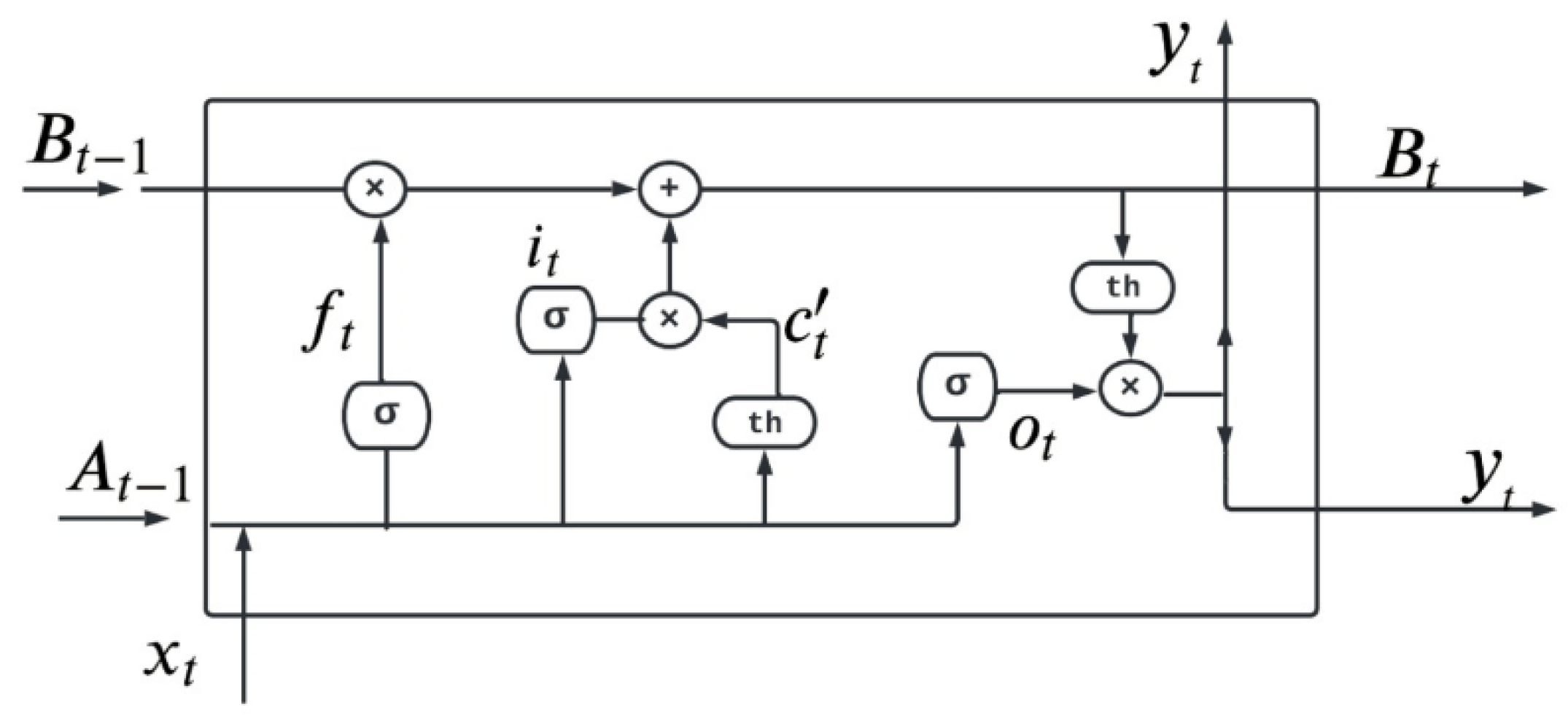
4.3.1. Long Short-Term Memory Network (LSTM)
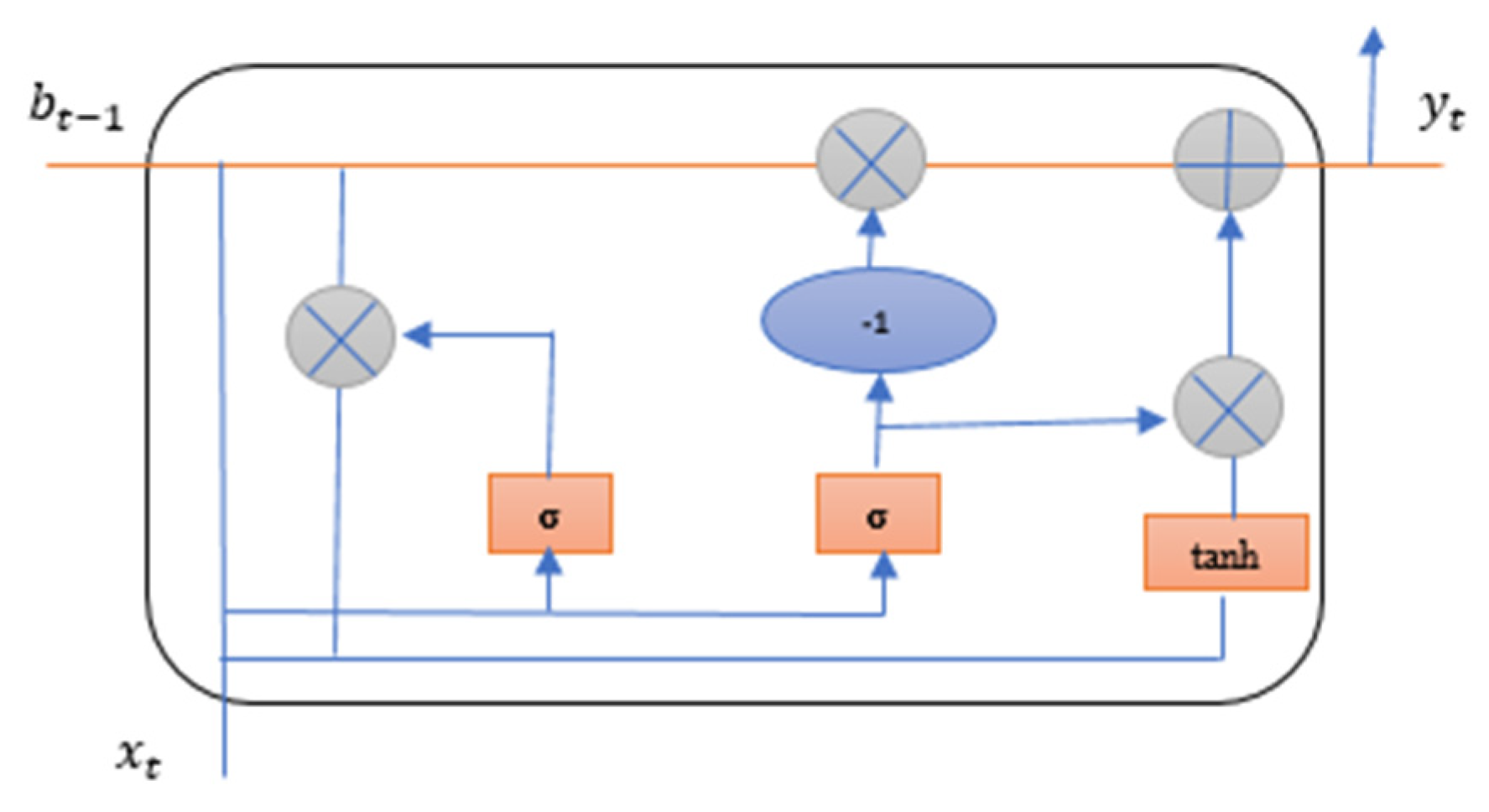
4.3.2. Gated Recurrent Unit (GRU)
4.3.3. Bi-Directional LSTM
4.3.4. Ensemble Learning Models
5. Conclusions and Future Work
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
- Databases used in this study
- Step by step process of extracting dataset
References
- Smith, T. Breast Cancer Surveillance Guidelines. J. Oncol. Pract. 2013, 9, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Breast Cancer—Statistics. Available online: https://www.cancer.net/cancer-types/breast-cancer/statistics (accessed on 17 August 2022).
- Biopsy. 2021. Available online: https://www.cancer.net/navigating-cancer-care/diagnosing-cancer/tests-and-procedures/biopsy (accessed on 16 August 2022).
