
Effects of Anti-Cancer Drug Sensitivity-Related Genetic Differences on Therapeutic Approaches in Refractory Papillary Thyroid Cancer

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
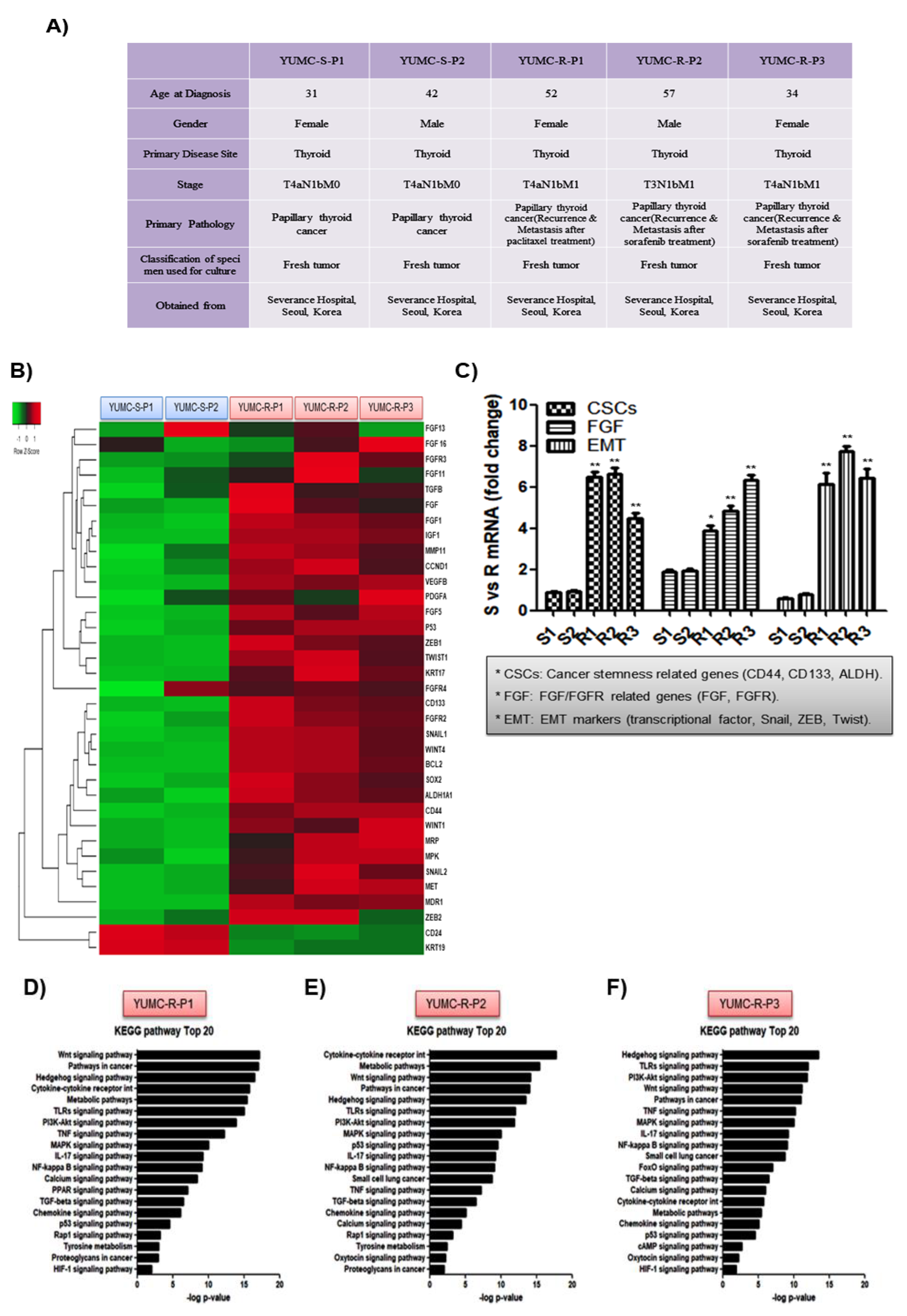
2.1. Patient Disease Characteristics
2.2. Characteristics of Patient-Derived Drug Resistant PTC Cell Lines
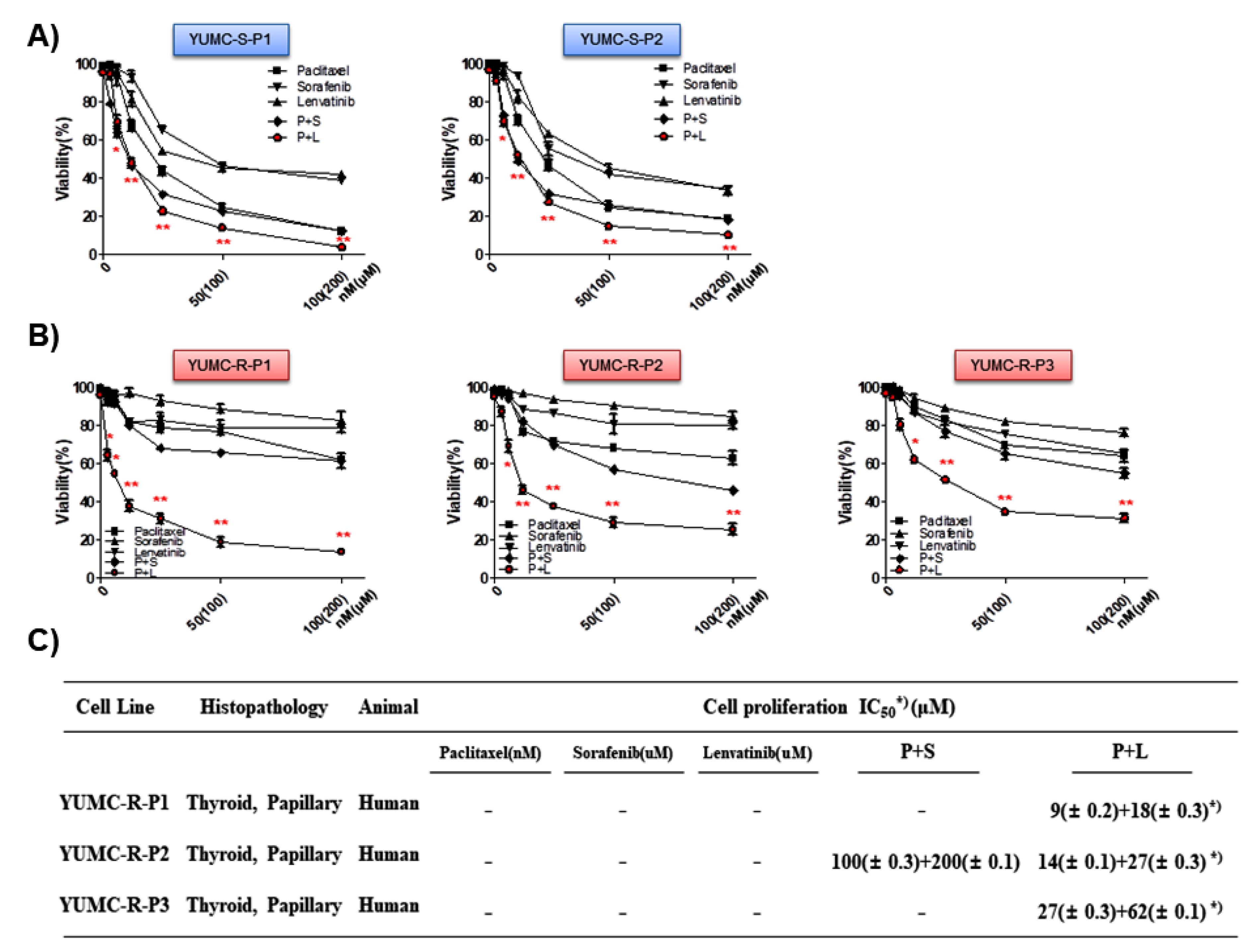
2.3. Combination of Paclitaxel and Lenvatinib Was More Effective Than Either Agent Alone or a Combination of Paclitaxel and Sorafenib
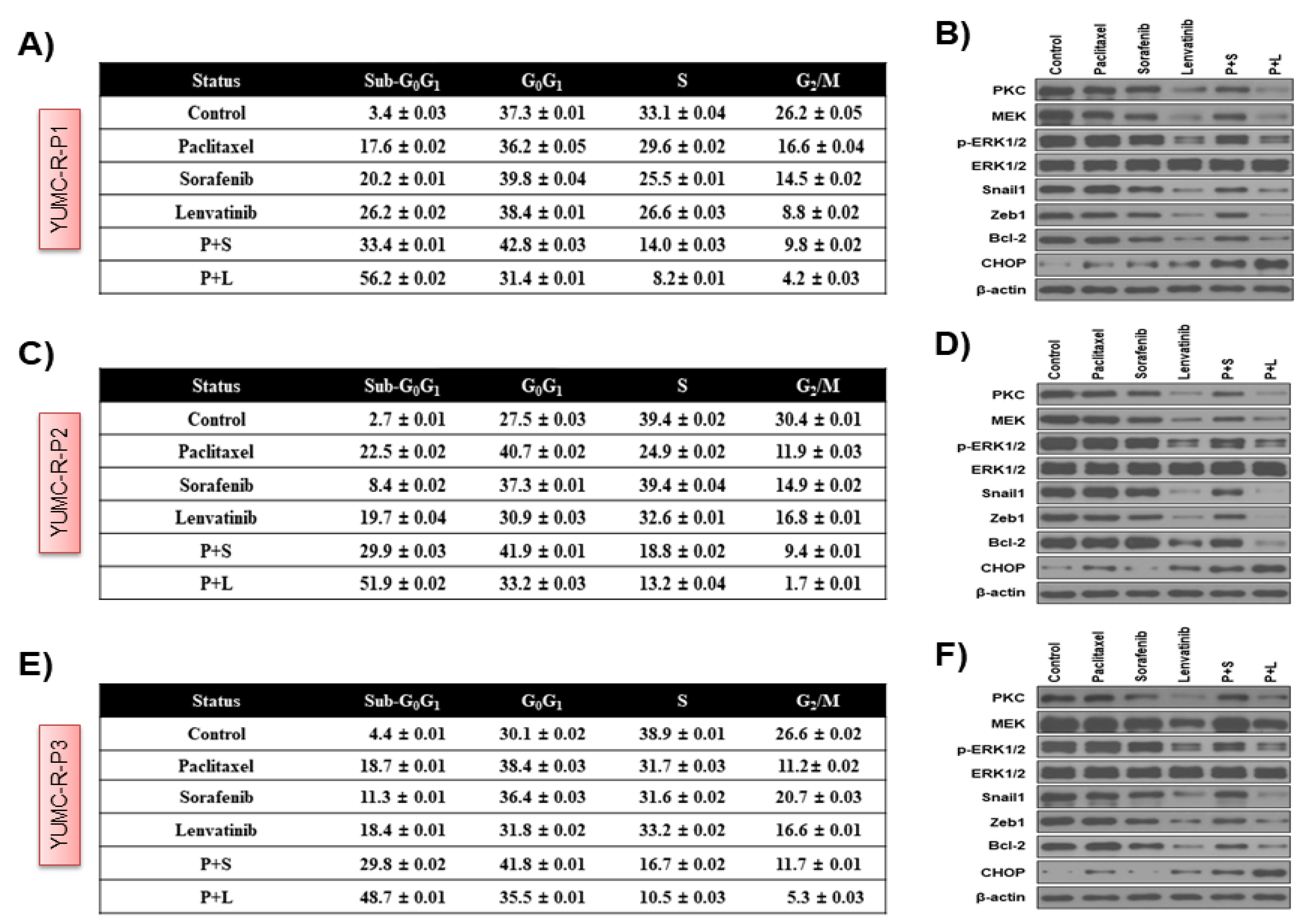
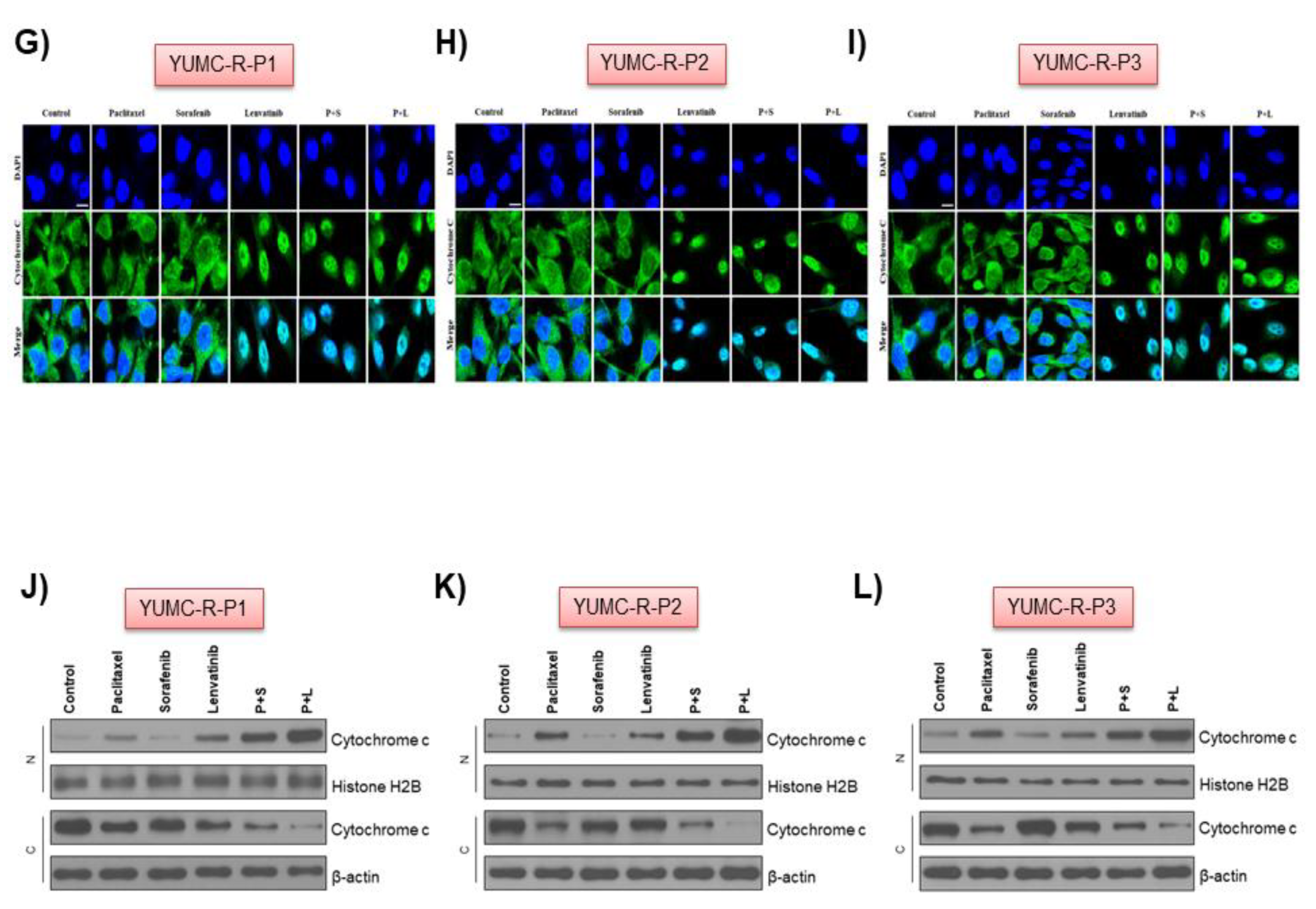
2.4. Paclitaxel and Lenvatinib Cotreatment Induced Nuclear Translocation-Mediated Apoptosis in YUMC-R-P1, YUMC-R-P2, and YUMC-R-P3 Cells
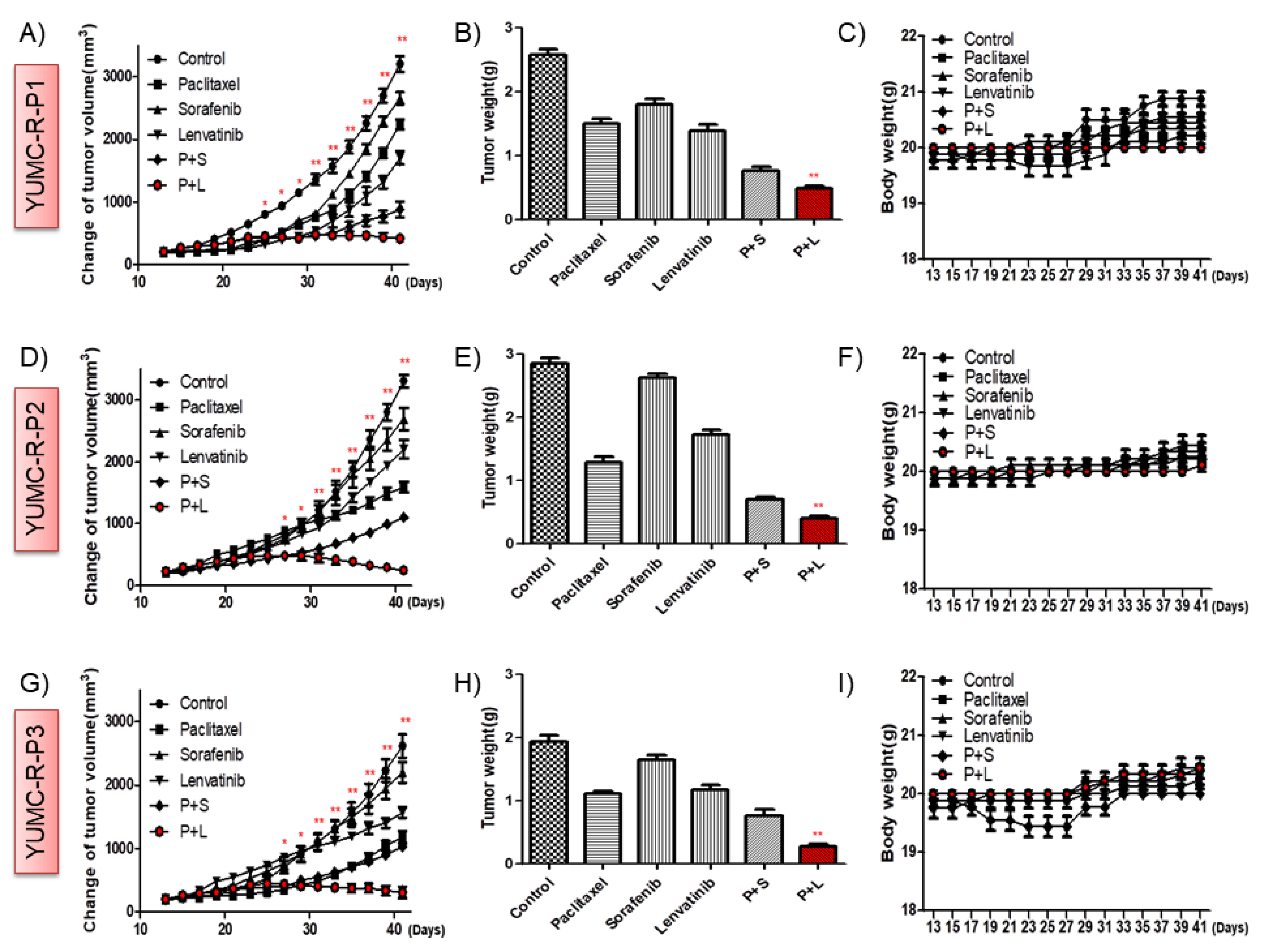
2.5. Paclitaxel and Lenvatinib Cotreatment Remarkably Suppressed Tumor Growth in a Mouse Xenograft Model with Patient-Derived Drug-Resistant PTC Cell Lines
3. Discussion
4. Materials and Methods
4.1. Study Design and Ethical Considerations
4.2. Patients
4.2.1. Patient 1
4.2.2. Patient 2
4.2.3. Patient 3
4.3. Specimens of Patients
4.4. Tumor Cell Isolation and Primary Culture
4.5. mRNA-Seq Data
4.6. Statistical Aanalysis of Gene Expression Level
4.7. Hierarchical Clustering
4.8. Cell Culture
4.9. Cell Viability Assay
4.10. Cell CycleAnalysis Using Flow Cytometry
4.11. Immunofluorescence Analysis and Confocal Imaging
4.12. Cellular Fractionation
4.13. Immunoblot Analysis
4.14. Human PTC Cell Xenograft
4.15. Immunohistochemistry
4.16. Image Analysis
4.17. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nikiforov, Y.E. Thyroid carcinoma: Molecular pathways and therapeutic targets. Mod. Pathol. Off. J. United States Can. Acad. Pathol. 2008, 21, S37–S43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, Q.T.; Lee, E.J.; Huang, M.G.; Park, Y.I.; Khullar, A.; Plodkowski, R.A. Diagnosis and treatment of patients with thyroid cancer. Am. Health Drug Benefits 2015, 8, 30–40. [Google Scholar]
- Fahiminiya, S.; de Kock, L.; Foulkes, W.D. Biologic and clinical perspectives on thyroid cancer. N. Engl. J. Med. 2016, 375, 2306–2307. [Google Scholar]
