
Scalable Generation of Nanovesicles from Human-Induced Pluripotent Stem Cells for Cardiac Repair

 , , and
, , and
Abstract
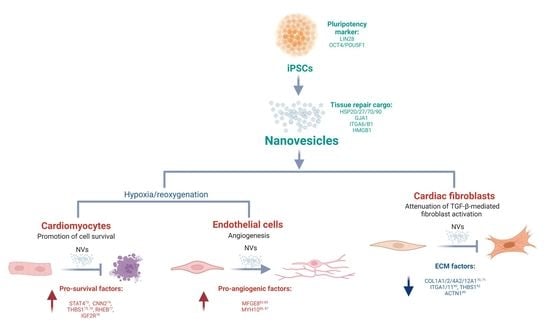
:
1. Introduction
2. Results
2.1. Validation of Human Induced Pluripotent Stem Cell Models

2.2. Generation and Characterization of Nanovesicles from iPSCs
2.3. Proteome Analysis Identify Stem Cell Markers and Tissue Repair Proteins in NVs
2.4. Uptake of iPSC NVs by Different Cardiac Cells
2.5. iPSC NVs Functionally Regulates Cell Survival, Angiogenesis, and Fibroblast Activation In Vitro
2.6. NVs Reprogram Target Cell Proteome to Support Cardiac Reparative and Protective Phenotype

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | NV Treatment | ID | Reactome Enrichment Pathways | Proteins Mapped | p-Val | Description | References |
|---|---|---|---|---|---|---|---|
| Cardiomyocyte survival | Target cells increased expression following iPSC NV treatments | R-HSA-75105 | Fatty acyl-CoA biosynthesis | ACSL3, ACSL4, HACD3 | 2.20 × 10−4 | NVs restore fatty acid metabolism; alterations affect the remodelling and functional capacity of cardiac cell/tissue | [84,85] |
| R-HSA-8978868 | Fatty acid metabolism | ACSL3, ACSL4, HACD3, HSD17B4 | 2.40 × 10−3 | ||||
| R-HSA-191273 | Cholesterol biosynthesis | FDFT1, MSMO1 | 3.36 × 10−3 | ||||
| HUVECs tube formation (angiogenesis) | Target cells increased expression following iPSC NV treatments | R-HSA-70326 | Glucose metabolism | PCK2, PFKL, SLC25A10, SLC25A11, SLC25A13 | 6.39 × 10−4 | NVs restore basal glycolytic metabolism of endothelial cells | [92,93] |
| Cardiac fibroblast (TGFβ-mediated) activation | Target cells decreased expression following iPSC NV treatments | R-HSA-446353 | Cell-extracellular matrix interactions | ACTN1, FBLIM1, FERMT2, FLNA, FLNC, RSU1VASP | 9.45 × 10−10 | NVs attenuate ECM remodelling, deposition, organization capabilities of hCFs | [75,96,97,98] |
| R-HSA-1474244 | Extracellular matrix organization | ACTN1, COL12A1, COL1A1, COL1A2, COL4A2, CTSD, ICAM1, ITGA1, ITGA11, ITGAV, P3H1, P3H3, PPIB, THBS1, TIMP2 | 3.54 × 10−6 | ||||
| R-HSA-216083 | Integrin cell surface interactions | COL1A1, COL1A2, COL4A2, ICAM1, ITGA1, ITGA11, ITGAV, THBS1 | 8.39 × 10−6 | ||||
| R-HSA-1650814 | Collagen biosynthesis and modifying enzymes | COL12A1, COL1A1, COL1A2, COL4A2, P3H1, P3H3, PPIB | 1.58 × 10−5 | ||||
| R-HSA-3000171 | Non-integrin membrane-ECM interactions | ACTN1, COL1A1, COL1A2, COL4A2, ITGAV, THBS1 | 7.61 × 10−5 | ||||
| R-HSA-1474290 | Collagen formation | COL12A1, COL1A1, COL1A2, COL4A2, P3H1, P3H3, PPIB | 1.07 × 10−4 | ||||
| R-HSA-446728 | Cell junction organization | ACTN1, FBLIM1, FERMT2, FLNA, FLNC, RSU1, VASP | 1.15 × 10−4 | ||||
| R-HSA-8948216 | Collagen chain trimerization | COL12A1, COL1A1, COL1A2, COL4A2 | 1.94 × 10−3 |
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Differentiation
4.1.1. Human Induced Pluripotent Stem Cells (hiPSCs)
4.1.2. Human iPSCs Cardiomyocytes Differentiation and Culture
4.1.3. Human Umbilical Vein Endothelial Cells (HUVEC) Culture
4.1.4. Human Cardiac Fibroblasts (hCF) Culture
4.2. Cell Proliferation Assay
4.3. iPSC Nanovesicle Generation and Purification
4.4. Cryo-Electron Microscopy
4.5. Nanoparticle Tracking Analysis
4.6. NV Recipient Cell Uptake
4.7. In Vitro Model of Hypoxia/Reoxygenation
4.8. iPSCs and Cardiomyocyte Survival Assay
4.9. Tube Formation Assay
4.10. Fibroblast TGF-β-Mediated Activation Assay
4.11. Proteomics: Solid-Phase-Enhanced Sample Preparation
4.12. Proteomics: Nano Liquid Chromatography–Tandem Mass Spectrometry
4.13. Proteomics: Data Processing and Informatics/Visualisation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, X.; Gu, H.; Huang, W.; Peng, J.; Li, Y.; Yang, L.; Qin, D.; Essandoh, K.; Wang, Y.; Peng, T.; et al. Hsp20-Mediated Activation of Exosome Biogenesis in Cardiomyocytes Improves Cardiac Function and Angiogenesis in Diabetic Mice. Diabetes 2016, 65, 3111–3128. [Google Scholar] [CrossRef] [Green Version]
- Saha, B.; Momen-Heravi, F.; Furi, I.; Kodys, K.; Catalano, D.; Gangopadhyay, A.; Haraszti, R.; Satishchandran, A.; Iracheta-Vellve, A.; Adejumo, A.; et al. Extracellular vesicles from mice with alcoholic liver disease carry a distinct protein cargo and induce macrophage activation through heat shock protein 90. Hepatology 2018, 67, 1986–2000. [Google Scholar] [CrossRef] [Green Version]
- Yue, Y.; Wang, C.; Benedict, C.; Huang, G.; Truongcao, M.; Roy, R.; Cimini, M.; Garikipati, V.N.S.; Cheng, Z.; Koch, W.J.; et al. Interleukin-10 Deficiency Alters Endothelial Progenitor Cell-Derived Exosome Reparative Effect on Myocardial Repair via Integrin-Linked Kinase Enrichment. Circ. Res. 2020, 126, 315–329. [Google Scholar] [CrossRef]
- Zhou, M.; Wen, Z.; Cheng, F.; Ma, J.; Li, W.; Ren, H.; Sheng, Y.; Dong, H.; Lu, L.; Hu, H.M.; et al. Tumor-released autophagosomes induce IL-10-producing B cells with suppressive activity on T lymphocytes via TLR2-MyD88-NF-kappaB signal pathway. Oncoimmunology 2016, 5, e1180485. [Google Scholar] [CrossRef]
- Li, L.; Guan, Q.; Dai, S.; Wei, W.; Zhang, Y. Integrin beta1 Increases Stem Cell Survival and Cardiac Function after Myocardial Infarction. Front. Pharmacol. 2017, 8, 135. [Google Scholar] [PubMed] [Green Version]