- Fitzgerald, D.; Rosenberg, S. What is mutation? A chapter in the series: How microbes “jeopardize” the modern synthesis. PLOS Genet. 2019, 15, e1007995. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, S.; Sansón, J.; Hidalgo, A. Theoretical Study of Adenine to Guanine Transition Assisted by Water and Formic Acid Using Steered Molecular Dynamic Simulations. Front. Chem. 2019, 7, 414. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A. Multifaceted Roles of Alkyltransferase and Related Proteins in DNA Repair, DNA Damage, Resistance to Chemotherapy, and Research Tools. Chem. Res. Toxicol. 2011, 24, 618–639. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Lee, H.; Perry, G.; Smith, M. Alzheimer disease, the two-hit hypothesis: An update. Biochim. Biophys. Acta-Mol. Basis Dis. 2007, 1772, 494–502. [Google Scholar] [CrossRef]
- Zhu, X.; Raina, A.; Perry, G.; Smith, M. Alzheimer’s disease: The two-hit hypothesis. Lancet Neurol. 2004, 3, 219–226. [Google Scholar] [CrossRef]
- Akbugday, B. Classification of Breast Cancer Data Using Machine Learning Algorithms. In Proceedings of the 2019 Medical Technologies Congress (TIPTEKNO), Izmir, Turkey, 3–5 October 2019. [Google Scholar]
- Chaurasia, V.; Pal, S. Data Mining Techniques: To Predict and Resolve Breast Cancer Survivability. Int. J. Comput. Sci. Mob. Comput. 2014, 3, 10–22. [Google Scholar]
- Chang, J.; Hilsenbeck, S.; Fuqua, S. Genomic approaches in the management and treatment of breast cancer. Br. J. Cancer 2005, 92, 618–624. [Google Scholar] [CrossRef]
- Khourdifi, Y.; Bahaj, M. Feature Selection with Fast Correlation-Based Filter for Breast Cancer Prediction and Classification Using Machine Learning Algorithms. In Proceedings of the 2018 International Symposium on Advanced Electrical and Communication Technologies (ISAECT), Rabat, Morocco, 21–23 November 2018. [Google Scholar]
- Bakr, M.; Al-Attar, H.; Mahra, N.; Abu-Naser, S. Breast Cancer Prediction Using JNN. Int. J. Acad. Inf. Syst. Res. 2020, 4, 1–8. [Google Scholar]
- Leclerc, Y.; Luong, Q.; Fua, P. Self-Consistency: A Novel Approach to Characterizing the Accuracy and Reliability of Point Correspondence Algorithms. In Proceedings of the 1998 Image Understanding Workshop, Monterey, CA, USA, 20–23 November 1998. [Google Scholar]
- Usmanova, D.; Bogatyreva, N.; Ariño Bernad, J.; Eremina, A.; Gorshkova, A.; Kanevskiy, G.; Lonishin, L.; Meister, A.; Yakupova, A.; Kondrashov, F.; et al. Self-consistency test reveals systematic bias in programs for prediction change of stability upon mutation. Bioinformatics 2018, 34, 3653–3658. [Google Scholar] [CrossRef]
- Shah, A.; Malik, H.; Mohammad, A.; Khan, Y.; Alourani, A. Machine learning techniques for identification of carcinogenic mutations, which cause breast adenocarcinoma. Sci. Rep. 2022, 12, 11738. [Google Scholar] [CrossRef]
- Malebary, S.; Khan, R.; Khan, Y. ProtoPred: Advancing Oncological Research Through Identification of Proto-Oncogene Proteins. IEEE Access 2021, 9, 68788–68797. [Google Scholar] [CrossRef]
- Arnastauskaitė, J.; Ruzgas, T.; Bražėnas, M. An Exhaustive Power Comparison of Normality Tests. Mathematics 2021, 9, 788. [Google Scholar] [CrossRef]
- Erlemann, R.; Lindqvist, B.H. Conditional Goodness-of-Fit Tests for Discrete Distributions. J. Stat. Theory Pract. 2022, 16, 8. [Google Scholar] [CrossRef]
- Holy, T.; Jakubek, J.; Pospisil, S.; Uher, J.; Vavrik, D.; Vykydal, Z. Data acquisition and processing software package for Medipix2. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrom. Detect. Assoc. Equip. 2006, 563, 254–258. [Google Scholar] [CrossRef]
- Gene: TP53 (ENSG00000141510)—Summary—Homo_Sapiens—Ensembl Genome Browser 107. 2022. Available online: http://asia.ensembl.org/Homo_sapiens/Gene/Summary?g=ENSG00000141510;r=17:7661779-7687538 (accessed on 18 August 2022).