- Owonikoko, T.K.; Chowdry, R.P.; Chen, Z.; Kim, S.; Saba, N.F.; Shin, D.M.; Khuri, F.R. Clinical efficacy of targeted biologic agents as second-line therapy of advanced thyroid cancer. Oncology 2013, 18, 1262–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raue, F.; Frank-Raue, K. Thyroid cancer: Risk-stratified management and individualized therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 5012–5021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahimpasic, T.; Ghossein, R.; Shah, J.P.; Ganly, I. Poorly differentiated carcinoma of the thyroid gland: Current status and future prospects. Thyroid Off. J. Am. Thyroid Assoc. 2019, 29, 311–321. [Google Scholar] [CrossRef] [PubMed]
- De Castroneves, L.A.; Negrao, M.V.; de Freitas, R.M.; Papadia, C.; Lima, J.V., Jr.; Fukushima, J.T.; Simao, E.F.; Kulcsar, M.A.; Tavares, M.R.; Jorge, A.A.; et al. Sorafenib for the treatment of progressive metastatic medullary thyroid cancer: Efficacy and safety analysis. Thyroid Off. J. Am. Thyroid Assoc. 2016, 26, 414–419. [Google Scholar] [CrossRef]
- Ito, Y.; Onoda, N.; Ito, K.I.; Sugitani, I.; Takahashi, S.; Yamaguchi, I.; Kabu, K.; Tsukada, K. Sorafenib in japanese patients with locally advanced or metastatic medullary thyroid carcinoma and anaplastic thyroid carcinoma. Thyroid Off. J. Am. Thyroid Assoc. 2017, 27, 1142–1148. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, T.; Yu, M.; Yin, D.; Sun, D.; Zhu, Y.; Bu, Y.; Sang, M. Impact of runx2 on drug-resistant human pancreatic cancer cells with p53 mutations. BMC Cancer 2018, 18, 309. [Google Scholar] [CrossRef] [Green Version]
- He, Y. Systematic response of staurosporine scaffold-based inhibitors to drug-resistant cancer kinase mutations. Arch. Der Pharm. 2020, 353, e1900320. [Google Scholar] [CrossRef]
- Smith, B.D.; Kaufman, M.D.; Lu, W.P.; Gupta, A.; Leary, C.B.; Wise, S.C.; Rutkoski, T.J.; Ahn, Y.M.; Al-Ani, G.; Bulfer, S.L.; et al. Ripretinib (dcc-2618) is a switch control kinase inhibitor of a broad spectrum of oncogenic and drug-resistant kit and pdgfra variants. Cancer Cell 2019, 35, 738–751.e739. [Google Scholar] [CrossRef]
- Cao, X.; Hou, J.; An, Q.; Assaraf, Y.G.; Wang, X. Towards the overcoming of anticancer drug resistance mediated by p53 mutations. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2020, 49, 100671. [Google Scholar] [CrossRef]