- Tzahor, E.; Dimmeler, S. A coalition to heal-the impact of the cardiac microenvironment. Science 2022, 377, eabm4443. [Google Scholar] [CrossRef]
- Laflamme, M.A.; Murry, C.E. Regenerating the heart. Nat. Biotechnol. 2005, 23, 845–856. [Google Scholar] [CrossRef]
- Segers, V.F.; Lee, R.T. Stem-cell therapy for cardiac disease. Nature 2008, 451, 937–942. [Google Scholar] [CrossRef]
- Menasche, P.; Hagège, A.A.; Vilquin, J.-T.; Desnos, M.; Abergel, E.; Pouzet, B.; Bel, A.; Sarateanu, S.; Scorsin, M.; Schwartz, K.; et al. Autologous skeletal myoblast transplantation for severe postinfarction left ventricular dysfunction. J. Am. Coll. Cardiol. 2003, 41, 1078–1083. [Google Scholar] [CrossRef] [Green Version]
- Loffredo, F.S.; Steinhauser, M.L.; Gannon, J.; Lee, R.T. Bone marrow-derived cell therapy stimulates endogenous cardiomyocyte progenitors and promotes cardiac repair. Cell Stem Cell 2011, 8, 389–398. [Google Scholar] [CrossRef]
- Smart, N.; Bollini, S.; Dubé, K.N.; Vieira, J.M.; Zhou, B.; Davidson, S.; Yellon, D.; Riegler, J.; Price, A.N.; Lythgoe, M.F.; et al. De novo cardiomyocytes from within the activated adult heart after injury. Nature 2011, 474, 640–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kompa, A.R.; Greening, D.W.; Kong, A.M.; McMillan, P.J.; Fang, H.; Saxena, R.; Wong, R.C.B.; Lees, J.G.; Sivakumaran, P.; Newcomb, A.E.; et al. Sustained subcutaneous delivery of secretome of human cardiac stem cells promotes cardiac repair following myocardial infarction. Cardiovasc. Res. 2021, 117, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Lee, B.W.; Nakanishi, K.; Villasante, A.; Williamson, R.; Metz, J.; Kim, J.; Kanai, M.; Bi, L.; Brown, K.; et al. Cardiac recovery via extended cell-free delivery of extracellular vesicles secreted by cardiomyocytes derived from induced pluripotent stem cells. Nat. Biomed. Eng. 2018, 2, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Adamiak, M.; Cheng, G.; Bobis-Wozowicz, S.; Zhao, L.; Kedracka-Krok, S.; Samanta, A.; Karnas, E.; Xuan, Y.-T.; Skupien-Rabian, B.; Chen, X.; et al. Induced Pluripotent Stem Cell (iPSC)-Derived Extracellular Vesicles Are Safer and More Effective for Cardiac Repair Than iPSCs. Circ. Res. 2018, 122, 296–309. [Google Scholar] [CrossRef]
- Gao, L.; Wang, L.; Wei, Y.; Krishnamurthy, P.; Walcott, G.P.; Menasche, P.; Zhang, J. Exosomes secreted by hiPSC-derived cardiac cells improve recovery from myocardial infarction in swine. Sci. Transl. Med. 2020, 12, eaay1318. [Google Scholar] [CrossRef]
- Lee, W.H.; Chen, W.-Y.; Shao, N.-Y.; Xiao, D.; Qin, X.; Baker, N.; Bae, H.R.; Wei, T.-T.; Wang, Y.; Shukla, P.; et al. Comparison of Non-Coding RNAs in Exosomes and Functional Efficacy of Human Embryonic Stem Cell- versus Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cells 2017, 35, 2138–2149. [Google Scholar] [CrossRef] [Green Version]
- Yates, A.G.; Pink, R.C.; Erdbrugger, U.; Siljander, P.R.; Dellar, E.R.; Pantazi, P.; Akbar, N.; Cooke, W.R.; Vatish, M.; Dias-Neto, E.; et al. In sickness and in health: The functional role of extracellular vesicles in physiology and pathology in vivo: Part I: Health and Normal Physiology: Part I: Health and Normal Physiology. J. Extracell. Vesicles 2022, 11, e12151. [Google Scholar]
- O’Grady, T.; Njock, M.-S.; Lion, M.; Bruyr, J.; Mariavelle, E.; Galvan, B.; Boeckx, A.; Struman, I.; Dequiedt, F. Sorting and packaging of RNA into extracellular vesicles shape intracellular transcript levels. BMC Biol. 2022, 20, 72. [Google Scholar] [CrossRef]
- Greening, D.W.; Simpson, R.J. Understanding extracellular vesicle diversity—Current status. Expert Rev. Proteom. 2018, 15, 887–910. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Qiu, H.; Yan, L.; Tian, B.; Yang, G.; Shen, Y.; Vatner, D.E.; Vatner, S.F.; Depre, C. Increased expression of genes promoting cell survival after myocardial infarction in monkeys. FASEB J. 2006, 20, A1190. [Google Scholar] [CrossRef]
- Schafer, S.; Viswanathan, S.; Widjaja, A.A.; Lim, W.-W.; Moreno-Moral, A.; DeLaughter, D.M.; Ng, B.; Patone, G.; Chow, K.; Khin, E.; et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 2017, 552, 110–115. [Google Scholar] [CrossRef]
- Ren, S.; Pan, L.; Yang, L.; Niu, Z.; Wang, L.; Feng, H.; Yuan, M. miR-29a-3p transferred by mesenchymal stem cells-derived extracellular vesicles protects against myocardial injury after severe acute pancreatitis. Life Sci. 2021, 272, 119189. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Pasquino, C.; Sanchez, M.B.H.; Tapparo, M.; Figliolini, F.; Grange, C.; Chiabotto, G.; Cedrino, M.; Deregibus, M.C.; Tetta, C.; et al. HLSC-Derived Extracellular Vesicles Attenuate Liver Fibrosis and Inflammation in a Murine Model of Non-alcoholic Steatohepatitis. Mol. Ther. 2020, 28, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Lou, G.; Yang, Y.; Liu, F.; Ye, B.; Chen, Z.; Zheng, M.; Liu, Y. MiR-122 modification enhances the therapeutic efficacy of adipose tissue-derived mesenchymal stem cells against liver fibrosis. J. Cell. Mol. Med. 2017, 21, 2963–2973. [Google Scholar] [CrossRef]
- Mansouri, N.; Willis, G.R.; Fernandez-Gonzalez, A.; Reis, M.; Nassiri, S.; Mitsialis, S.A.; Kourembanas, S. Mesenchymal stromal cell exosomes prevent and revert experimental pulmonary fibrosis through modulation of monocyte phenotypes. JCI Insight 2019, 4, e128060. [Google Scholar]
