
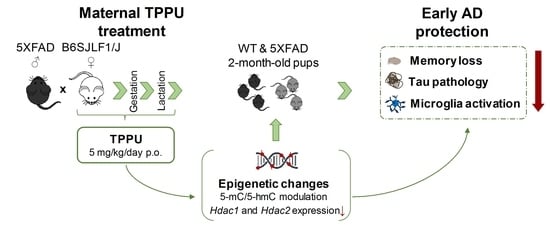
Neuroprotective Epigenetic Changes Induced by Maternal Treatment with an Inhibitor of Soluble Epoxide Hydrolase Prevents Early Alzheimer′s Disease Neurodegeneration

 , , , ,
, , , ,  ,
,  and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Maternal TPPU Treatment Prevented Cognitive and Behavioral Changes of 5XFAD Offspring
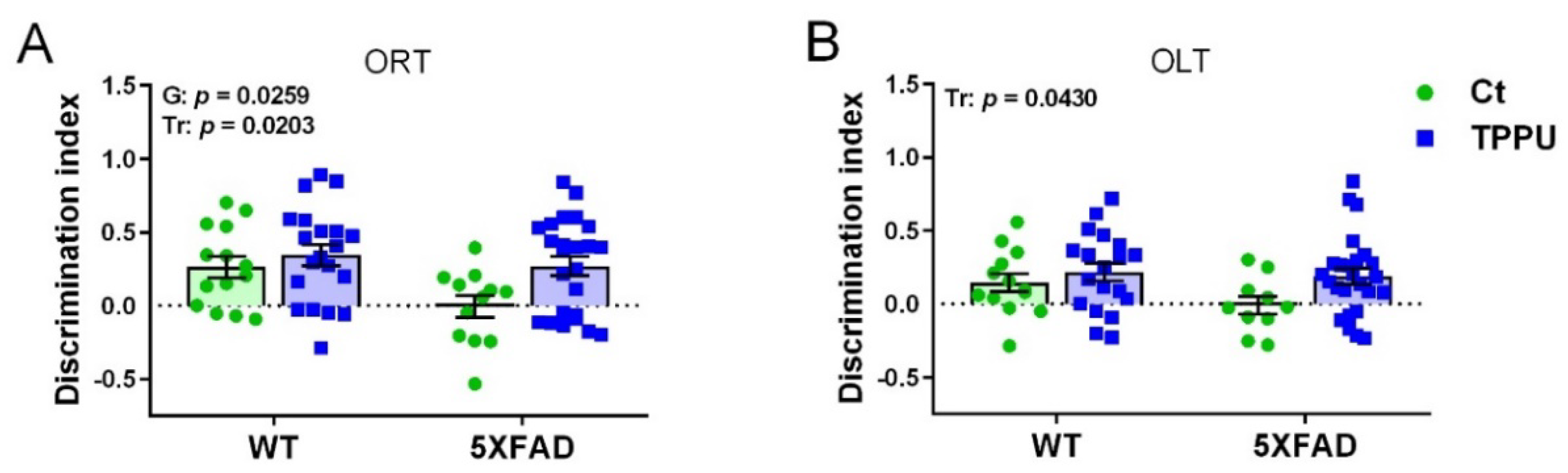
2.1.1. Protection against Memory Loss