- Liu, Q.; Yu, S.; Zhao, W.; Qin, S.; Chu, Q.; Wu, K. Egfr-tkis resistance via egfr-independent signaling pathways. Mol. Cancer 2018, 17, 53. [Google Scholar] [CrossRef] [Green Version]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Liu, Y.; Lin, Y.; Liang, J. Radioactive iodine-refractory differentiated thyroid cancer and redifferentiation therapy. Endocrinol. Metab. 2019, 34, 215–225. [Google Scholar] [CrossRef]
- Brose, M.S.; Cabanillas, M.E.; Cohen, E.E.; Wirth, L.J.; Riehl, T.; Yue, H.; Sherman, S.I.; Sherman, E.J. Vemurafenib in patients with braf(v600e)-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: A non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1272–1282. [Google Scholar] [CrossRef] [Green Version]
- Miftari, R.; Topciu, V.; Nura, A.; Haxhibeqiri, V. Management of the patient with aggressive and resistant papillary thyroid carcinoma. Med. Arch. 2016, 70, 314–317. [Google Scholar] [CrossRef] [Green Version]
- Grygielewicz, P.; Dymek, B.; Bujak, A.; Gunerka, P.; Stanczak, A.; Lamparska-Przybysz, M.; Wieczorek, M.; Dzwonek, K.; Zdzalik, D. Epithelial-mesenchymal transition confers resistance to selective fgfr inhibitors in snu-16 gastric cancer cells. Gastric Cancer Off. J. Int. Gastric Cancer Assoc. Jpn. Gastric Cancer Assoc. 2016, 19, 53–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The role of snail in emt and tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef]
- Gloushankova, N.A.; Rubtsova, S.N.; Zhitnyak, I.Y. Cadherin-mediated cell-cell interactions in normal and cancer cells. Tissue Barriers 2017, 5, e1356900. [Google Scholar] [CrossRef] [Green Version]
- Stuhlmiller, T.J.; Miller, S.M.; Zawistowski, J.S.; Nakamura, K.; Beltran, A.S.; Duncan, J.S.; Angus, S.P.; Collins, K.A.; Granger, D.A.; Reuther, R.A.; et al. Inhibition of lapatinib-induced kinome reprogramming in erbb2-positive breast cancer by targeting bet family bromodomains. Cell Rep. 2015, 11, 390–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amato, V.; Raimondo, L.; Formisano, L.; Giuliano, M.; De Placido, S.; Rosa, R.; Bianco, R. Mechanisms of lapatinib resistance in her2-driven breast cancer. Cancer Treat. Rev. 2015, 41, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Otte, J.; Dizdar, L.; Behrens, B.; Goering, W.; Knoefel, W.T.; Wruck, W.; Stoecklein, N.H.; Adjaye, J. Fgf