
Autophagy Mediates Escherichia Coli-Induced Cellular Inflammatory Injury by Regulating Calcium Mobilization, Mitochondrial Dysfunction, and Endoplasmic Reticulum Stress

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
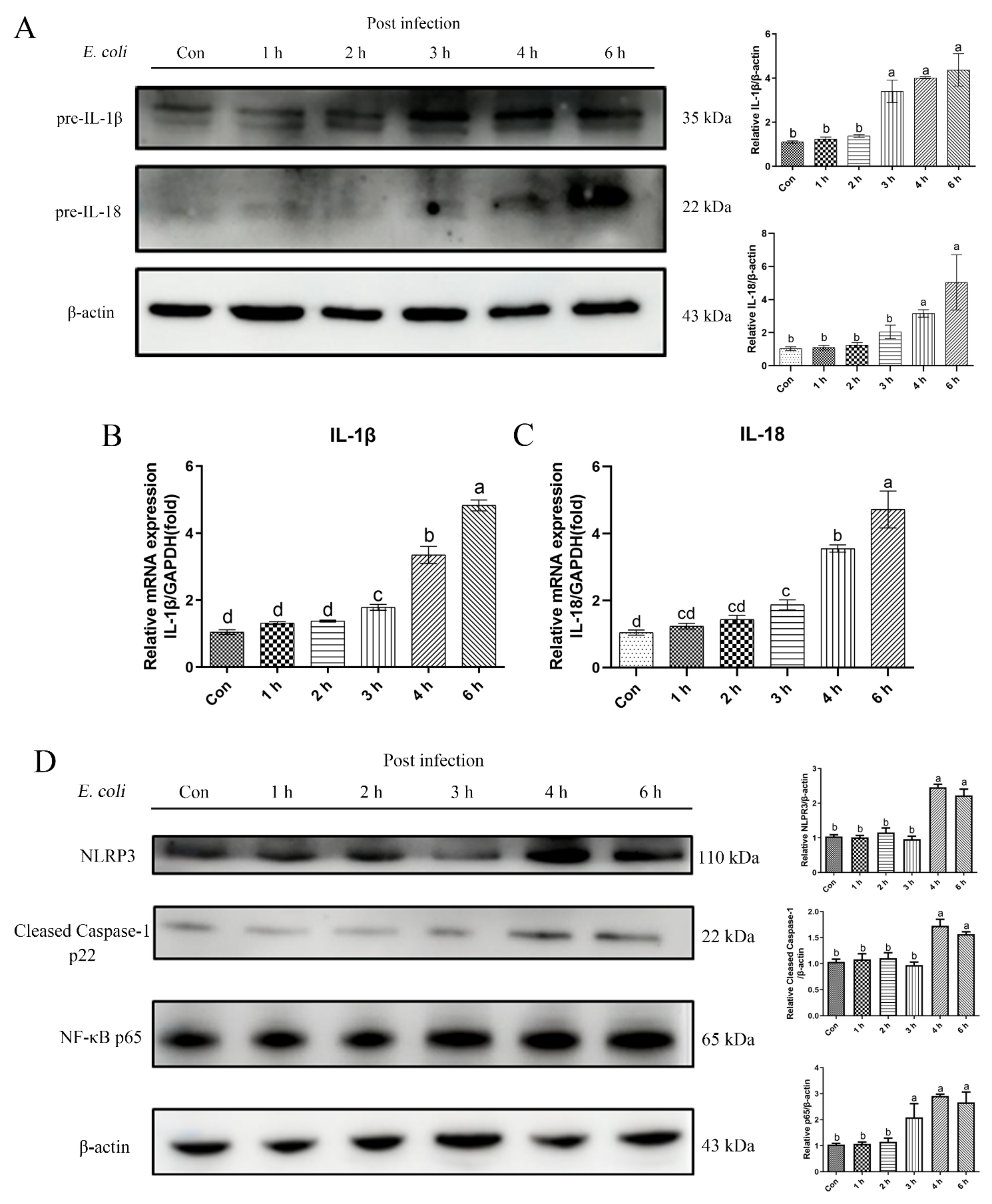
2.1. E. Coli Activated Inflammatory Response in bEECs
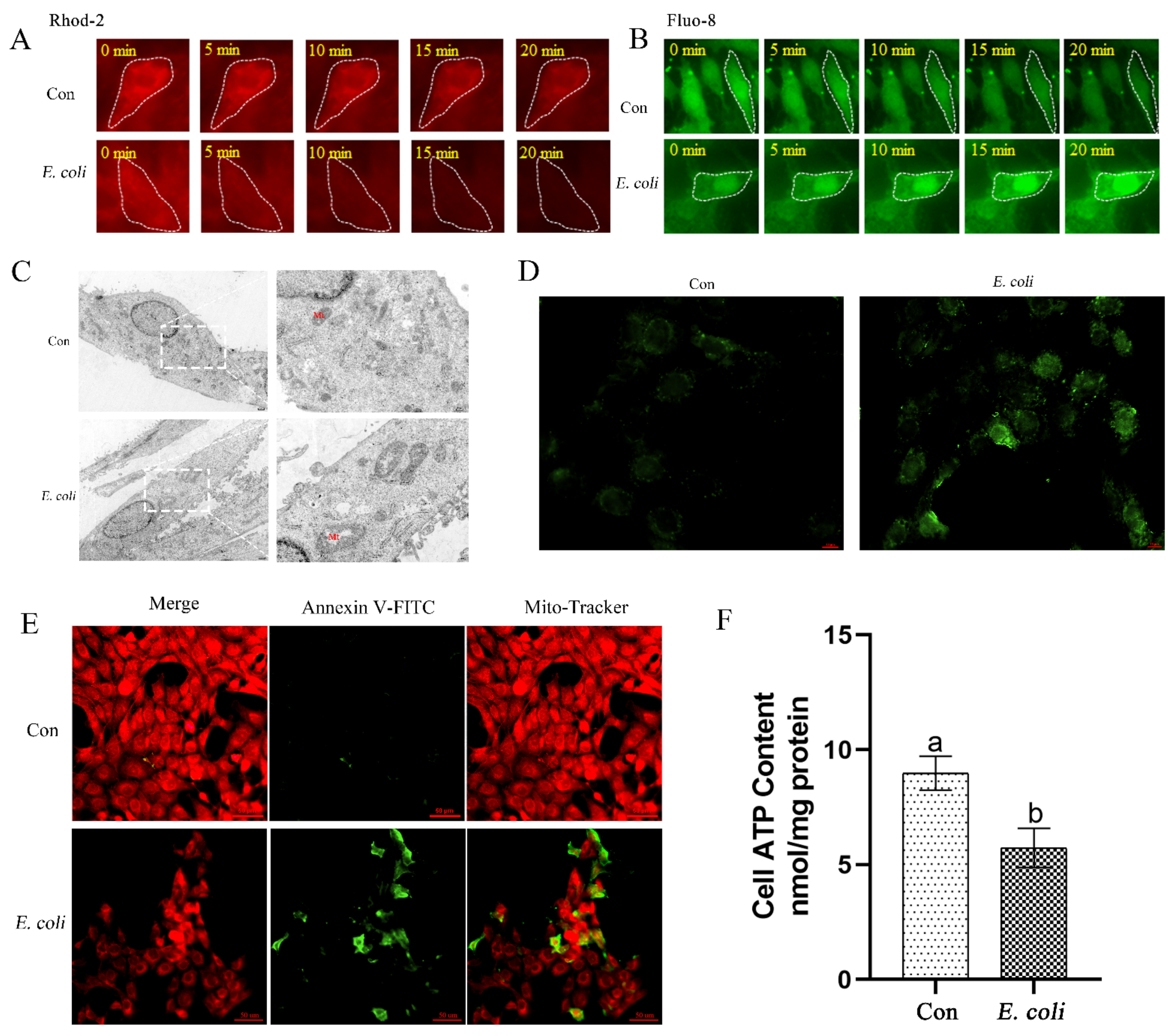
2.2. E. Coli Increases Cellular Ca2+ and Promotes Mitochondrial Dysfunction
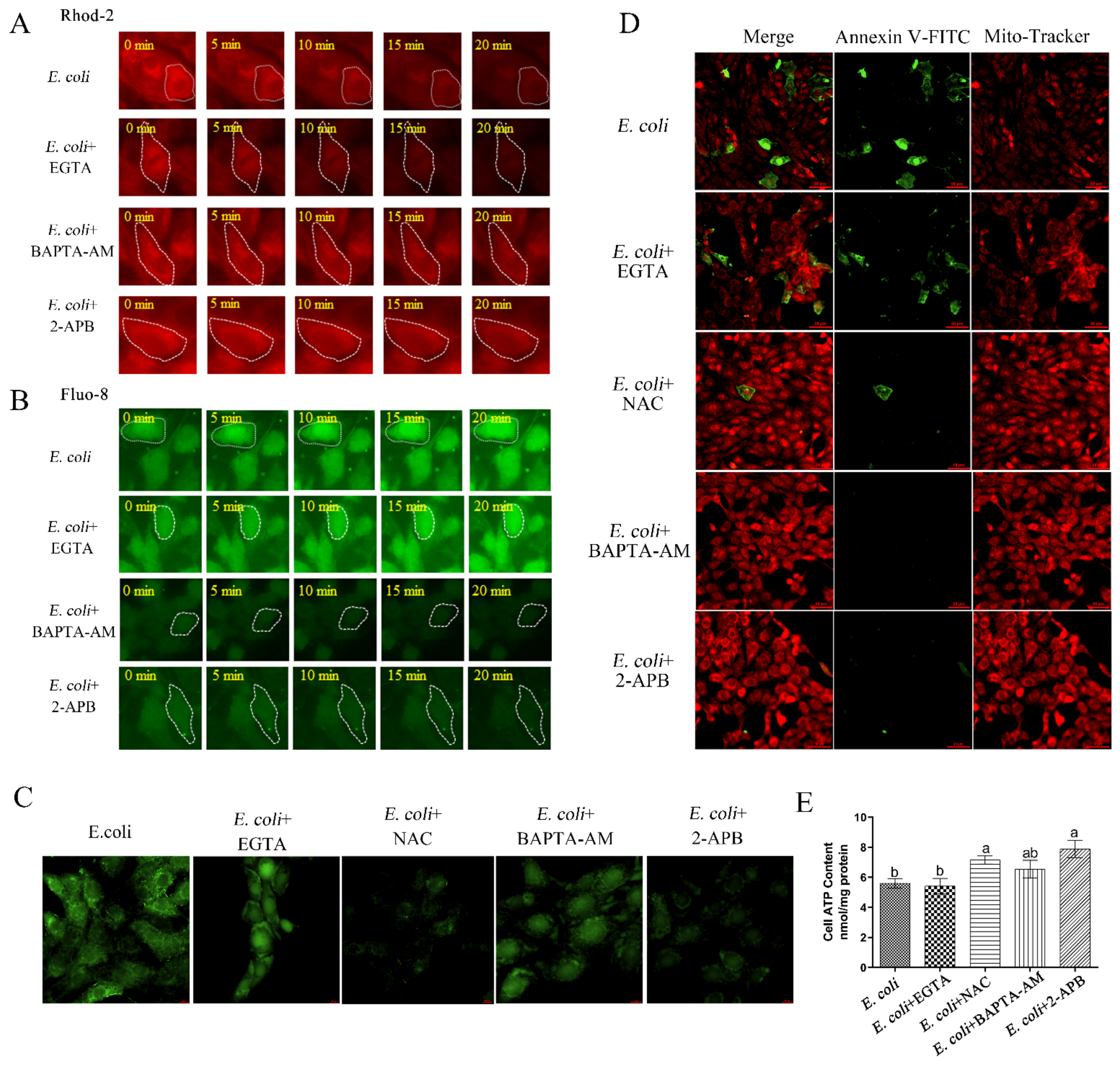
2.3. Cytoplasmic Ca2+ Regulates Mitochondrial Function
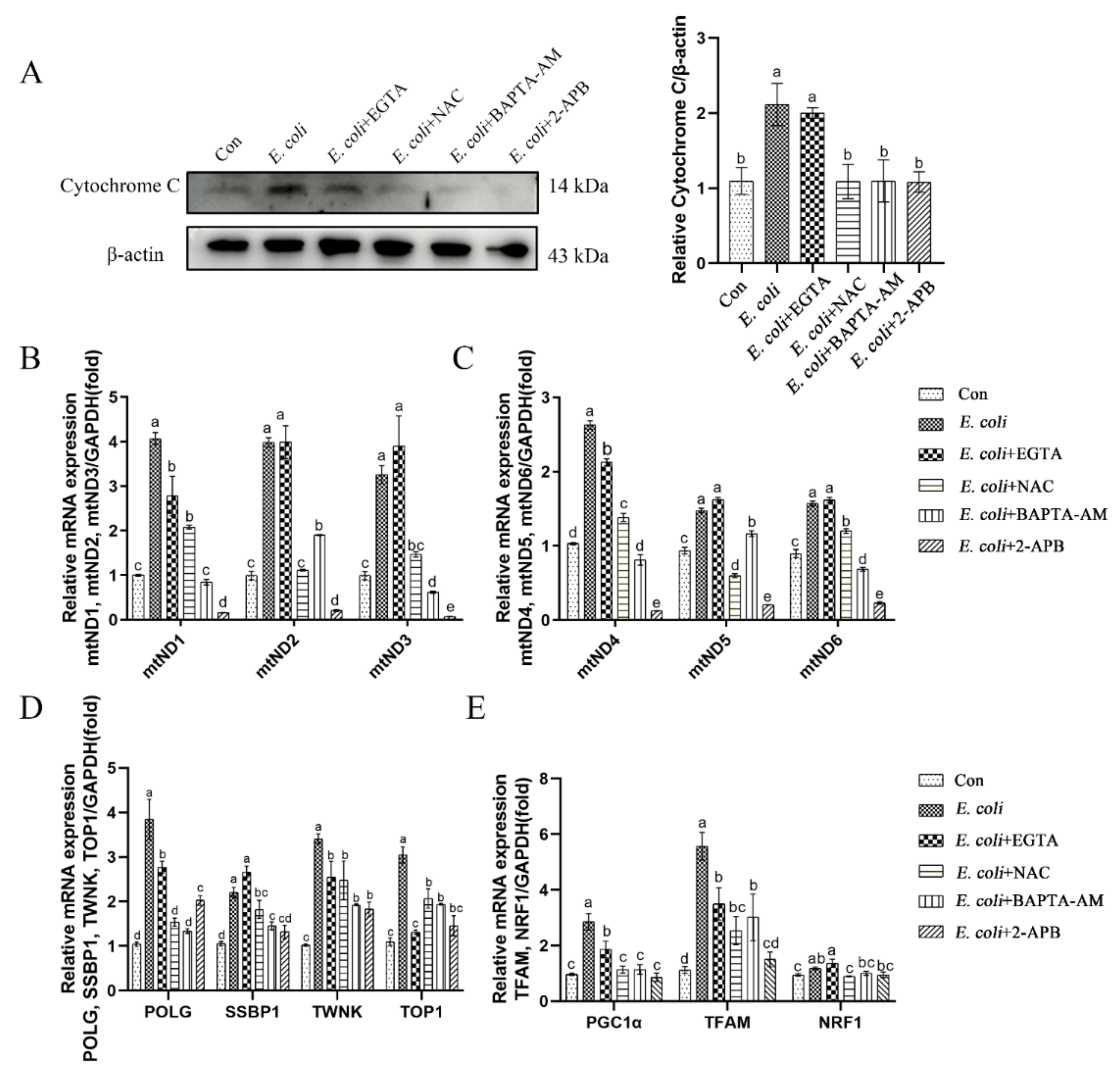
2.4. Cytoplasmic Ca2+ Promotes Mitochondrial Damage Caused by E. Coli
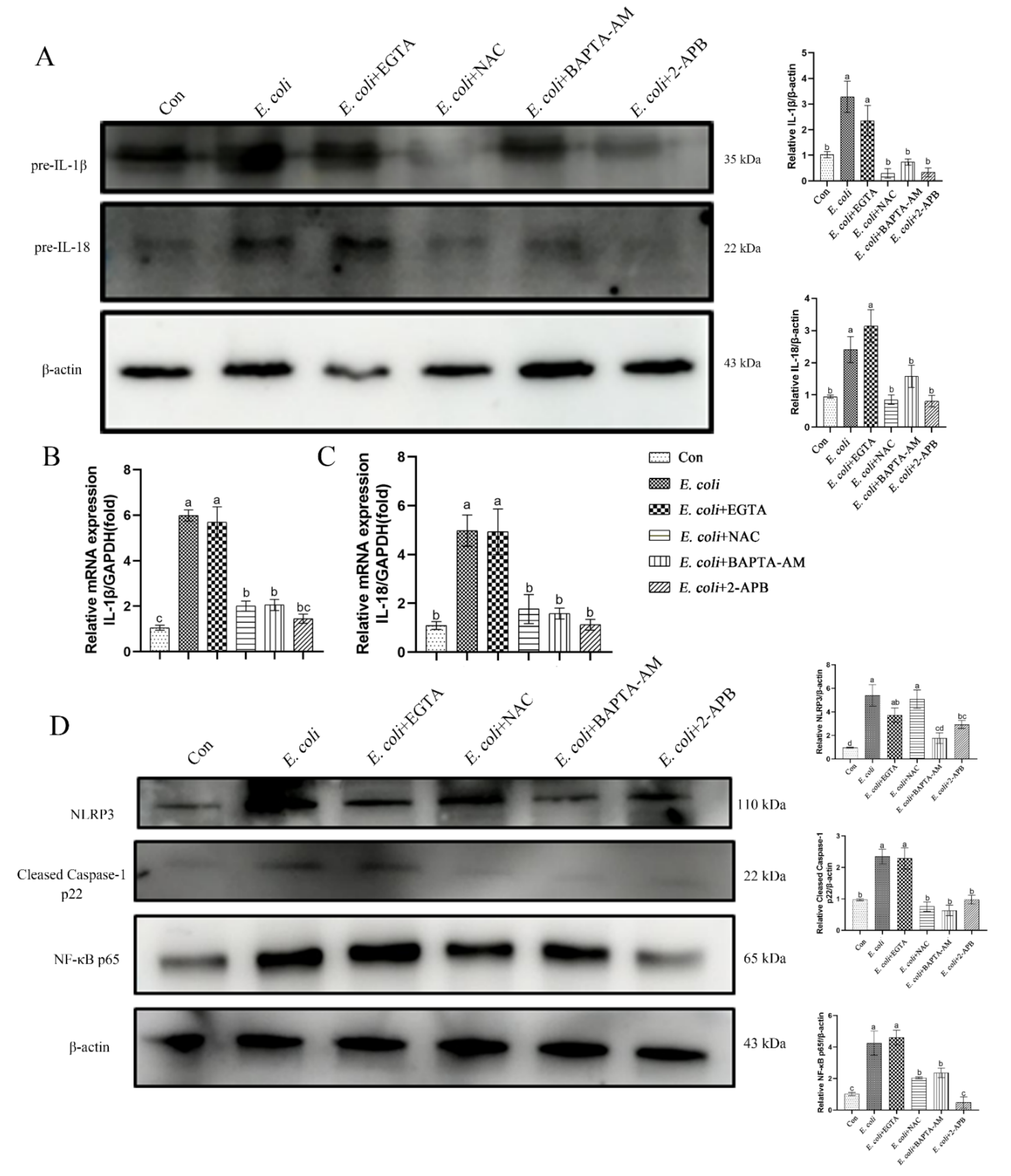
2.5. The Inflammatory Response Induced by E. Coli Requires ROS Production and Release of Ca2+
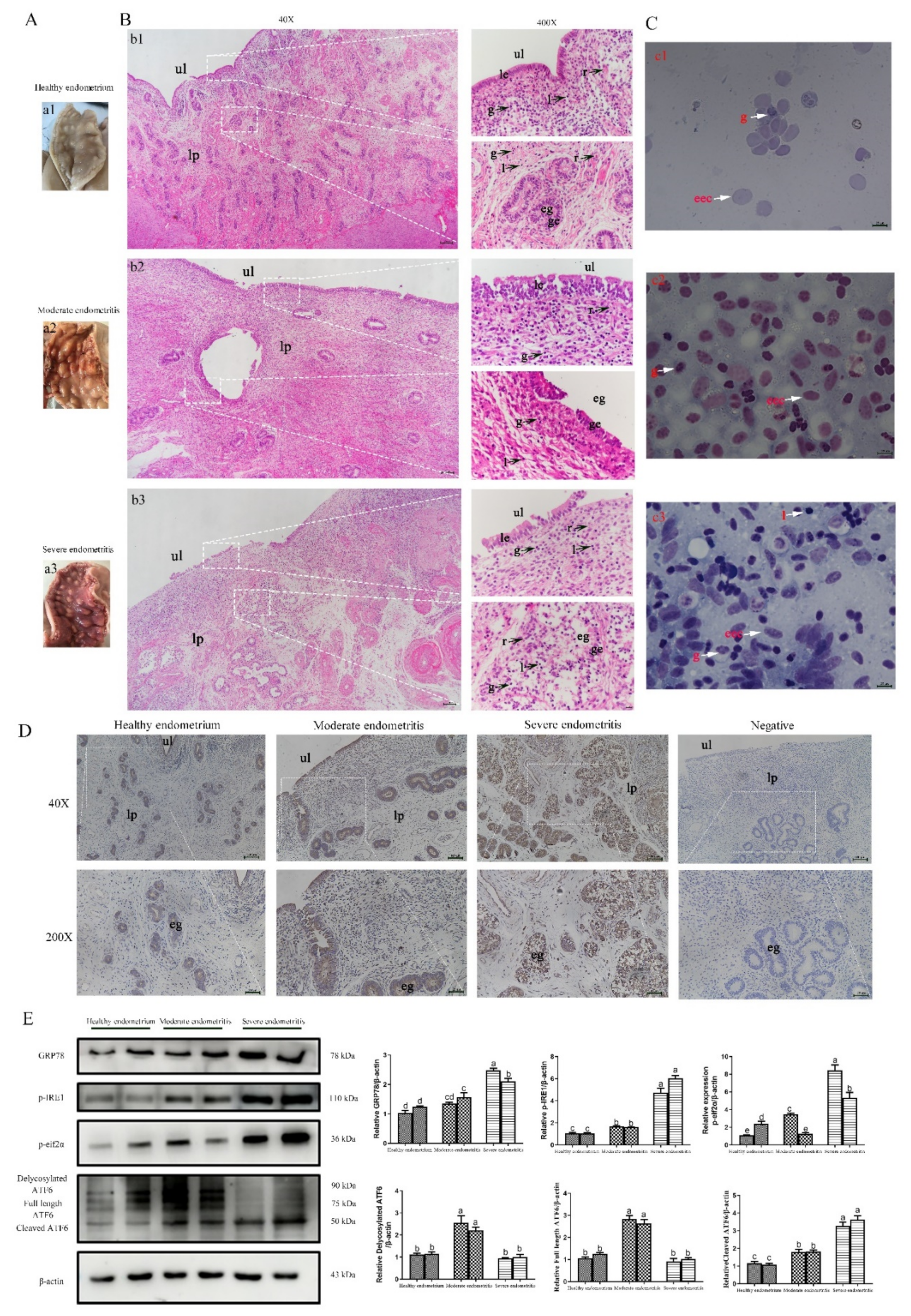
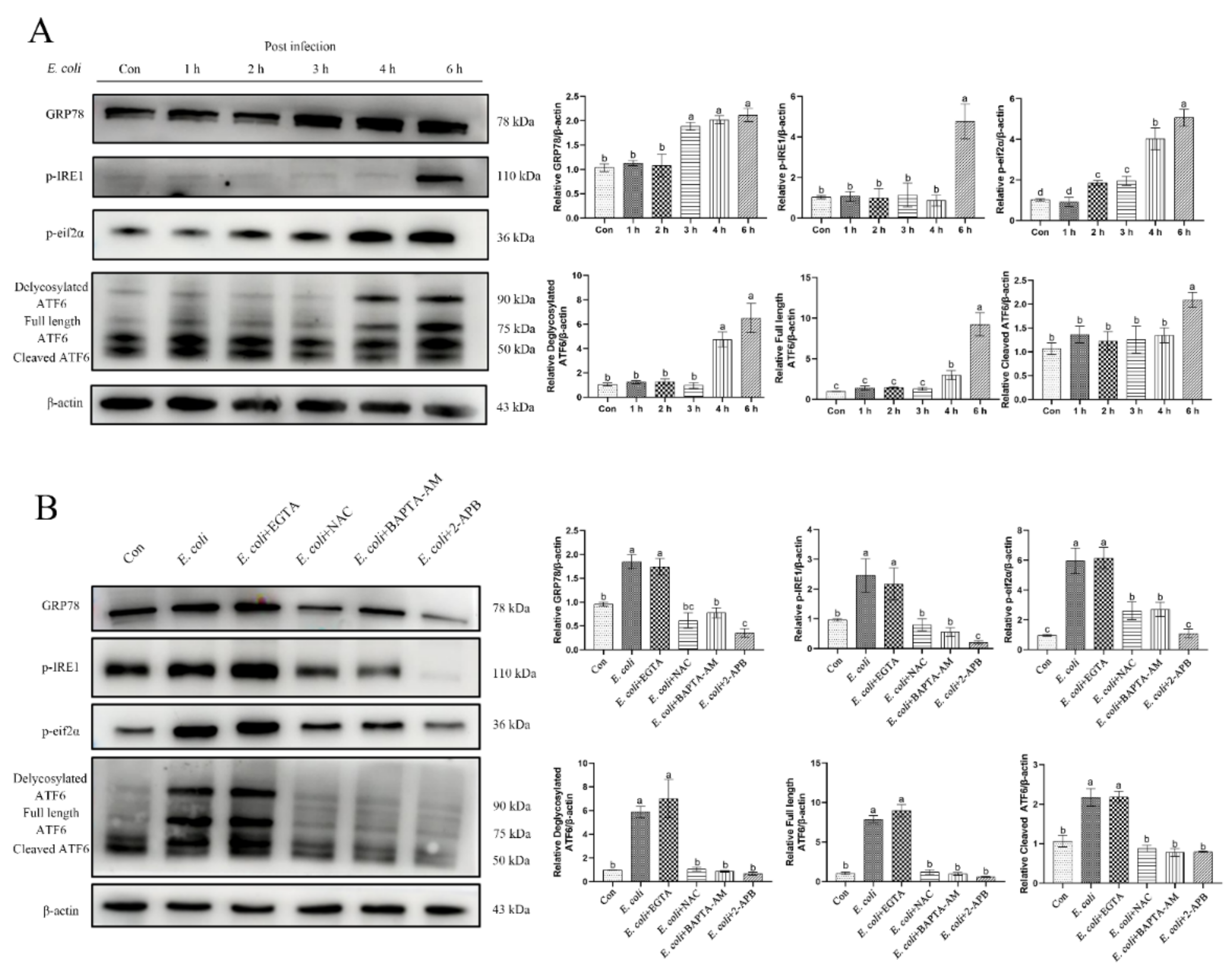
2.6. ERS Is Involved in the Regulation of Bovine Endometritis
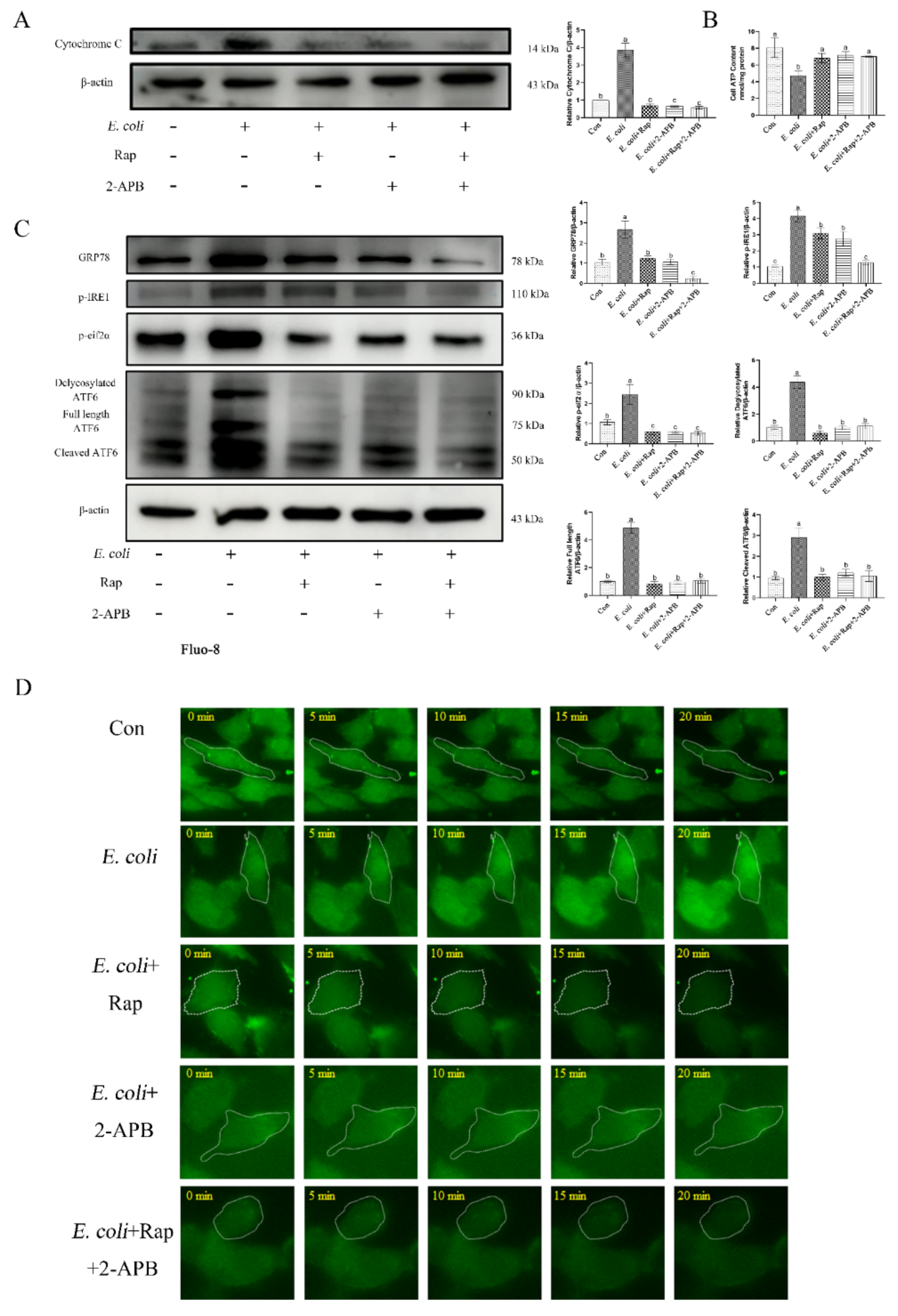
2.7. ERS Involvement in Regulating the E. Coli-Induced Inflammatory Response Depends on Cytoplasmic Ca2+ Levels
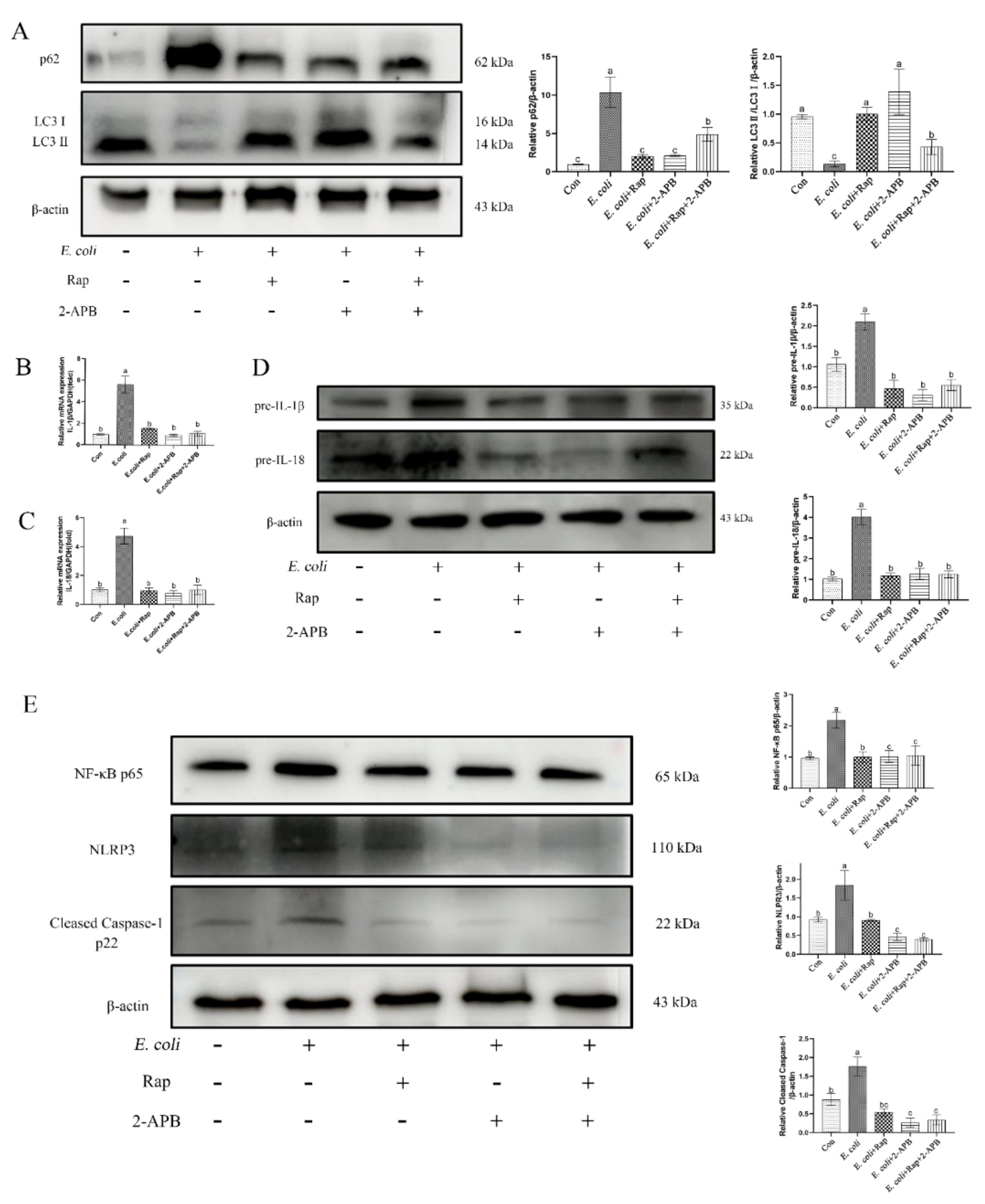
2.8. Enhancement of Autophagic Activity with 2-APB Reduces the E. Coli-Induced Damage Response
3. Discussion
4. Materials and Methods
4.1. Bovine Uterine Tissue Collection
4.2. Bacterial Strains, Cell Culture and Growth Conditions
4.3. Real-Time Quantitative PCR
4.4. Western Blot Analysis
4.5. Real-Time Cellular Imaging
4.6. Transmission Electron Microscopy
4.7. Cellular ROS Detection
4.8. Mitochondrial Membrane Potential and Apoptosis Detection
4.9. Cellular ATP Detection
4.10. Diff Staining
4.11. Hematoxylin-Eosin (HE) Stain
4.12. Immunohistochemistry
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sheldon, I.M.; Cronin, J.G.; Bromfield, J.J. Tolerance and Innate Immunity Shape the Development of Postpartum Uterine Disease and the Impact of Endometritis in Dairy Cattle. Annu. Rev. Anim. Biosci. 2019, 7, 361–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagener, K.; Prunner, I.; Pothmann, H.; Drillich, M.; Ehling-Schulz, M. Diversity and health status specific fluctuations of intrauterine microbial communities in postpartum dairy cows. Vet. Microbiol. 2015, 175, 286–293. [Google Scholar] [CrossRef]