- Collino, F.; Lopes, J.A.; Tapparo, M.; Tortelote, G.G.; Kasai-Brunswick, T.H.; Lopes, G.M.; Almeida, D.B.; Skovronova, R.; Wendt, C.H.C.; de Miranda, K.R.; et al. Extracellular Vesicles Derived from Induced Pluripotent Stem Cells Promote Renoprotection in Acute Kidney Injury Model. Cells 2020, 9, 453. [Google Scholar] [CrossRef] [Green Version]
- Povero, D.; Pinatel, E.M.; Leszczynska, A.; Goyal, N.P.; Nishio, T.; Kim, J.; Kneiber, D.; Horcel, L.D.A.; Eguchi, A.; Ordonez, P.M.; et al. Human induced pluripotent stem cell-derived extracellular vesicles reduce hepatic stellate cell activation and liver fibrosis. JCI Insight 2019, 5, e125652. [Google Scholar] [CrossRef]
- Paganini, C.; Palmiero, U.C.; Pocsfalvi, G.; Touzet, N.; Bongiovanni, A.; Arosio, P. Scalable Production and Isolation of Extracellular Vesicles: Available Sources and Lessons from Current Industrial Bioprocesses. Biotechnol. J. 2019, 14, e1800528. [Google Scholar] [CrossRef]
- Claridge, B.; Lozano, J.; Poh, Q.H.; Greening, D.W. Development of Extracellular Vesicle Therapeutics: Challenges, Considerations, and Opportunities. Front. Cell Dev. Biol. 2021, 9, 734720. [Google Scholar] [CrossRef]
- Guo, S.; Debbi, L.; Zohar, B.; Samuel, R.; Arzi, R.S.; Fried, A.I.; Carmon, T.; Shevach, D.; Redenski, I.; Schlachet, I.; et al. Stimulating Extracellular Vesicles Production from Engineered Tissues by Mechanical Forces. Nano Lett. 2021, 21, 2497–2504. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Bhang, S.H. Stem Cell-Engineered Nanovesicles Exert Proangiogenic and Neuroprotective Effects. Materials 2021, 14, 1078. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Fu, Q.; Soliwoda, A.; Zhang, S.; Zheng, M.; Mao, W.; Wan, Y. Cell-derived nanovesicles prepared by membrane extrusion are good substitutes for natural extracellular vesicles. Extracell. Vesicle 2022, 1, 100004. [Google Scholar] [CrossRef]
- Lee, J.R.; Kyung, J.W.; Kumar, H.; Kwon, S.P.; Song, S.Y.; Han, I.-B.; Kim, B.-S. Targeted Delivery of Mesenchymal Stem Cell-Derived Nanovesicles for Spinal Cord Injury Treatment. Int. J. Mol. Sci. 2020, 21, 4185. [Google Scholar] [CrossRef]
- Hu, L.; Wang, J.; Lin, D.; Shen, Y.; Huang, H.; Cao, Y.; Li, Y.; Li, K.; Yu, Y.; Yu, Y.; et al. Mesenchymal Stem Cell-derived Nanovesicles as a Credible Agent for Therapy of Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2022, 67, 61–75. [Google Scholar] [CrossRef]
- Ko, K.W.; Yoo, Y.-I.; Kim, J.Y.; Choi, B.; Park, S.-B.; Park, W.; Rhim, W.-K.; Han, D.K. Attenuation of Tumor Necrosis Factor-alpha Induced Inflammation by Umbilical Cord-Mesenchymal Stem Cell Derived Exosome-Mimetic Nanovesicles in Endothelial Cells. Tissue Eng. Regen. Med. 2020, 17, 155–163. [Google Scholar] [CrossRef]
- Lee, J.R.; Park, B.-W.; Kim, J.; Choo, Y.W.; Kim, H.Y.; Yoon, J.-K.; Kim, H.; Hwang, J.-W.; Kang, M.; Kwon, S.P.; et al. Nanovesicles derived from iron oxide nanoparticles-incorporated mesenchymal stem cells for cardiac repair. Sci. Adv. 2020, 6, eaaz0952. [Google Scholar] [CrossRef]
- Wang, X.; Hu, S.; Li, J.; Zhu, D.; Wang, Z.; Cores, J.; Cheng, K.; Liu, G.; Huang, K. Extruded Mesenchymal Stem Cell Nanovesicles Are Equally Potent to Natural Extracellular Vesicles in Cardiac Repair. ACS Appl. Mater. Interfaces 2021, 13, 55767–55779. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gu, H.; Qin, D.; Yang, L.; Huang, W.; Essandoh, K.; Wang, Y.; Caldwell, C.C.; Peng, T.; Zingarelli, B.; et al. Exosomal miR-223 Contributes to Mesenchymal Stem Cell-Elicited Cardioprotection in Polymicrobial Sepsis. Sci. Rep. 2015, 5, 13721. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.; Kim, H.W.; Gong, M.; Wang, J.; Millard, R.W.; Wang, Y.; Ashraf, M.; Xu, M. Exosomes secreted from GATA-4 overexpressing mesenchymal stem cells serve as a reservoir of anti-apoptotic microRNAs for cardioprotection. Int. J. Cardiol. 2015, 182, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Jiang, W.-J.; Sun, C.; Hou, C.-Z.; Yang, X.-M.; Gao, J.-G. Induced pluripotent stem cells: Origins, applications, and future perspectives. J. Zhejiang Univ. Sci. B 2013, 14, 1059–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geuder, J.; Wange, L.E.; Janjic, A.; Radmer, J.; Janssen, P.; Bagnoli, J.W.; Müller, S.; Kaul, A.; Ohnuki, M.; Enard, W. A non-invasive method to generate induced pluripotent stem cells from primate urine. Sci. Rep. 2021, 11, 3516. [Google Scholar] [CrossRef]
- Zhou, T.; Benda, C.; Duzinger, S.; Huang, Y.; Li, X.; Li, Y.; Guo, X.; Cao, G.; Chen, S.; Hao, L.; et al. Generation of induced pluripotent stem cells from urine. J. Am. Soc. Nephrol. 2011, 22, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Dukers, D.F.; Meij, P.; Vervoort, M.B.H.J.; Vos, W.; Scheper, R.J.; Meijer, C.J.L.M.; Bloemena, E.; Middeldorp, J. Direct immunosuppressive effects of EBV-encoded latent membrane protein 1. J. Immunol. 2000, 165, 663–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wilson, G.F.; Soerens, A.G.; Koonce, C.H.; Yu, J.; Palecek, S.P.; Thomson, J.A.; Kamp, T. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ. Res. 2009, 104, e30–e41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Miyagawa, S.; Miki, K.; Saito, A.; Fukushima, S.; Higuchi, T.; Kawamura, M.; Kawamura, T.; Ito, E.; Kawaguchi, N.; et al. In vivo differentiation of induced pluripotent stem cell-derived cardiomyocytes. Circ. J. 2013, 77, 1297–1306. [Google Scholar] [CrossRef] [Green Version]