signalling in the self-renewal of colon cancer organoids. Sci. Rep. 2019, 9, 17365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, P.; Konno, Y.; Watari, H.; Hosaka, M.; Noguchi, M.; Sakuragi, N. The impact of microrna-mediated pi3k/akt signaling on epithelial-mesenchymal transition and cancer stemness in endometrial cancer. J. Transl. Med. 2014, 12, 231. [Google Scholar] [CrossRef]
- Garg, M. Urothelial cancer stem cells and epithelial plasticity: Current concepts and therapeutic implications in bladder cancer. Cancer Metastasis Rev. 2015, 34, 691–701. [Google Scholar] [CrossRef]
- Brown, W.S.; Akhand, S.S.; Wendt, M.K. Fgfr signaling maintains a drug persistent cell population following epithelial-mesenchymal transition. Oncotarget 2016, 7, 83424–83436. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target 2020, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Kweon, S.S.; Shin, M.H.; Chung, I.J.; Kim, Y.J.; Choi, J.S. Thyroid cancer is the most common cancer in women, based on the data from population-based cancer registries, South Korea. Jpn. J. Clin. Oncol. 2013, 43, 1039–1046. [Google Scholar] [CrossRef] [Green Version]
- Cabanillas, M.E.; McFadden, D.G.; Durante, C. Thyroid cancer. Lancet 2016, 388, 2783–2795. [Google Scholar] [CrossRef]
- Kim, J.; Gosnell, J.E.; Roman, S.A. Geographic influences in the global rise of thyroid cancer. Nat. Rev. Endocrinol. 2020, 16, 17–29. [Google Scholar] [CrossRef]
- Prete, A.; Borges de Souza, P.; Censi, S.; Muzza, M.; Nucci, N.; Sponziello, M. Update on fundamental mechanisms of thyroid cancer. Front. Endocrinol. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Wiltshire, J.J.; Drake, T.M.; Uttley, L.; Balasubramanian, S.P. Systematic review of trends in the incidence rates of thyroid cancer. Thyroid Off. J. Am. Thyroid Assoc. 2016, 26, 1541–1552. [Google Scholar] [CrossRef]
- Pellegriti, G.; De Vathaire, F.; Scollo, C.; Attard, M.; Giordano, C.; Arena, S.; Dardanoni, G.; Frasca, F.; Malandrino, P.; Vermiglio, F.; et al. Papillary thyroid cancer incidence in the volcanic area of sicily. J. Natl. Cancer Inst. 2009, 101, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Bergdorf, K.; Ferguson, D.C.; Mehrad, M.; Ely, K.; Stricker, T.; Weiss, V.L. Papillary thyroid carcinoma behavior: Clues in the tumor microenvironment. Endocr.-Relat. Cancer 2019, 26, 601–614. [Google Scholar] [CrossRef]