- IntOGen—Cancer Driver Mutations in Breast Adenocarcinoma. 2020. Available online: https://intogen.org/search?cancer=BRCA (accessed on 18 August 2022).
- Zhao, B. Web Scraping. Encycl. Big Data 2020, 5, 1–3. [Google Scholar]
- Kumar, S.; Warrell, J.; Li, S.; McGillivray, P.; Meyerson, W.; Salichos, L.; Harmanci, A.; Martinez-Fundichely, A.; Chan, C.; Nielsen, M.; et al. Passenger Mutations in More Than 2,500 Cancer Genomes: Overall Molecular Functional Impact and Consequences. Cell 2020, 180, 915–927.e16. [Google Scholar] [CrossRef]
- Bozic, I.; Antal, T.; Ohtsuki, H.; Carter, H.; Kim, D.; Chen, S.; Karchin, R.; Kinzler, K.; Vogelstein, B.; Nowak, M. Accumulation of driver and passenger mutations during tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 18545–18550. [Google Scholar] [CrossRef]
- Stratton, M.; Campbell, P.; Futreal, P. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef]
- Kaur, P.; Gosain, A. Comparing the Behavior of Oversampling and Undersampling Approach of Class Imbalance Learning by Combining Class Imbalance Problem with Noise. Adv. Intell. Syst. Comput. 2017, 310, 23–30. [Google Scholar]
- Chawla, N.; Bowyer, K.; Hall, L.; Kegelmeyer, W. SMOTE: Synthetic Minority Over-sampling Technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Shah, A.; Khan, Y. Identification of 4-carboxyglutamate residue sites based on position based statistical feature and multiple classification. Sci. Rep. 2020, 10, 16913. [Google Scholar] [CrossRef]
- Levine, M. Feature extraction: A survey. Proc. IEEE 1969, 57, 1391–1407. [Google Scholar] [CrossRef]
- Ghoraani, B.; Krishnan, S. Time-Frequency Matrix Feature Extraction and Classification of Environmental Audio Signals. IEEE Trans. Audio Speech Lang. Process. 2011, 19, 2197–2209. [Google Scholar] [CrossRef]
- Amanat, S.; Ashraf, A.; Hussain, W.; Rasool, N.; Khan, Y. Identification of lysine carboxylation sites in proteins by integrating statistical moments and position relative features via general PseAAC. Curr. Bioinform. 2020, 15, 396–407. [Google Scholar] [CrossRef]
- Hussain, W.; Rasool, N.; Khan, Y. Insights into Machine Learning-based approaches for Virtual Screening in Drug Discovery: Existing strategies and streamlining through FP-CADD. Curr. Drug Discov. Technol. 2021, 18, 463–472. [Google Scholar] [CrossRef]
- Hussain, W.; Rasool, N.; Khan, Y.; Screening, H. A sequence-based predictor of Zika virus proteins developed by integration of PseAAC and statistical moments. Comb. Chem. High Throughput Screen. 2020, 23, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Khan, Y.; Alzahrani, E.; Alghamdi, W.; Ullah, M. Sequence-based identification of allergen proteins developed by integration of PseAAC and statistical moments via 5-step rule. Curr. Bioinform. 2020, 15, 1046–1055. [Google Scholar] [CrossRef]
- Mahmood, M.; Ehsan, A.; Khan, Y.; Chou, K. iHyd-LysSite (EPSV): Identifying hydroxylysine sites in protein using statistical formulation by extracting enhanced position and sequence variant feature technique. Curr. Genom. 2020, 21, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Naseer, S.; Hussain, W.; Khan, Y.; Rasool, N. Optimization of serine phosphorylation prediction in proteins by comparing human engineered features and deep representations. Anal. Biochem. 2021, 615, 114069. [Google Scholar] [CrossRef]