- Gorecka, J.; Kostiuk, V.; Fereydooni, A.; Gonzalez, L.; Luo, J.; Dash, B.; Isaji, T.; Ono, S.; Liu, S.; Lee, S.R.; et al. The potential and limitations of induced pluripotent stem cells to achieve wound healing. Stem Cell Res. Ther. 2019, 10, 87. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, D.; Millard, R.; Sivakumaran, P.; Wong, R.C.B.; Crombie, D.E.; Hewitt, A.W.; Liang, H.; Hung, S.S.C.; Pébay, A.; Shepherd, R.K.; et al. Electrical Stimulation Promotes Cardiac Differentiation of Human Induced Pluripotent Stem Cells. Stem Cells Int. 2016, 2016, 1718041. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.-I.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Piao, Y.; Hung, S.S.; Lim, S.Y.; Wong, R.C.; Ko, M.S. Efficient generation of integration-free human induced pluripotent stem cells from keratinocytes by simple transfection of episomal vectors. Stem Cells Transl. Med. 2014, 3, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.D.; Lee, J.; Kim, H.-S.; Lee, M.-O.; Son, M.-Y.; Yoo, C.H.; Choi, J.-K.; Lee, S.C.; Cho, Y.S. The unique spliceosome signature of human pluripotent stem cells is mediated by SNRPA1, SNRPD1, and PNN. Stem Cell Res. 2017, 22, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Goncharov, N.V.; Popova, P.I.; Avdonin, P.P.; Kudryavtsev, I.V.; Serebryakova, M.K.; Korf, E.A.; Avdonin, P.V. Markers of Endothelial Cells in Normal and Pathological Conditions. Biochem. Suppl. Ser. A Membr. Cell Biol. 2020, 14, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Brooimans, R.A.; Van der Ark, A.A.; Tomita, M.; Van Es, L.A.; Daha, M.R. CD59 expressed by human endothelial cells functions as a protective molecule against complement-mediated lysis. Eur. J. Immunol. 1992, 22, 791–797. [Google Scholar] [CrossRef]
- Nasiri Kenari, A.; Kastaniegaard, K.; Greening, D.; Shambrook, M.; Stensballe, A.; Cheng, L.; Hill, A.F. Proteomic and Post-Translational Modification Profiling of Exosome-Mimetic Nanovesicles Compared to Exosomes. Proteomics 2019, 19, e1800161. [Google Scholar] [CrossRef] [Green Version]
- Rai, A.; Greening, D.W.; Xu, R.; Chen, M.; Suwakulsiri, W.; Simpson, R.J. Secreted midbody remnants are a class of extracellular vesicles molecularly distinct from exosomes and microparticles. Commun. Biol. 2021, 4, 400. [Google Scholar] [CrossRef]
- Roefs, M.T.; Sluijter, J.P.G.; Vader, P. Extracellular Vesicle-Associated Proteins in Tissue Repair. Trends Cell Biol. 2020, 30, 990–1013. [Google Scholar] [CrossRef]
- Mohammadi, H.; Pinto, V.I.; Wang, Y.; Hinz, B.; Janmey, P.A.; McCulloch, C.A. Filamin A Mediates Wound Closure by Promoting Elastic Deformation and Maintenance of Tension in the Collagen Matrix. J. Investig. Dermatol. 2015, 135, 2852–2861. [Google Scholar] [CrossRef] [Green Version]
- Hamill, K.J.; Hiroyasu, S.; Colburn, Z.T.; Ventrella, R.V.; Hopkinson, S.B.; Skalli, O.; Jones, J.C.R. Alpha actinin-1 regulates cell-matrix adhesion organization in keratinocytes: Consequences for skin cell motility. J. Investig. Dermatol. 2015, 135, 1043–1052. [Google Scholar] [CrossRef] [Green Version]
- Vicente-Manzanares, M.; Ma, X.; Adelstein, R.S.; Horwitz, A.R. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 2009, 10, 778–790. [Google Scholar] [CrossRef] [Green Version]
- Pollard, T.D.; Cooper, J.A. Actin, a central player in cell shape and movement. Science 2009, 326, 1208–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, I.; Díez-Marqués, M.L.; Rodríguez-Puyol, M.; Herrero-Fresneda, I.; García, D.M.R.; Dedhar, S.; Ruiz-Torres, M.P.; Rodríguez-Puyol, D. Integrin-linked kinase (ILK) modulates wound healing through regulation of hepatocyte growth factor (HGF). Exp. Cell Res. 2012, 318, 2470–2481. [Google Scholar] [CrossRef] [PubMed]
- Ayer, A.; Zarjou, A.; Agarwal, A.; Stocker, R. Heme Oxygenases in Cardiovascular Health and Disease. Physiol. Rev. 2016, 96, 1449–1508. [Google Scholar] [CrossRef] [Green Version]
- Glass, J.J.; Phillips, P.A.; Gunning, P.W.; Stehn, J.R. Hypoxia alters the recruitment of tropomyosins into the actin stress fibres of neuroblastoma cells. BMC Cancer 2015, 15, 712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlando, U.D.; Castillo, A.F.; Dattilo, M.A.; Solano, A.R.; Maloberti, P.M.; Podesta, E.J. Acyl-CoA synthetase-4, a new regulator of mTOR and a potential therapeutic target for enhanced estrogen receptor function in receptor-positive and -negative breast cancer. Oncotarget 2015, 6, 42632–42650. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Umetsu, A.; Tamanoi, F. Characterization of the Rheb-mTOR signaling pathway in mammalian cells: Constitutive active mutants of Rheb and mTOR. Methods Enzymol. 2008, 438, 307–320. [Google Scholar]
- You, K.; Wang, L.; Chou, C.-H.; Liu, K.; Nakata, T.; Jaiswal, A.; Yao, J.; Lefkovith, A.; Omar, A.; Perrigoue, J.G.; et al. QRICH1 dictates the outcome of ER stress through transcriptional control of proteostasis. Science 2021, 371, eabb6896. [Google Scholar] [CrossRef]
- Chen, W.; Zheng, P.; Hong, T.; Wang, Y.; Liu, N.; He, B.; Zou, S.; Ren, D.; Duan, J.; Zhao, L.; et al. Astrocytes-derived exosomes induce neuronal recovery after traumatic brain injury via delivering gap junction alpha 1-20 k. J. Tissue Eng. Regen. Med. 2020, 14, 412–423. [Google Scholar] [CrossRef]