- Lewinski, A.; Adamczewski, Z. Papillary thyroid carcinoma: A cancer with an extremely diverse genetic background and prognosis. Pol. Arch. Intern. Med. 2017, 127, 388–389. [Google Scholar] [CrossRef] [Green Version]
- Colombo, C.; Minna, E.; Gargiuli, C.; Muzza, M.; Dugo, M.; De Cecco, L.; Pogliaghi, G.; Tosi, D.; Bulfamante, G.; Greco, A.; et al. The molecular and gene/mirna expression profiles of radioiodine resistant papillary thyroid cancer. J. Exp. Clin. Cancer Res. CR 2020, 39, 245. [Google Scholar] [CrossRef] [PubMed]
- Zelinskaya, A. Immunocytochemical characteristics of thyrocytes in radioiodine refractory metastases of papillary thyroid cancer. Exp. Oncol. 2019, 41, 342–345. [Google Scholar] [CrossRef]
- Stassi, G.; Todaro, M.; Zerilli, M.; Ricci-Vitiani, L.; Di Liberto, D.; Patti, M.; Florena, A.; Di Gaudio, F.; Di Gesu, G.; De Maria, R. Thyroid cancer resistance to chemotherapeutic drugs via autocrine production of interleukin-4 and interleukin-10. Cancer Res. 2003, 63, 6784–6790. [Google Scholar]
- Giuffrida, R.; Adamo, L.; Iannolo, G.; Vicari, L.; Giuffrida, D.; Eramo, A.; Gulisano, M.; Memeo, L.; Conticello, C. Resistance of papillary thyroid cancer stem cells to chemotherapy. Oncol. Lett. 2016, 12, 687–691. [Google Scholar] [CrossRef] [Green Version]
- Jemal, A.; Siegel, R.; Ward, E.; Murray, T.; Xu, J.; Thun, M.J. Cancer statistics, 2007. CA Cancer J. Clin. 2007, 57, 43–66. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.; Welch, H.G. Increasing incidence of thyroid cancer in the United States, 1973–2002. JAMA 2006, 295, 2164–2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Groot, J.W.; Links, T.P.; Plukker, J.T.; Lips, C.J.; Hofstra, R.M. Ret as a diagnostic and therapeutic target in sporadic and hereditary endocrine tumors. Endocr. Rev. 2006, 27, 535–560. [Google Scholar] [CrossRef] [Green Version]
- Kondo, T.; Ezzat, S.; Asa, S.L. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat. Rev. Cancer 2006, 6, 292–306. [Google Scholar] [CrossRef]