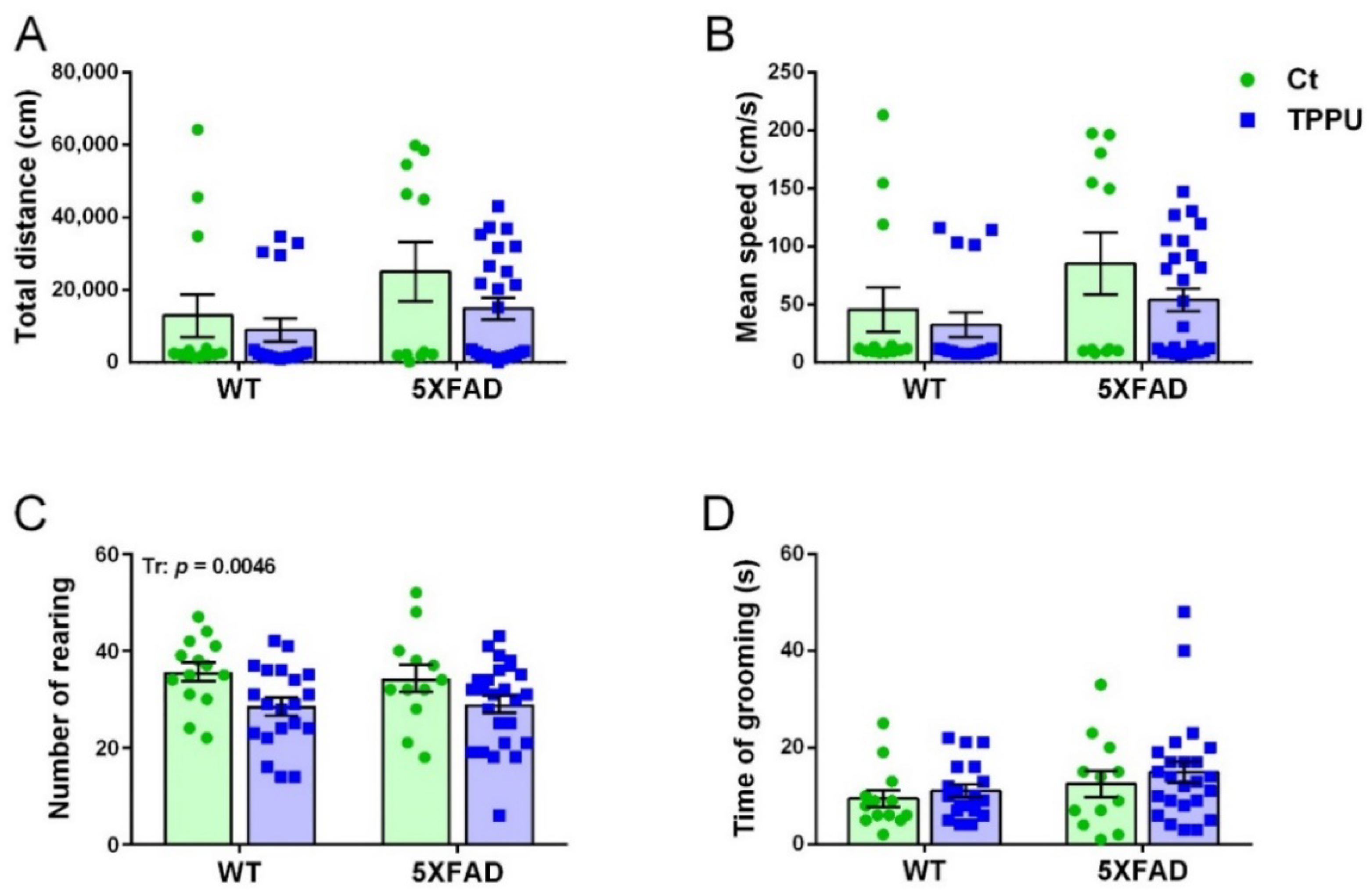
2.1.2. Maternal Treatment with TPPU Rescued Exploratory Activity in the 5XFAD Offspring
2.2. Maternal TPPU Treatment Induced Neuroprotective Epigenetic Changes in 5XFAD and WT Offspring