- Naseer, S.; Hussain, W.; Khan, Y.; Rasool, N. Sequence-based identification of arginine amidation sites in proteins using deep representations of proteins and PseAAC. Curr. Bioinform. 2020, 15, 937–948. [Google Scholar] [CrossRef]
- Naseer, S.; Hussain, W.; Khan, Y.; Rasool, N. NPalmitoylDeep-PseAAC: A predictor of N-palmitoylation sites in proteins using deep representations of proteins and PseAAC via modified 5-steps rule. Curr. Bioinform. 2021, 16, 294–305. [Google Scholar] [CrossRef]
- Naseer, S.; Hussain, W.; Khan, Y.; Rasool, N. Bioinformatics IPhosS (Deep)-PseAAC: Identify phosphoserine sites in proteins using deep learning on general pseudo amino acid compositions via modified 5-Steps rule. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 19, 1703–1714. [Google Scholar]
- Hall, A.R. Generalized Method of Moments; Oxford University Press: Oxford, UK, 2005. [Google Scholar]
- Zhu, H.; Shu, H.; Zhou, J.; Luo, L.; Coatrieux, J. Image analysis by discrete orthogonal dual Hahn moments. Pattern Recognit. Lett. 2007, 28, 1688–1704. [Google Scholar] [CrossRef] [Green Version]
- Malebary, S.; Khan, Y. Evaluating machine learning methodologies for identification of cancer driver genes. Sci. Rep. 2021, 11, 12281. [Google Scholar] [CrossRef]
- Sohail, M.; Shabbir, J.; Sohil, F. Imputation of Missing Values by Using Raw Moments. Stat. Transit. New Ser. 2019, 20, 21–40. [Google Scholar] [CrossRef]
- Butt, A.; Khan, Y. CanLect-Pred: A Cancer Therapeutics Tool for Prediction of Target Cancerlectins Using Experiential Annotated Proteomic Sequences. IEEE Access 2020, 8, 9520–9531. [Google Scholar] [CrossRef]
- Akmal, M.; Rasool, N.; Khan, Y. Prediction of N-linked glycosylation sites using position relative features and statistical moments. PLoS ONE 2017, 12, e0181966. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Wang, H.; Chen, S.; Xu, F.; Jin, Y. Application of deep-learning algorithms to mstar data. In Proceedings of the 2015 IEEE International Geoscience and Remote Sensing Symposium (IGARSS), Milan, Italy, 26–31 July 2015; pp. 3743–3745. [Google Scholar]
- Hochreiter, S. The Vanishing Gradient Problem During Learning Recurrent Neural Nets and Problem Solutions. Int. J. Uncertain. Fuzziness Knowl.-Based Syst. 1998, 6, 107–116. [Google Scholar] [CrossRef]
- Sundermeyer, M.; Schlüter, R.; Ney, H. LSTM neural networks for language processing. In Proceedings of the Interspeech 2012, ISCA’s 13th Annual Conference, Portland, OR, USA, 9–13 September 2012; pp. 194–197. [Google Scholar]
- Rengasamy, D.; Jafari, M.; Rothwell, B.; Chen, X.; Figueredo, G. Deep Learning with Dynamically Weighted Loss Function for Sensor-Based Prognostics and Health Management. Sensors 2020, 20, 723. [Google Scholar] [CrossRef]
- Gu, J.; Wang, Z.; Kuen, J.; Ma, L.; Shahroudy, A.; Shuai, B.; Liu, T.; Wang, X.; Wang, G.; Cai, J.; et al. Recent advances in convolutional neural networks. Pattern Recognit. 2018, 77, 354–377. [Google Scholar] [CrossRef]