- Smallridge, R.C.; Marlow, L.A.; Copland, J.A. Anaplastic thyroid cancer: Molecular pathogenesis and emerging therapies. Endocr.-Relat. Cancer 2009, 16, 17–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivan, M.; Bond, J.A.; Prat, M.; Comoglio, P.M.; Wynford-Thomas, D. Activated ras and ret oncogenes induce over-expression of c-met (hepatocyte growth factor receptor) in human thyroid epithelial cells. Oncogene 1997, 14, 2417–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvatore, D.; Barone, M.V.; Salvatore, G.; Melillo, R.M.; Chiappetta, G.; Mineo, A.; Fenzi, G.; Vecchio, G.; Fusco, A.; Santoro, M. Tyrosines 1015 and 1062 are in vivo autophosphorylation sites in ret and ret-derived oncoproteins. J. Clin. Endocrinol. Metab. 2000, 85, 3898–3907. [Google Scholar]
- Wilson, M.M.; Weinberg, R.A.; Lees, J.A.; Guen, V.J. Emerging mechanisms by which emt programs control stemness. Trends Cancer 2020, 6, 775–780. [Google Scholar] [CrossRef]
- Miyoshi, N.; Haraguchi, N.; Mizushima, T.; Ishii, H.; Yamamoto, H.; Mori, M. Targeting cancer stem cells in refractory cancer. Regen. Ther. 2021, 17, 13–19. [Google Scholar] [CrossRef]
- Fujita, Y.; Kojima, K.; Ohhashi, R.; Hamada, N.; Nozawa, Y.; Kitamoto, A.; Sato, A.; Kondo, S.; Kojima, T.; Deguchi, T.; et al. Mir-148a attenuates paclitaxel resistance of hormone-refractory, drug-resistant prostate cancer pc3 cells by regulating msk1 expression. J. Biol. Chem. 2010, 285, 19076–19084. [Google Scholar] [CrossRef] [Green Version]
- Oien, D.B.; Pathoulas, C.L.; Ray, U.; Thirusangu, P.; Kalogera, E.; Shridhar, V. Repurposing quinacrine for treatment-refractory cancer. Semin. Cancer Biol. 2021, 68, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Kang, Y. Epithelial-mesenchymal plasticity in cancer progression and metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef]
- Tsubakihara, Y.; Moustakas, A. Epithelial-mesenchymal transition and metastasis under the control of transforming growth factor beta. Int. J. Mol. Sci. 2018, 19, 3672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicari, L.; Colarossi, C.; Giuffrida, D.; De Maria, R.; Memeo, L. Cancer stem cells as a potential therapeutic target in thyroid carcinoma. Oncol. Lett. 2016, 12, 2254–2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Hardin, H.; Lloyd, R.V. Cancer stem-like cells and thyroid cancer. Endocr.