- Vicencio, J.M.; Yellon, D.M.; Sivaraman, V.; Das, D.; Boi-Doku, C.; Arjun, S.; Zheng, Y.; Riquelme, J.A.; Kearney, J.; Sharma, V.; et al. Plasma exosomes protect the myocardium from ischemia-reperfusion injury. J. Am. Coll. Cardiol. 2015, 65, 1525–1536. [Google Scholar] [CrossRef] [Green Version]
- Rai, A.; Fang, H.; Claridge, B.; Simpson, R.J.; Greening, D.W. Proteomic dissection of large extracellular vesicle surfaceome unravels interactive surface platform. J. Extracell. Vesicles 2021, 10, e12164. [Google Scholar] [CrossRef]
- Teng, F.; Fussenegger, M. Shedding Light on Extracellular Vesicle Biogenesis and Bioengineering. Adv. Sci. 2020, 8, 2003505. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Olson, E.N.; Bassel-Duby, R. Therapeutic approaches for cardiac regeneration and repair. Nat. Rev. Cardiol. 2018, 15, 585–600. [Google Scholar] [CrossRef]
- Verma, S.; Fedak, P.W.; Weisel, R.D.; Butany, J.; Rao, V.; Maitland, A.; Li, R.-K.; Dhillon, B.; Yau, T.M. Fundamentals of reperfusion injury for the clinical cardiologist. Circulation 2002, 105, 2332–2336. [Google Scholar] [CrossRef]
- Fan, D.; Takawale, A.; Lee, J.; Kassiri, Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenes. Tissue Repair 2012, 5, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zile, M.R.; Baicu, C.F.; Ikonomidis, J.S.; Stroud, R.E.; Nietert, P.J.; Bradshaw, A.D.; Slater, R.; Palmer, B.M.; Van Buren, P.; Meyer, M.; et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: Contributions of collagen and titin. Circulation 2015, 131, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Nakerakanti, S.S.; Bujor, A.M.; Trojanowska, M. CCN2 is required for the TGF-beta induced activation of Smad1-Erk1/2 signaling network. PLoS ONE 2011, 6, e21911. [Google Scholar] [CrossRef]
- Kishore, R.; Verma, S.K. Roles of STATs signaling in cardiovascular diseases. JAKSTAT 2012, 1, 118–124. [Google Scholar] [CrossRef] [Green Version]
- Ritschel, V.; Shetelig, C.; Seljeflot, I.; Limalanathan, S.; Hoffmann, P.; Halvorsen, S.; Arnesen, H.; Eritsland, J.; Andersen, G.O. Evaluation of circulating levels of CCN2/connective tissue growth factor in patients with ST-elevation myocardial infarction. Sci. Rep. 2017, 7, 11945. [Google Scholar] [CrossRef] [Green Version]
- Isenberg, J.S.; Roberts, D.D. THBS1 (thrombospondin-1). Atlas Genet. Cytogenet. Oncol. Haematol. 2020, 24, 291–299. [Google Scholar] [CrossRef]
- Forbes, T.; Pauza, A.G.; Adams, J.C. In the balance: How do thrombospondins contribute to the cellular pathophysiology of cardiovascular disease? Am. J. Physiol. Cell Physiol. 2021, 321, C826–C845. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz Del Moral, S.; Benaouicha, M.; Munoz-Chapuli, R.; Carmona, R. The Insulin-like Growth Factor Signalling Pathway in Cardiac Development and Regeneration. Int. J. Mol. Sci. 2021, 23, 234. [Google Scholar] [CrossRef]
- Yamamoto, T.; Sano, M. Deranged Myocardial Fatty Acid Metabolism in Heart Failure. Int. J. Mol. Sci. 2022, 23, 996. [Google Scholar] [CrossRef]
- DeBosch, B.; Sambandam, N.; Weinheimer, C.; Courtois, M.; Muslin, A.J. Akt2 regulates cardiac metabolism and cardiomyocyte survival. J. Biol. Chem. 2006, 281, 32841–32851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Ding, G.; Qin, Q.; Huang, Y.; Lewis, W.; He, N.; Evans, R.M.; Schneider, M.D.; Brako, F.A.; Xiao, Y.; et al. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat. Med. 2004, 10, 1245–1250. [Google Scholar] [CrossRef]
- Uchiyama, A.; Yamada, K.; Ogino, S.; Yokoyama, Y.; Takeuchi, Y.; Udey, M.C.; Ishikawa, O.; Motegi, S.-I. MFG-E8 regulates angiogenesis in cutaneous wound healing. Am. J. Pathol. 2014, 184, 1981–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocca, S.; Anderson, S.; Oehninger, S. Milk fat globule epithelial growth factor 8 (MFG-E8) regulates human endometrial endothelial cell adhesion and proliferation. Fertil. Steril. 2010, 94, S215–S216. [Google Scholar] [CrossRef]
- Howangyin, K.Y.; Zlatanova, I.; Pinto, C.; Ngkelo, A.; Cochain, C.; Rouanet, M.; Vilar, J.; Lemitre, M.; Stockmann, C.; Fleischmann, B.K.; et al. Myeloid-Epithelial-Reproductive Receptor Tyrosine Kinase and Milk Fat Globule Epidermal Growth Factor 8 Coordinately Improve Remodeling After Myocardial Infarction via Local Delivery of Vascular Endothelial Growth Factor. Circulation 2016, 133, 826–839. [Google Scholar] [CrossRef]
- Wang, B.; Ge, Z.; Wu, Y.; Zha, Y.; Zhang, X.; Yan, Y.; Xie, Y. MFGE8 is down-regulated in cardiac fibrosis and attenuates endothelial-mesenchymal transition through Smad2/3-Snail signalling pathway. J. Cell. Mol. Med. 2020, 24, 12799–12812. [Google Scholar] [CrossRef]
- Ridge, L.A.; Mitchell, K.; Al-Anbaki, A.; Qureshi, W.M.S.; Stephen, L.A.; Tenin, G.; Lu, Y.; Lupu, I.-E.; Clowes, C.; Robertson, A.; et al. Non-muscle myosin IIB (Myh10) is required for epicardial function and coronary vessel formation during mammalian development. PLoS Genet. 2017, 13, e1007068. [Google Scholar] [CrossRef] [Green Version]
- Shankar, T.S.; Ramadurai, D.K.A.; Steinhorst, K.; Sommakia, S.; Badolia, R.; Krokidi, A.T.; Calder, D.; Navankasattusas, S.; Sander, P.; Kwon, O.S.; et al. Cardiac-specific deletion of voltage dependent anion channel 2 leads to dilated cardiomyopathy by altering calcium homeostasis. Nat. Commun. 2021, 12, 4583. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Ren, L.; Hamblin, M.H.; Fan, Y. Endothelial Cell Glucose Metabolism and Angiogenesis. Biomedicines 2021, 9, 147. [Google Scholar] [CrossRef] [PubMed]