- Lin, G.; Shen, W. Research on convolutional neural network based on improved Relu piecewise activation function. Procedia Comput. Sci. 2018, 131, 977–984. [Google Scholar] [CrossRef]
- Guo, H.; Tang, R.; Ye, Y.; Li, Z.; He, X.; Dong, Z. DeepFM: An End-to-End Wide & Deep Learning Framework for CTR Prediction. arXiv 2018, arXiv:1804.04950. [Google Scholar]
- Gao, Y.; Glowacka, D. Deep gate recurrent neural network. J. Mach. Learn. Res. 2016, 63, 350–365. [Google Scholar]
- Graves, A.; Schmidhuber, J. Framewise phoneme classification with bidirectional LSTM and other neural network architectures. Neural Netw. 2005, 18, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Basaldella, M.; Antolli, E.; Serra, G.; Tasso, C. Bidirectional LSTM Recurrent Neural Network for Keyphrase Extraction. Commun. Comput. Inf. Sci. 2017, 180–187. [Google Scholar] [CrossRef]
- Mendes-Moreira, J.; Soares, C.; Jorge, A.; Sousa, J. Ensemble approaches for regression: A survey. ACM Comput. Surv. 2012, 45, 1–40. [Google Scholar] [CrossRef]
- Breiman, L. Bagging predictors. Mach. Learn. 1996, 2, 123–140. [Google Scholar] [CrossRef]
- Schapire, R. The strength of weak learnability. Mach. Learn. 1990, 5, 197–227. [Google Scholar] [CrossRef]
- Stefenon, S.; Ribeiro, M.; Nied, A.; Mariani, V.; Coelho, L.; Leithardt, V.; Silva, L.; Seman, L. Hybrid Wavelet Stacking Ensemble Model for Insulators Contamination Forecasting. IEEE Access 2021, 9, 66387–66397. [Google Scholar] [CrossRef]
- Feng, P.; Yang, H.; Ding, H.; Lin, H.; Chen, W.; Chou, K. iDNA6mA-PseKNC: Identifying DNA N6-methyladenosine sites by incorporating nucleotide physicochemical properties into PseKNC. Genomics 2019, 111, 96–102. [Google Scholar] [CrossRef]
- Piovesan, D.; Hatos, A.; Minervini, G.; Quaglia, F.; Monzon, A.; Tosatto, S. Assessing predictors for new post translational modification sites: A case study on hydroxylation. PLoS Comput. Biol. 2020, 16, e1007967. [Google Scholar] [CrossRef]
- Hoo, Z.; Candlish, J.; Teare, D. What is an ROC curve? Emerg. Med. J. 2017, 34, 357–359. [Google Scholar] [CrossRef]
- Xu, W.; Hao, D.; Hou, F.; Zhang, D.; Wang, H. Soft Tissue Sarcoma: Preoperative MRI-Based Radiomics and Machine Learning May Be Accurate Predictors of Histopathologic Grade. Am. J. Roentgenol. 2020, 215, 963–969. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Evaluation Matrices | Values | Evaluation Matrices | Values |
|---|---|---|---|
| Accuracy (%) | 97.65 | Precision (%) | 97.65 |
| Sensitivity (%) | 97.81 | Recall (%) | 97.65 |
| Specificity (%) | 97.50 | F1 Score (%) | 97.65 |
| MCC | 0.95 | Cohens Kappa (%) | 95.31 |
| AUC | 1.00 | Training Accuracy (%) | 78.29 |
| Training Loss | 0.3649 | Testing Accuracy (%) | 78.51 |
| Evaluation Matrices | Values | Evaluation Matrices | Values |
|---|---|---|---|
| Accuracy (%) | 99.57 | Precision (%) | 99.57 |
| Sensitivity (%) | 99.50 | Recall (%) | 99.57 |
| Specificity (%) | 99.63 | F1 Score (%) | 99.57 |
| MCC | 0.99 | Cohens Kappa (%) | 99.14 |
| AUC | 1.00 | Training Accuracy (%) | 99.79 |