-Relat. Cancer 2014, 21, T285–T300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borah, A.; Raveendran, S.; Rochani, A.; Maekawa, T.; Kumar, D.S. Targeting self-renewal pathways in cancer stem cells: Clinical implications for cancer therapy. Oncogenesis 2015, 4, e177. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, F.H.; Li, Y.; Wang, Z.; Kong, D. Pancreatic cancer stem cells and emt in drug resistance and metastasis. Minerva Chir. 2009, 64, 489–500. [Google Scholar]
- Singh, A.; Settleman, J. Emt, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Kim, S.M.; Kim, B.W.; Chang, H.J.; Kim, S.Y.; Park, C.S.; Park, K.C.; Chang, H.S. Anti-cancer effects of hnha and lenvatinib by the suppression of emt-mediated drug resistance in cancer stem cells. Neoplasia 2018, 20, 197–206. [Google Scholar] [CrossRef]
- Du, B.; Shim, J.S. Targeting epithelial-mesenchymal transition (emt) to overcome drug resistance in cancer. Molecules 2016, 21(7), 965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.C. Cancer stem cells: Role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine 2016, 95, S20–S25. [Google Scholar] [CrossRef] [PubMed]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer stem cells (cscs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018, 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G. The proto-oncogene bcl-2 and its role in regulating apoptosis. Nat. Med. 1997, 3, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, E.F.; Lavik, A.R.; Distelhorst, C.W. Bcl-2 regulation of the inositol 1,4,5-trisphosphate receptor and calcium signaling in normal and malignant lymphocytes: Potential new target for cancer treatment. Biochim. Biophys. Acta 2014, 1843, 2205–2210. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Lv, J.; Li, A.; Qiao, J.; Fang, L.; Li, Z.; Li, B.; Zhao, W.; Chen, H.; Wang, L. Constitutive expression of bcl-2 induces epithelial-mesenchymal transition in mammary epithelial cells. BMC Cancer 2015, 15, 476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar] [CrossRef] [Green Version]