- Carneiro, L.C.; Cronin, J.G.; Sheldon, I.M. Mechanisms linking bacterial infections of the bovine endometrium to disease and infertility. Reprod. Biol. 2016, 16, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sicsic, R.; Goshen, T.; Dutta, R.; Kedem-Vaanunu, N.; Kaplan-Shabtai, V.; Pasternak, Z.; Gottlieb, Y.; Shpigel, N.Y.; Raz, T. Microbial communities and inflammatory response in the endometrium differ between normal and metritic dairy cows at 5–10 days post-partum. Vet. Res. 2018, 49, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piersanti, R.L.; Zimpel, R.; Molinari, P.C.C.; Dickson, M.J.; Ma, Z.; Jeong, K.C.; Santos, J.E.P.; Sheldon, I.M.; Bromfield, J.J. A model of clinical endometritis in Holstein heifers using pathogenic Escherichia coli and Trueperella pyogenes. J. Dairy Sci. 2019, 102, 2686–2697. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Lemaitre, B.; Nicolas, E.; Michaut, L.; Reichhart, J.M.; Hoffmann, J.A. The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 1996, 86, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Xu, L.; Park, H.B.; Hwang, J.; Kwak, M.; Lee, P.C.W.; Liang, G.; Zhang, X.; Xu, J.; Jin, J.O. Escherichia coli adhesion portion FimH functions as an adjuvant for cancer immunotherapy. Nat. Commun. 2020, 11, 1187. [Google Scholar] [CrossRef]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca2+ homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef]