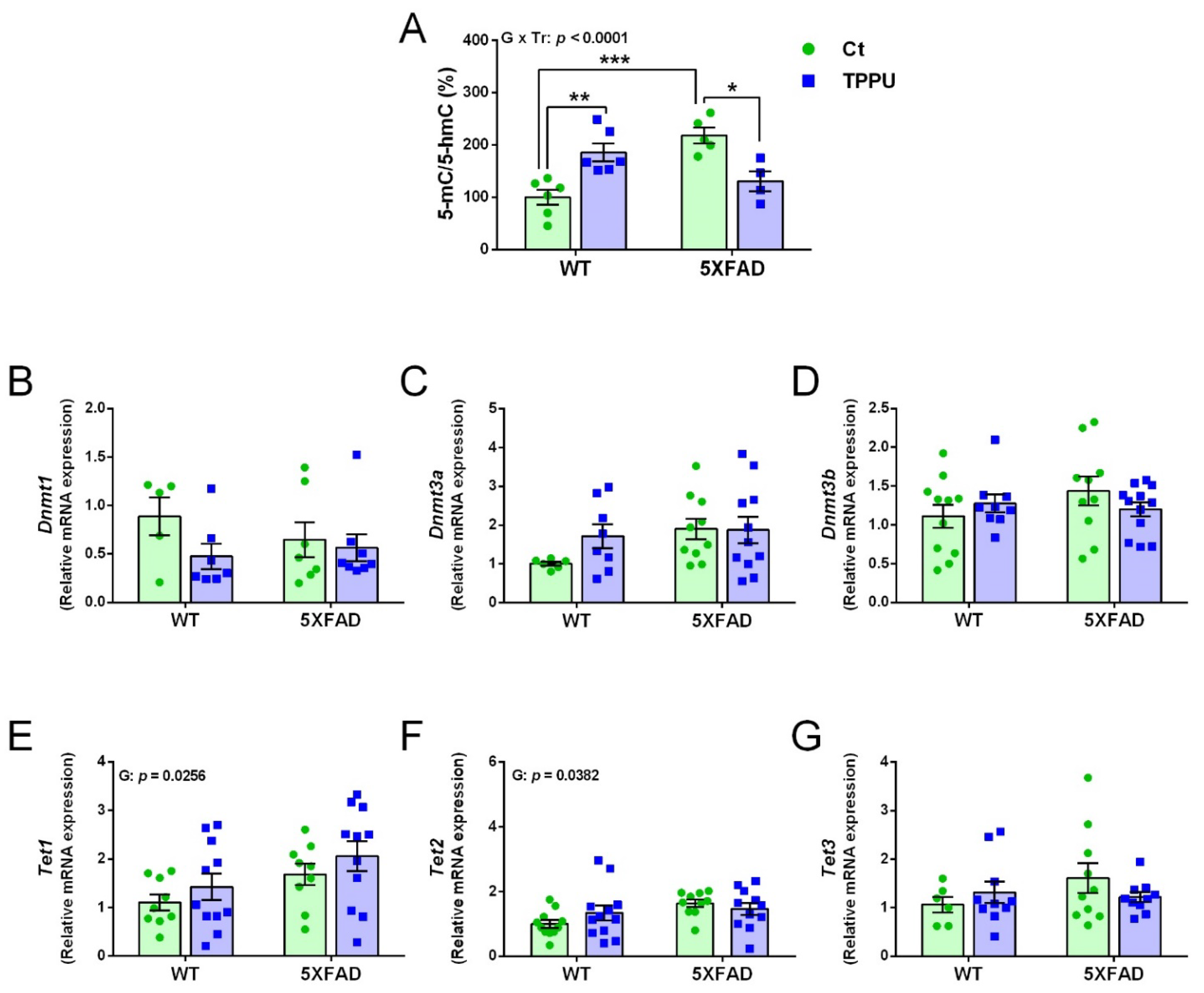
2.2.1. Changes in the Global DNA Methylation Landscape
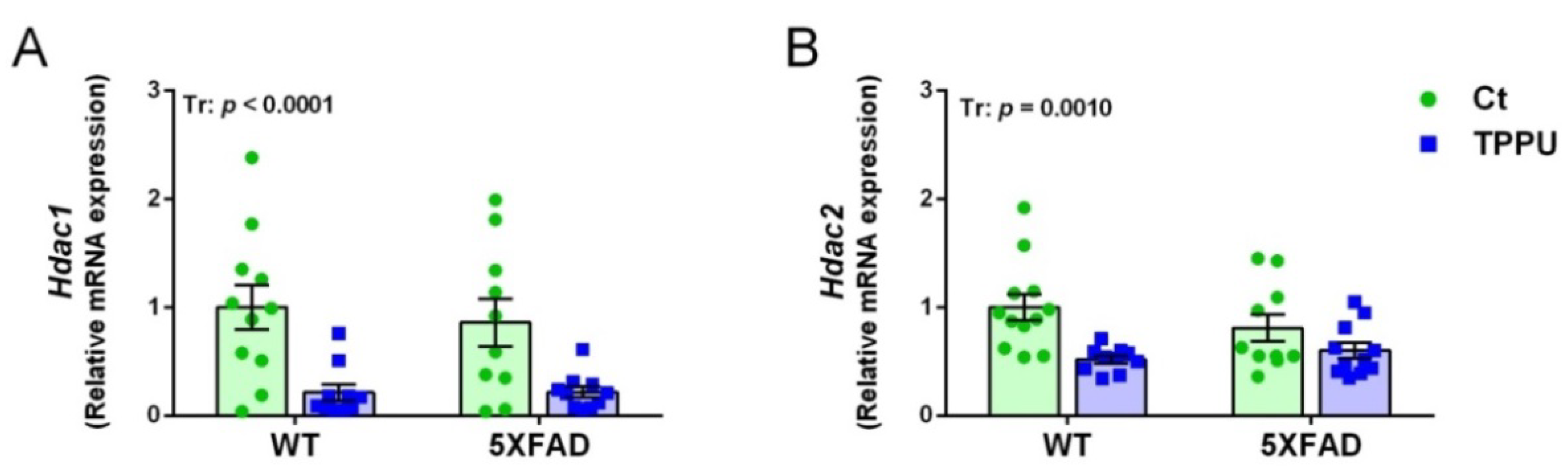
2.2.2. Changes in the Histone Deacetylation Machinery
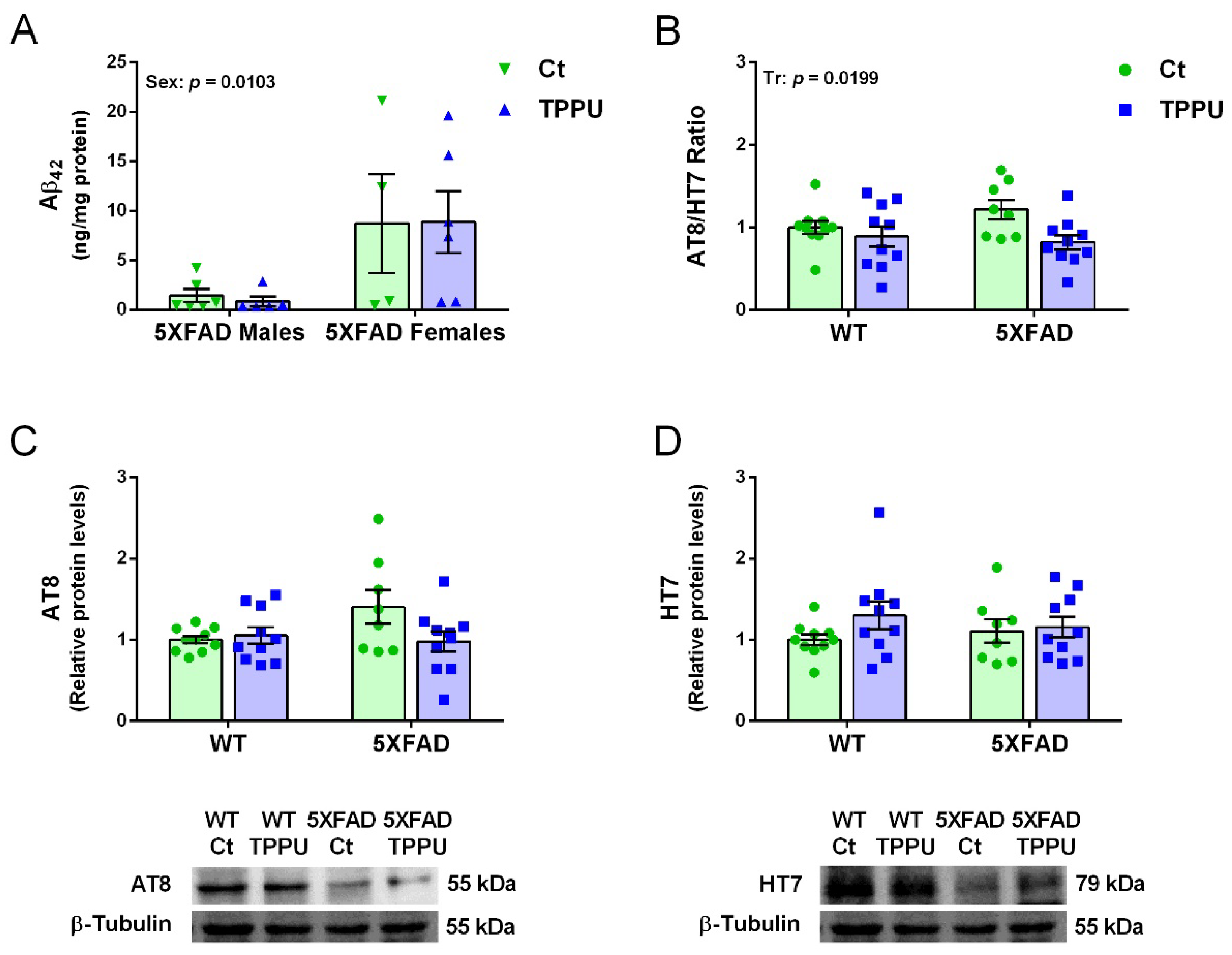