- Leung, S.W.S.; Shi, Y. The glycolytic process in endothelial cells and its implications. Acta Pharmacol. Sin. 2022, 43, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Wettlaufer, S.H.; Scott, J.P.; McEachin, R.C.; Peters-Golden, M.; Huang, S.K. Reversal of the Transcriptome by Prostaglandin E2 during Myofibroblast Dedifferentiation. Am. J. Respir. Cell Mol. Biol. 2016, 54, 114–127. [Google Scholar] [CrossRef] [Green Version]
- Bansal, R.; Nakagawa, S.; Yazdani, S.; Van Baarlen, J.; Venkatesh, A.; Koh, A.P.; Song, W.-M.; Goossens, N.; Watanabe, H.; Beasley, M.B.; et al. Integrin alpha 11 in the regulation of the myofibroblast phenotype: Implications for fibrotic diseases. Exp. Mol. Med. 2017, 49, e396. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, F.; Liu, X.; Liu, G.; Qiu, H.; He, Y.; Hu, H.; Jiang, P. Long non-coding RNA RNF7 promotes the cardiac fibrosis in rat model via miR-543/THBS1 axis and TGFbeta1 activation. Aging 2020, 12, 996–1010. [Google Scholar] [CrossRef]
- Jiang, D.; Guo, B.; Lin, F.; Hui, Q.; Tao, K. Effect of THBS1 on the Biological Function of Hypertrophic Scar Fibroblasts. BioMed Res. Int. 2020, 2020, 8605407. [Google Scholar] [CrossRef]
- Sweetwyne, M.T.; Murphy-Ullrich, J.E. Thrombospondin1 in tissue repair and fibrosis: TGF-beta-dependent and independent mechanisms. Matrix Biol. 2012, 31, 178–186. [Google Scholar] [CrossRef] [Green Version]
- Hohn, J.; Tan, W.; Carver, A.; Barrett, H.; Carver, W. Roles of Exosomes in Cardiac Fibroblast Activation and Fibrosis. Cells 2021, 10, 2933. [Google Scholar] [CrossRef]
- Gangadaran, P.; Oh, E.J.; Rajendran, R.L.; Kim, H.M.; Oh, J.M.; Kwak, S.; Hong, C.M.; Choi, K.Y.; Chung, H.Y.; Ahn, B.-C. Identification of Angiogenic Cargoes in Human Fibroblasts-Derived Extracellular Vesicles and Induction of Wound Healing. Pharmaceuticals 2022, 15, 702. [Google Scholar] [CrossRef]
- Lu, Y.; Wen, H.; Huang, J.; Liao, P.; Liao, H.; Tu, J.; Zeng, Y. Extracellular vesicle-enclosed miR-486-5p mediates wound healing with adipose-derived stem cells by promoting angiogenesis. J. Cell. Mol. Med. 2020, 24, 9590–9604. [Google Scholar] [PubMed]
- Monguio-Tortajada, M.; Prat-Vidal, C.; Martínez-Falguera, D.; Teis, A.; Soler-Botija, C.; Courageux, Y.; Munizaga-Larroudé, M.; Moron-Font, M.; Bayes-Genis, A.; Borràs, F.E.; et al. Acellular cardiac scaffolds enriched with MSC-derived extracellular vesicles limit ventricular remodelling and exert local and systemic immunomodulation in a myocardial infarction porcine model. Theranostics 2022, 12, 4656–4670. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, J.A.; Kumar, N.; Noor, M.; Angelos, M.G.; Khan, M.; Chen, C.-A.; Khan, M. Extracellular Vesicles Released by Human Induced-Pluripotent Stem Cell-Derived Cardiomyocytes Promote Angiogenesis. Front. Physiol. 2018, 9, 1794. [Google Scholar] [CrossRef]
- Abolgheit, S.; Abdelkader, S.; Aboushelib, M.; Omar, E.; Mehanna, R. Bone marrow-derived mesenchymal stem cells and extracellular vesicles enriched collagen chitosan scaffold in skin wound healing (a rat model). J. Biomater. Appl. 2021, 36, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Lyon, C.J.; Fletcher, J.K.; Tang, W.; Wan, M.; Hu, T.Y. Extracellular vesicle activities regulating macrophage- and tissue-mediated injury and repair responses. Acta Pharm. Sin. B 2021, 11, 1493–1512. [Google Scholar] [CrossRef]
- Ge, L.; Xun, C.; Li, W.; Jin, S.; Liu, Z.; Zhuo, Y.; Da Duan, D.; Hu, Z.; Chen, P.; Lu, M. Extracellular vesicles derived from hypoxia-preconditioned olfactory mucosa mesenchymal stem cells enhance angiogenesis via miR-612. J. Nanobiotechnol. 2021, 19, 380. [Google Scholar] [CrossRef]
- Brennan, K.; Martin, K.; Fitzgerald, S.P.; O’Sullivan, J.; Wu, Y.; Blanco, A.; Richardson, C.; Mc Gee, M.M. A comparison of methods for the isolation and separation of extracellular vesicles from protein and lipid particles in human serum. Sci. Rep. 2020, 10, 1039. [Google Scholar] [CrossRef] [Green Version]
- Rai, A.; Fang, H.; Fatmous, M.; Claridge, B.; Poh, Q.H.; Simpson, R.J.; Greening, D.W. A Protocol for Isolation, Purification, Characterization, and Functional Dissection of Exosomes. Methods Mol. Biol. 2021, 2261, 105–149. [Google Scholar]
- Cheng, L.; Hill, A.F. Therapeutically harnessing extracellular vesicles. Nat. Rev. Drug Discov. 2022, 21, 379–399. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e18. [Google Scholar] [CrossRef] [Green Version]
- Kugeratski, F.G.; Hodge, K.; Lilla, S.; McAndrews, K.M.; Zhou, X.; Hwang, R.F.; Zanivan, S.; Kalluri, R. Quantitative proteomics identifies the core proteome of exosomes with syntenin-1 as the highest abundant protein and a putative universal biomarker. Nat. Cell Biol. 2021, 23, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Vandergriff, A.; Huang, K.; Shen, D.; Hu, S.; Hensley, M.T.; Caranasos, T.G.; Qian, L.; Cheng, K. Targeting regenerative exosomes to myocardial infarction using cardiac homing peptide. Theranostics 2018, 8, 1869–1878. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, L.; Li, Y.; Chen, L.; Wang, X.; Guo, W.; Zhang, X.; Qin, G.; He, S.-H.; Zimmerman, A.; et al. Exosomes/microvesicles from induced pluripotent stem cells deliver cardioprotective miRNAs and prevent cardiomyocyte apoptosis in the ischemic myocardium. Int. J. Cardiol. 2015, 192, 61–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagai, A.; Dangas, G.D.; Stone, G.W.; Granger, C.B. Reperfusion strategies in acute coronary syndromes. Circ. Res. 2014, 114, 1918–1928. [Google Scholar] [CrossRef] [PubMed]