- Bravo-Sagua, R.; Parra, V.; López-Crisosto, C.; Díaz, P.; Quest, A.F.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. Compr. Physiol. 2017, 7, 623–634. [Google Scholar] [PubMed]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biczo, G.; Vegh, E.T.; Shalbueva, N.; Mareninova, O.A.; Elperin, J.; Lotshaw, E.; Gretler, S.; Lugea, A.; Malla, S.R.; Dawson, D.; et al. Mitochondrial Dysfunction, Through Impaired Autophagy, Leads to Endoplasmic Reticulum Stress, Deregulated Lipid Metabolism, and Pancreatitis in Animal Models. Gastroenterology 2018, 154, 689–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morishita, H.; Mizushima, N. Diverse Cellular Roles of Autophagy. Annu. Rev. Cell Dev. Biol. 2019, 35, 453–475. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Clarke, A.J.; Simon, A.K. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat. Rev. Immunol. 2019, 19, 170–183. [Google Scholar] [CrossRef]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Deretic, V.; Levine, B. Autophagy balances inflammation in innate immunity. Autophagy 2018, 14, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, A.A.A.; Yang, D.; Liu, S.; Lin, P.; Mohamad, O.A.A.; Jin, Y. Endoplasmic reticulum stress is involved in lipopolysaccharide-induced inflammatory response and apoptosis in goat endometrial stromal cells. Mol. Reprod. Dev. 2019, 86, 908–921. [Google Scholar] [CrossRef]
- Hu, X.; Li, D.; Wang, J.; Guo, J.; Li, Y.; Cao, Y.; Zhang, N.; Fu, Y. Melatonin inhibits endoplasmic reticulum stress-associated TXNIP/NLRP3 inflammasome activation in lipopolysaccharide-induced endometritis in mice. Int. Immunopharmacol. 2018, 64, 101–109. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Brodzki, P.; Kostro, K.; Krakowski, L.; Marczuk, J. Inflammatory cytokine and acute phase protein concentrations in the peripheral blood and uterine washings of cows with subclinical endometritis in the late postpartum period. Vet. Res. Commun. 2015, 39, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Salilew-Wondim, D.; Ibrahim, S.; Gebremedhn, S.; Tesfaye, D.; Heppelmann, M.; Bollwein, H.; Pfarrer, C.; Tholen, E.; Neuhoff, C.; Schellander, K.; et al. Clinical and subclinical endometritis induced alterations in bovine endometrial transcriptome and miRNome profile. BMC Genom. 