- Morales-Cruz, M.; Figueroa, C.M.; Gonzalez-Robles, T.; Delgado, Y.; Molina, A.; Mendez, J.; Morales, M.; Griebenow, K. Activation of caspase-dependent apoptosis by intracellular delivery of cytochrome c-based nanoparticles. J. Nanobiotechnol. 2014, 12, 33. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.H.; Choi, K.H.; Kim, S.Y.; Park, C.S.; Kim, S.M.; Park, K.C. Patient-derived, drug-resistant colon cancer cells evade chemotherapeutic drug effects via the induction of epithelial-mesenchymal transition-mediated angiogenesis. Int. J. Mol. Sci. 2020, 21, 7469. [Google Scholar] [CrossRef]
- Han, J.Y.; Ahn, K.S.; Kim, T.S.; Kim, Y.H.; Cho, K.B.; Shin, D.W.; Baek, W.K.; Suh, S.I.; Jang, B.C.; Kang, K.J. Liquid biopsy from bile-circulating tumor DNA in patients with biliary tract cancer. Cancers 2021, 13, 4581. [Google Scholar] [CrossRef]
- Kim, S.M.; Kim, S.Y.; Park, C.S.; Chang, H.S.; Park, K.C. Impact of age-related genetic differences on the therapeutic outcome of papillary thyroid cancer. Cancers 2020, 12, 448. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yun, H.J.; Kim, M.; Kim, S.Y.; Fang, S.; Kim, Y.; Chang, H.-S.; Chang, H.-J.; Park, K.C. Effects of Anti-Cancer Drug Sensitivity-Related Genetic Differences on Therapeutic Approaches in Refractory Papillary Thyroid Cancer. Int. J. Mol. Sci. 2022, 23, 699. https://doi.org/10.3390/ijms23020699
Yun HJ, Kim M, Kim SY, Fang S, Kim Y, Chang H-S, Chang H-J, Park KC. Effects of Anti-Cancer Drug Sensitivity-Related Genetic Differences on Therapeutic Approaches in Refractory Papillary Thyroid Cancer. International Journal of Molecular Sciences. 2022; 23(2):699. https://doi.org/10.3390/ijms23020699
Chicago/Turabian StyleYun, Hyeok Jun, Minki Kim, Sang Yong Kim, Sungsoon Fang, Yonjung Kim, Hang-Seok Chang, Ho-Jin Chang, and Ki Cheong Park. 2022. "Effects of Anti-Cancer Drug Sensitivity-Related Genetic Differences on Therapeutic Approaches in Refractory Papillary Thyroid Cancer" International Journal of Molecular Sciences 23, no. 2: 699. https://doi.org/10.3390/ijms23020699
APA StyleYun, H. J., Kim, M., Kim, S. Y., Fang, S., Kim, Y., Chang, H. -S., Chang, H. -J., & Park, K. C. (2022). Effects of Anti-Cancer Drug Sensitivity-Related Genetic Differences on Therapeutic Approaches in Refractory Papillary Thyroid Cancer. International Journal of Molecular Sciences, 23(2), 699. https://doi.org/10.3390/ijms23020699