| Training Loss | 0.2027 | Testing Accuracy (%) | 99.82 |
| Evaluation Matrices | Values | Evaluation Matrices | Values |
|---|---|---|---|
| Accuracy (%) | 98.26 | MCC | 0.9852 |
| Sensitivity (%) | 98.02 | AUC | 0.99 |
| Specificity (%) | 98.50 |
| Evaluation Matrices | Ensemble Learning Approach | LSTM | GRU | Bi-Directional LSTM |
|---|---|---|---|---|
| Accuracy (%) | 99.57 | 99.02 | 97.12 | 96.51 |
| Sensitivity (%) | 99.50 | 98.89 | 96.94 | 96.32 |
| Specificity (%) | 99.63 | 99.14 | 97.31 | 96.69 |
| MCC | 0.99 | 0.98 | 0.94 | 93.02 |
| AUC | 1.00 | 1.00 | 1.00 | 1.00 |
| Training Loss | 0.2027 | 0.0235 | 0.1199 | 0.2122 |
| Precision (%) | 99.57 | 99.02 | 97.12 | 96.51 |
| Recall (%) | 99.57 | 99.02 | 97.12 | 96.51 |
| F1 Score (%) | 99.57 | 99.02 | 97.12 | 96.51 |
| Cohens Kappa (%) | 99.14 | 98.04 | 94.25 | 93.02 |
| Training Accuracy (%) | 99.79 | 99.30 | 95.60 | 97.70 |
| Testing Accuracy (%) | 99.82 | 99.44 | 97.43 | 96.94 |
| Gene | Mutation | Gene | Mutation | Gene | Mutation |
|---|---|---|---|---|---|
| TP53 | 846 | KMT2C | 205 | ERBB4 | 43 |
| GATA3 | 63 | CDI | 176 | MDM4 | 14 |
| ESR1 | 129 | PTEN | 105 | GATA1 | 15 |
| AKT1 | 88 | NCOR1 | 89 | USP6 | 19 |
| FOXA1 | 72 | TBX3 | 54 | EGFR | 45 |
| NF1 | 85 | ERBB2 | 83 | MEN1 | 28 |
| RB1 | 60 | CFFB | 64 | GNAS | 29 |
| SF3B1 | 56 | KMT2D | 99 | KDM6A | 30 |
| FAT3 | 112 | ERBB3 | 55 | FAT4 | 78 |
| PREX2 | 73 | CTFC | 47 | KAT6B | 37 |
| LRP1B | 114 | RUNX1 | 37 | JAK2 | 19 |
| ATM | 64 | SPEN | 74 | ALK | 33 |
| FGFR2 | 37 | BRCA1 | 49 | BAP1 | 25 |
| CASP8 | 28 | FBXW7 | 29 | CUX1 | 29 |
| BRCA2 | 52 | PTPRD | 64 | KLF4 | 8 |
| MYH11 | 59 | RGS7 | 32 | FAT1 | 61 |
| KRAS | 15 | NCOA1 | 21 | DDX3X | 23 |
| MYH9 | 59 | ABL2 | 31 | NONO | 9 |
| EPHA3 | 31 | NCOR2 | 44 | MTOR | 57 |
| AFF3 | 37 | ETV5 | 16 | ASXL1 | 36 |
| BRAF | 22 | ELN | 26 | MYOSA | 19 |
| ZXBD | 18 | NTRK1 | 26 | POLD1 | 18 |
| SALL4 | 17 | SMAD2 | 17 | PLAG1 | 15 |
| EPAS1 | 25 | RHPN2 | 18 | NIN | 44 |
| SMAD4 | 17 | MAX | 9 | NUMA1 | 33 |
| HAS | 10 | ZFHX3 | 72 | CLTC | 31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, A.A.; Alturise, F.; Alkhalifah, T.; Khan, Y.D. Deep Learning Approaches for Detection of Breast Adenocarcinoma Causing Carcinogenic Mutations. Int. J. Mol. Sci. 2022, 23, 11539. https://doi.org/10.3390/ijms231911539
Shah AA, Alturise F, Alkhalifah T, Khan YD. Deep Learning Approaches for Detection of Breast Adenocarcinoma Causing Carcinogenic Mutations. International Journal of Molecular Sciences. 2022; 23(19):11539. https://doi.org/10.3390/ijms231911539
Chicago/Turabian StyleShah, Asghar Ali, Fahad Alturise, Tamim Alkhalifah, and Yaser Daanial Khan. 2022. "Deep Learning Approaches for Detection of Breast Adenocarcinoma Causing Carcinogenic Mutations" International Journal of Molecular Sciences 23, no. 19: 11539. https://doi.org/10.3390/ijms231911539
APA StyleShah, A. A., Alturise, F., Alkhalifah, T., & Khan, Y. D. (2022). Deep Learning Approaches for Detection of Breast Adenocarcinoma Causing Carcinogenic Mutations. International Journal of Molecular Sciences, 23(19), 11539. https://doi.org/10.3390/ijms231911539