2016, 17, 218. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.L.; Song, L.Q.; Huang, Y.M.; Xiong, Y.W.; Zhang, X.A.; Sun, H.; Zhu, X.P.; Meng, G.X.; Xu, J.G.; Ren, Z.H. Effect of enterohaemorrhagic Escherichia coli O157:H7-specific enterohaemolysin on interleukin-1β production differs between human and mouse macrophages due to the different sensitivity of NLRP3 activation. Immunology 2015, 145, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Nold-Petry, C.A.; Nold, M.F.; Joosten, L.A.; Opitz, B.; van der Meer, J.H.; van de Veerdonk, F.L.; Ferwerda, G.; Heinhuis, B.; Devesa, I.; et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 2009, 113, 2324–2335. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Fernandes-Alnemri, T.; Alnemri, E.S. Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J. Clin. Immunol. 2010, 30, 693–702. [Google Scholar] [CrossRef] [Green Version]
- Rathinam, V.A.; Vanaja, S.K.; Waggoner, L.; Sokolovska, A.; Becker, C.; Stuart, L.M.; Leong, J.M.; Fitzgerald, K.A. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 2012, 150, 606–619. [Google Scholar] [CrossRef] [Green Version]
- Rawat, M.; Nighot, M.; Al-Sadi, R.; Gupta, Y.; Viszwapriya, D.; Yochum, G.; Koltun, W.; Ma, T.Y. IL1B Increases Intestinal Tight Junction Permeability by Up-regulation of MIR200C-3p, Which Degrades Occludin mRNA. Gastroenterology 2020, 159, 1375–1389. [Google Scholar] [CrossRef]
- Chen, X.; Wang, N.; Zhu, Y.; Lu, Y.; Liu, X.; Zheng, J. The Antimalarial Chloroquine Suppresses LPS-Induced NLRP3 Inflammasome Activation and Confers Protection against Murine Endotoxic Shock. Mediat. Inflamm. 2017, 2017, 6543237. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.S.; Lee, S.; Park, M.A.; Siempos, I.I.; Haslip, M.; Lee, P.J.; Yun, M.; Kim, C.K.; Howrylak, J.; Ryter, S.W.; et al. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J. Clin. Invest. 2015, 125, 665–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Pace, J.; Hayman, M.J.; Galán, J.E. Signal transduction and invasion of epithelial cells by S. typhimurium. Cell 1993, 72, 505–514. [Google Scholar] [CrossRef]