- Boersma, E.; Maas, A.C.; Deckers, J.W.; Simoons, M.L. Early thrombolytic treatment in acute myocardial infarction: Reappraisal of the golden hour. Lancet 1996, 348, 771–775. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.J.; Anderson, J.L.; Antman, E.M.; Braniff, B.A.; Brooks, N.H.; Califf, R.M.; Hillis, L.D.; Hiratzka, L.F.; Rapaport, E.; Riegel, B.J.; et al. ACC/AHA guidelines for the management of patients with acute myocardial infarction: Executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on Management of Acute Myocardial Infarction). Circulation 1996, 94, 2341–2350. [Google Scholar]
- Mohsin, S.; Khan, M.; Nguyen, J.; Alkatib, M.; Siddiqi, S.; Hariharan, N.; Wallach, K.; Monsanto, M.; Gude, N.; Dembitsky, W.; et al. Rejuvenation of human cardiac progenitor cells with Pim-1 kinase. Circ. Res. 2013, 113, 1169–1179. [Google Scholar] [CrossRef] [Green Version]
- Mohsin, S.; Khan, M.; Nguyen, J.; Alkatib, M.; Siddiqi, S.; Hariharan, N.; Wallach, K.; Monsanto, M.; Gude, N.; Dembitsky, W.; et al. Nucleostemin rejuvenates cardiac progenitor cells and antagonizes myocardial aging. J. Am. Coll. Cardiol. 2015, 65, 133–147. [Google Scholar] [CrossRef] [Green Version]
- Nasiri Kenari, A.; Cheng, L.; Hill, A.F. Methods for loading therapeutics into extracellular vesicles and generating extracellular vesicles mimetic-nanovesicles. Methods 2020, 177, 103–113. [Google Scholar] [CrossRef]
- Hu, S.; Wang, X.; Li, Z.; Zhu, D.; Cores, J.; Wang, Z.; Li, J.; Mei, X.; Cheng, X.; Su, T.; et al. Platelet membrane and stem cell exosome hybrid enhances cellular uptake and targeting to heart injury. Nano Today 2021, 39, 101210. [Google Scholar] [CrossRef]
- Zhou, X.; Miao, Y.; Wang, Y.; He, S.; Guo, L.; Mao, J.; Chen, M.; Yang, Y.; Zhang, X.; Gan, Y. Tumour-derived extracellular vesicle membrane hybrid lipid nanovesicles enhance siRNA delivery by tumour-homing and intracellular freeway transportation. J. Extracell. Vesicles 2022, 11, e12198. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Fan, M.; Huang, D.; Li, B.; Xu, R.; Gao, F.; Chen, Y. Clodronate-loaded liposomal and fibroblast-derived exosomal hybrid system for enhanced drug delivery to pulmonary fibrosis. Biomaterials 2021, 271, 120761. [Google Scholar] [CrossRef] [PubMed]
- Munagala, R.; Aqil, F.; Jeyabalan, J.; Kandimalla, R.; Wallen, M.; Tyagi, N.; Wilcher, S.; Yan, J.; Schultz, D.J.; Spencer, W.; et al. Exosome-mediated delivery of RNA and DNA for gene therapy. Cancer Lett. 2021, 505, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Zhang, X.; Tang, J.; Lv, Q.; Liu, J. Gene-engineered exosomes-thermosensitive liposomes hybrid nanovesicles by the blockade of CD47 signal for combined photothermal therapy and cancer immunotherapy. Biomaterials 2021, 275, 120964. [Google Scholar] [CrossRef]
- Zhupanyn, P.; Cheng, L.; Zhang, X.; Tang, J.; Lv, Q.; Liu, J. Extracellular vesicle (ECV)-modified polyethylenimine (PEI) complexes for enhanced siRNA delivery in vitro and in vivo. J. Control. Release 2020, 319, 63–76. [Google Scholar] [CrossRef]
- Murphy, D.E.; De Jong, O.G.; Brouwer, M.; Wood, M.J.; Lavieu, G.; Schiffelers, R.M.; Vader, P. Extracellular vesicle-based therapeutics: Natural versus engineered targeting and trafficking. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, T.L.; Russell, A.J.; Riley, P. Experimental limitations of extracellular vesicle-based therapies for the treatment of myocardial infarction. Trends Cardiovasc. Med. 2021, 31, 405–415. [Google Scholar] [CrossRef]
- Hoshino, A.; Kim, H.S.; Bojmar, L.; Gyan, K.E.; Cioffi, M.; Hernandez, J.; Zambirinis, C.P.; Rodrigues, G.; Molina, H.; Heissel, S.; et al. Extracellular Vesicle and Particle Biomarkers Define Multiple Human Cancers. Cell 2020, 182, 1044–1061.e18. [Google Scholar] [CrossRef]
- Burbidge, K.; Zwikelmaier, V.; Cook, B.; Long, M.M.; Balva, B.; Lonigro, M.; Ispas, G.; Rademacher, D.J.; Campbell, E.M. Cargo and cell-specific differences in extracellular vesicle populations identified by multiplexed immunofluorescent analysis. J. Extracell. Vesicles 2020, 9, 1789326. [Google Scholar] [CrossRef]
- Garcia-Martin, R.; Brandao, B.B.; Thomou, T.; Altindis, E.; Kahn, C.R. Tissue differences in the exosomal/small extracellular vesicle proteome and their potential as indicators of altered tissue metabolism. Cell Rep. 2022, 38, 110277. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Tran, N.T.; Than, U.T.T.; Nguyen, M.Q.; Tran, A.M.; Do, P.T.X.; Chu, T.T.; Nguyen, T.D.; Bui, A.V.; Ngo, T.A.; et al. Optimization of human umbilical cord blood-derived mesenchymal stem cell isolation and culture methods in serum- and xeno-free conditions. Stem Cell Res. Ther. 2022, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.; Unger, M.; van Griensven, M.; Balmayor, E.R. Adipose-derived mesenchymal stem cells from liposuction and resected fat are feasible sources for regenerative medicine. Eur. J. Med. Res. 2017, 22, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudleviciene, Z.; Kundrotas, G.; Liudkeviciene, R.; Rascon, J.; Jurga, M. Quick and effective method of bone marrow mesenchymal stem cell extraction. Open Med. 2015, 10, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aasen, T.; Raya, A.; Barrero, M.; Garreta, E.; Consiglio, A.; Gonzalez, F.; Vassena, R.; Bilić, J.; Pekarik, V.; Tiscornia, G.; et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 2008, 26, 1276–1284. [Google Scholar] [CrossRef]