- Gekara, N.O.; Westphal, K.; Ma, B.; Rohde, M.; Groebe, L.; Weiss, S. The multiple mechanisms of Ca2+ signalling by listeriolysin O, the cholesterol-dependent cytolysin of Listeria monocytogenes. Cell Microbiol. 2007, 9, 2008–2021. [Google Scholar] [CrossRef]
- Ramachandran, R.P.; Spiegel, C.; Keren, Y.; Danieli, T.; Melamed-Book, N.; Pal, R.R.; Zlotkin-Rivkin, E.; Rosenshine, I.; Aroeti, B. Mitochondrial Targeting of the Enteropathogenic Escherichia coli Map Triggers Calcium Mobilization, ADAM10-MAP Kinase Signaling, and Host Cell Apoptosis. mBio 2020, 11, e01397-20. [Google Scholar] [CrossRef]
- Xue, Y.; Du, M.; Zhu, M.-J. Quercetin suppresses NLRP3 inflammasome activation in epithelial cells triggered by Escherichia coli O157:H7. Free Rad. Biol. Med. 2017, 108, 760–769. [Google Scholar] [CrossRef]
- Maekawa, H.; Inoue, T.; Ouchi, H.; Jao, T.M.; Inoue, R.; Nishi, H.; Fujii, R.; Ishidate, F.; Tanaka, T.; Tanaka, Y.; et al. Mitochondrial Damage Causes Inflammation via cGAS-STING Signaling in Acute Kidney Injury. Cell Rep. 2019, 29, 1261–1273. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Hernandez, A.; Asmat, T.M.; Tenenbaum, T.; Jonsson, A.-B.; Schwerk, C.; Schroten, H. Impact of Calcium Signaling during Infection of Neisseria meningitidis to Human Brain Microvascular Endothelial Cells. PLoS ONE 2014, 9, e114474. [Google Scholar]
- Liu, X.; Wang, N.; Zhu, Y.; Yang, Y.; Chen, X.; Chen, Q.; Zhou, H.; Zheng, J. Extracellular calcium influx promotes antibacterial autophagy in Escherichia coli infected murine macrophages via CaMKKβ dependent activation of ERK1/2, AMPK and FoxO1. Biochem. Biophys. Res. Commun. 2016, 469, 639–645. [Google Scholar] [CrossRef]
- de Boland, A.R.; Norman, A.W. Influx of extracellular calcium mediates 1,25-dihydroxyvitamin D3-dependent transcaltachia (the rapid stimulation of duodenal Ca2+ transport). Endocrinology 1990, 127, 2475–2480. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Kalpage, H.A.; Bazylianska, V.; Recanati, M.A.; Fite, A.; Liu, J.; Wan, J.; Mantena, N.; Malek, M.H.; Podgorski, I.; Heath, E.I.; et al. Tissue-specific regulation of cytochrome c by post-translational modifications: Respiration, the mitochondrial membrane potential, ROS, and apoptosis. Faseb J. 2019, 33, 1540–1553. [Google Scholar] [CrossRef]
- Feng, X.; Ching, C.B.; Chen, W.N. EBV up-regulates cytochrome c through VDAC1 regulations and decreases the release of cytoplasmic Ca2+ in the NPC cell line. Cell Biol Int. 2012, 36, 733–738. [Google Scholar] [CrossRef]
- Kalpage, H.A.; Wan, J.; Morse, P.T.; Zurek, M.P.; Turner, A.A.; Khobeir, A.; Yazdi, N.; Hakim, L.; Liu, J.; Vaishnav, A.; et al. Cytochrome c phosphorylation: Control of mitochondrial electron transport chain flux and apoptosis. Int. J. Biochem. Cell Biol. 2020, 121, 105704. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Huang, L.S.; Hong, Z.; Wu, W.; Xiong, S.; Zhong, M.; Gao, X.; Rehman, J.; Malik, A.B. mtDNA Activates cGAS Signaling and Suppresses the YAP-Mediated Endothelial Cell Proliferation Program to Promote Inflammatory Injury. Immunity 2020, 52, 475–486. [Google Scholar] [CrossRef]
- Yan, C.; Duanmu, X.; Zeng, L.; Liu, B.; Song, Z. Mitochondrial DNA: Distribution, Mutations, and Elimination. Cells 2019, 8, 379. [Google Scholar] [CrossRef] [Green Version]
- Pereira, C.V.; Gitschlag, B.L.; Patel, M.R. Cellular mechanisms of mtDNA heteroplasmy dynamics. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 510–525. [Google Scholar] [CrossRef]
- Russell, L.K.; Mansfield, C.M.; Lehman, J.J.; Kovacs, A.; Courtois, M.; Saffitz, J.E.; Medeiros, D.M.; Valencik, M.L.; McDonald, J.A.; Kelly, D.P. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ. Res. 2004, 94, 525–533. [Google Scholar] [CrossRef] [Green Version]