- Cai, J.; Li, W.; Su, H.; Qin, D.; Yang, J.; Zhu, F.; Xu, J.; He, W.; Guo, X.; Labuda, K.; et al. Generation of human induced pluripotent stem cells from umbilical cord matrix and amniotic membrane mesenchymal cells. J. Biol. Chem. 2010, 285, 11227–11234. [Google Scholar] [CrossRef] [Green Version]
- Loh, Y.H.; Hartung, O.; Li, H.; Guo, C.; Sahalie, J.M.; Manos, P.D.; Urbach, A.; Heffner, G.C.; Grskovic, M.; Vigneault, F.; et al. Reprogramming of T cells from human peripheral blood. Cell Stem Cell 2010, 7, 15–19. [Google Scholar] [CrossRef] [Green Version]
- Prakoso, D.; Lim, S.Y.; Erickson, J.R.; Wallace, R.S.; Lees, J.G.; Tate, M.; Kiriazis, H.; Donner, D.G.; Henstridge, D.C.; Davey, J.R.; et al. Fine-tuning the cardiac O-GlcNAcylation regulatory enzymes governs the functional and structural phenotype of the diabetic heart. Cardiovasc. Res. 2022, 118, 212–225. [Google Scholar] [CrossRef]
- Lozano, O.; Silva-Platas, C.; Chapoy-Villanueva, H.; Perez, B.E.; Lees, J.G.; Ramachandra, C.J.A.; Contreras-Torres, F.F.; Lazaro-Alfaro, A.; Luna-Figueroa, E.; Bernal-Ramirez, J.; et al. Amorphous SiO2 nanoparticles promote cardiac dysfunction via the opening of the mitochondrial permeability transition pore in rat heart and human cardiomyocytes. Part Fibre Toxicol. 2020, 17, 15. [Google Scholar] [CrossRef]
- Jang, S.C.; Kim, O.Y.; Yoon, C.M.; Choi, D.-S.; Roh, T.-Y.; Park, J.; Nilsson, J.; Lötvall, J.; Kim, Y.-K.; Gho, Y.S. Bioinspired exosome-mimetic nanovesicles for targeted delivery of chemotherapeutics to malignant tumors. ACS Nano 2013, 7, 7698–7710. [Google Scholar] [CrossRef]
- Xu, R.; Greening, D.W.; Rai, A.; Ji, H.; Simpson, R.J. Highly-purified exosomes and shed microvesicles isolated from the human colon cancer cell line LIM1863 by sequential centrifugal ultrafiltration are biochemically and functionally distinct. Methods 2015, 87, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Poh, Q.H.; Rai, A.; Carmichael, I.I.; Salamonsen, L.A.; Greening, D.W. Proteome reprogramming of endometrial epithelial cells by human trophectodermal small extracellular vesicles reveals key insights into embryo implantation. Proteomics 2021, 21, e2000210. [Google Scholar] [CrossRef] [PubMed]
- Claridge, B.; Rai, A.; Fang, H.; Matsumoto, A.; Luo, J.; McMullen, J.; Greening, D. Proteome characterisation of extracellular vesicles isolated from heart. Proteomics 2021, 21, e2100026. [Google Scholar] [CrossRef]
- Hughes, C.S.; Moggridge, S.; Müller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef] [PubMed]
- Greening, D.W.; Notaras, M.; Chen, M.; Xu, R.; Smith, J.D.; Cheng, L.; Simpson, R.J.; Hill, A.F.; van den Buuse, M. Chronic methamphetamine interacts with BDNF Val66Met to remodel psychosis pathways in the mesocorticolimbic proteome. Mol. Psychiatry 2021, 26, 4431–4447. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Evans, J.; Hutchison, J.; Salamonsen, L.A.; Greening, D.W. Proteomic Insights into Endometrial Receptivity and Embryo-Endometrial Epithelium Interaction for Implantation Reveal Critical Determinants of Fertility. Proteomics 2020, 20, e1900250. [Google Scholar] [CrossRef]
- Nagaraj, N.; Kulak, N.A.; Cox, J.; Neuhauser, N.; Mayr, K.; Hoerning, O.; Vorm, O.; Mann, M. System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top Orbitrap. Mol. Cell. Proteom. 2012, 11, M111.013722. [Google Scholar] [CrossRef] [Green Version]
- Tyanova, S.; Cox, J. Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research. Methods Mol. Biol. 2018, 1711, 133–148. [Google Scholar]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lozano, J.; Rai, A.; Lees, J.G.; Fang, H.; Claridge, B.; Lim, S.Y.; Greening, D.W. Scalable Generation of Nanovesicles from Human-Induced Pluripotent Stem Cells for Cardiac Repair. Int. J. Mol. Sci. 2022, 23, 14334. https://doi.org/10.3390/ijms232214334
Lozano J, Rai A, Lees JG, Fang H, Claridge B, Lim SY, Greening DW. Scalable Generation of Nanovesicles from Human-Induced Pluripotent Stem Cells for Cardiac Repair. International Journal of Molecular Sciences. 2022; 23(22):14334. https://doi.org/10.3390/ijms232214334
Chicago/Turabian StyleLozano, Jonathan, Alin Rai, Jarmon G. Lees, Haoyun Fang, Bethany Claridge, Shiang Y. Lim, and David W. Greening. 2022. "Scalable Generation of Nanovesicles from Human-Induced Pluripotent Stem Cells for Cardiac Repair" International Journal of Molecular Sciences 23, no. 22: 14334. https://doi.org/10.3390/ijms232214334
APA StyleLozano, J., Rai, A., Lees, J. G., Fang, H., Claridge, B., Lim, S. Y., & Greening, D. W. (2022). Scalable Generation of Nanovesicles from Human-Induced Pluripotent Stem Cells for Cardiac Repair. International Journal of Molecular Sciences, 23(22), 14334. https://doi.org/10.3390/ijms232214334