- Ciron, C.; Lengacher, S.; Dusonchet, J.; Aebischer, P.; Schneider, B.L. Sustained expression of PGC-1α in the rat nigrostriatal system selectively impairs dopaminergic function. Hum. Mol. Genet. 2012, 21, 1861–1876. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Chen, X.; Gao, C.; Jiao, L.; Wang, J.; Xu, G.; Fu, H.; Guo, X.; Zhao, Y. IL-6 induces lipolysis and mitochondrial dysfunction, but does not affect insulin-mediated glucose transport in 3T3-L1 adipocytes. J. Bioenerg. Biomembr. 2011, 43, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.; Chu, C.T.; Kaufman, B.A. The mitochondrial transcription factor TFAM in neurodegeneration: Emerging evidence and mechanisms. FEBS Lett. 2018, 592, 793–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umar, T.; Yin, B.; Umer, S.; Ma, X.; Jiang, K.; Umar, Z.; Akhtar, M.; Shaukat, A.; Deng, G. MicroRNA: Could It Play a Role in Bovine Endometritis? Inflammation 2021, 44, 1683–1695. [Google Scholar] [CrossRef] [PubMed]
- Salemi, Z.; Rezaie, A.; Goorani Nejad, S.; Mohammadian, B. Histopathological and cytological analyses of endometrium in water buffaloes (Bubalus bubalis) to detect estrus and endometritis. Vet. Res. Forum. 2020, 11, 409–414. [Google Scholar]
- Zhang, S.D.; Dong, S.W.; Wang, D.S.; Oguejiofor, C.F.; Fouladi-Nashta, A.A.; Yang, Z.Q.; Yan, Z.T. Differential proteomic profiling of endometrium and plasma indicate the importance of hydrolysis in bovine endometritis. J. Dairy Sci. 2017, 100, 9324–9337. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Song, Z.; Cao, X.; Fan, M.; Zhou, Y.; Zhang, G. Interleukin-33 regulates the endoplasmic reticulum stress of human myometrium via an influx of calcium during initiation of labor. eLife 2022, 11, e75072. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.P.; Ka, S.M.; Hsu, W.H.; Chen, A.; Chao, L.K.; Lin, C.C.; Hsieh, C.C.; Chen, M.C.; Chiu, H.W.; Ho, C.L.; et al. Resveratrol inhibits NLRP3 inflammasome activation by preserving mitochondrial integrity and augmenting autophagy. J. Cell Physiol. 2015, 230, 1567–1579. [Google Scholar] [CrossRef]
- Hu, Y.X.; Han, X.S.; Jing, Q. Ca(2+) Ion and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 151–166. [Google Scholar]
- Yang, D.; Jiang, T.; Liu, J.; Hong, J.; Lin, P.; Chen, H.; Zhou, D.; Tang, K.; Wang, A.; Jin, Y. Hormone regulates endometrial function via cooperation of endoplasmic reticulum stress and mTOR-autophagy. J. Cell Physiol. 2018, 233, 6644–6659. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Qiu, R.; Liu, R.; Song, P.; Lin, P.; Chen, H.; Zhou, D.; Wang, A.; Jin, Y. Autophagy Mediates Escherichia Coli-Induced Cellular Inflammatory Injury by Regulating Calcium Mobilization, Mitochondrial Dysfunction, and Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2022, 23, 14174. https://doi.org/10.3390/ijms232214174
Liu J, Qiu R, Liu R, Song P, Lin P, Chen H, Zhou D, Wang A, Jin Y. Autophagy Mediates Escherichia Coli-Induced Cellular Inflammatory Injury by Regulating Calcium Mobilization, Mitochondrial Dysfunction, and Endoplasmic Reticulum Stress. International Journal of Molecular Sciences. 2022; 23(22):14174. https://doi.org/10.3390/ijms232214174
Chicago/Turabian StyleLiu, Jianguo, Rendong Qiu, Ran Liu, Pengjie Song, Pengfei Lin, Huatao Chen, Dong Zhou, Aihua Wang, and Yaping Jin. 2022. "Autophagy Mediates Escherichia Coli-Induced Cellular Inflammatory Injury by Regulating Calcium Mobilization, Mitochondrial Dysfunction, and Endoplasmic Reticulum Stress" International Journal of Molecular Sciences 23, no. 22: 14174. https://doi.org/10.3390/ijms232214174
APA StyleLiu, J., Qiu, R., Liu, R., Song, P., Lin, P., Chen, H., Zhou, D., Wang, A., & Jin, Y. (2022). Autophagy Mediates Escherichia Coli-Induced Cellular Inflammatory Injury by Regulating Calcium Mobilization, Mitochondrial Dysfunction, and Endoplasmic Reticulum Stress. International Journal of Molecular Sciences, 23(22), 14174. https://doi.org/10.3390/ijms232214174