2.3. Maternal TPPU Treatment Did Not Modify Amyloid β42 but It Protected against Tau Hyperphosphorylation in 5XFAD Offspring
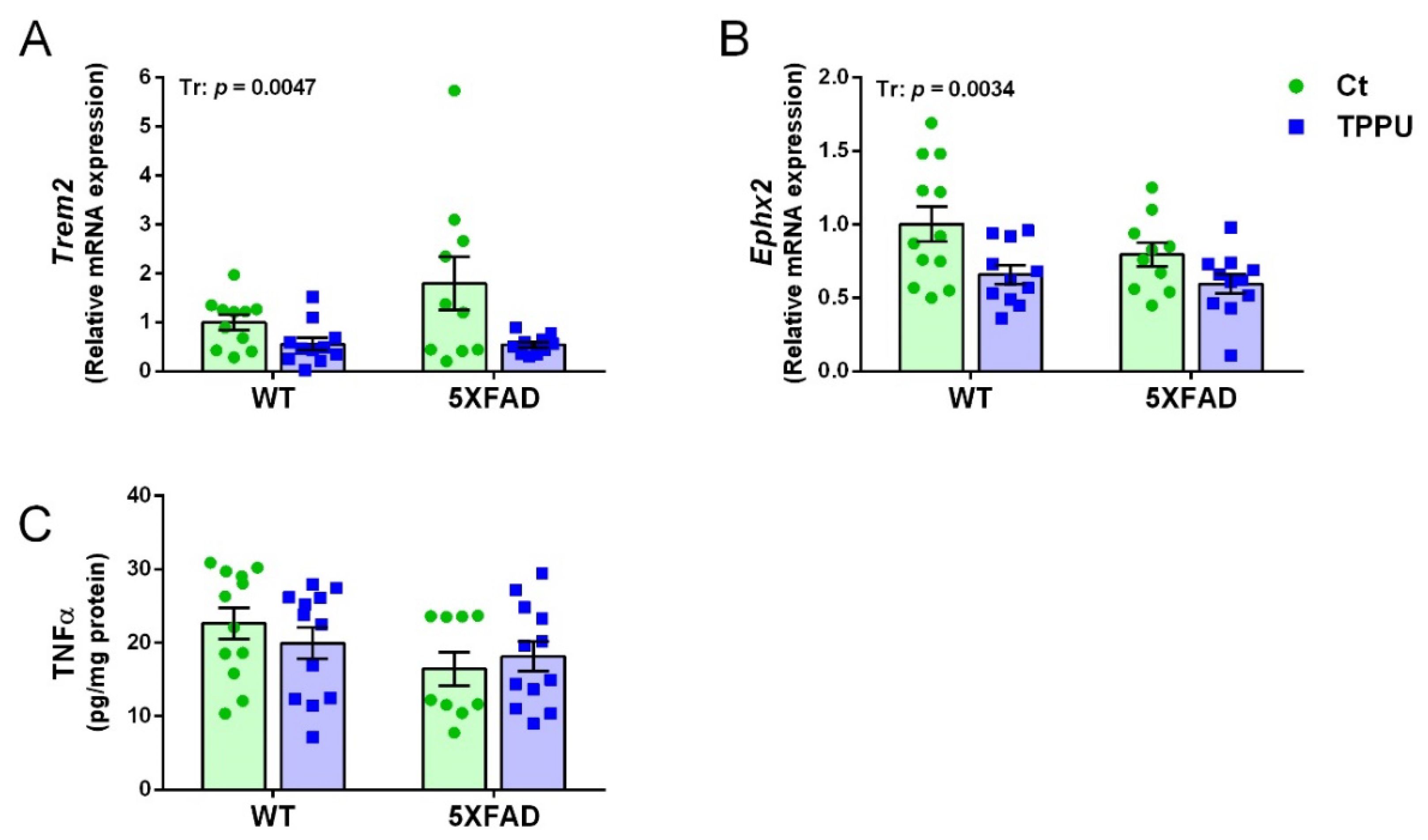
2.4. Maternal TPPU Treatment Decreased Neuroinflammation in the Offspring Mice
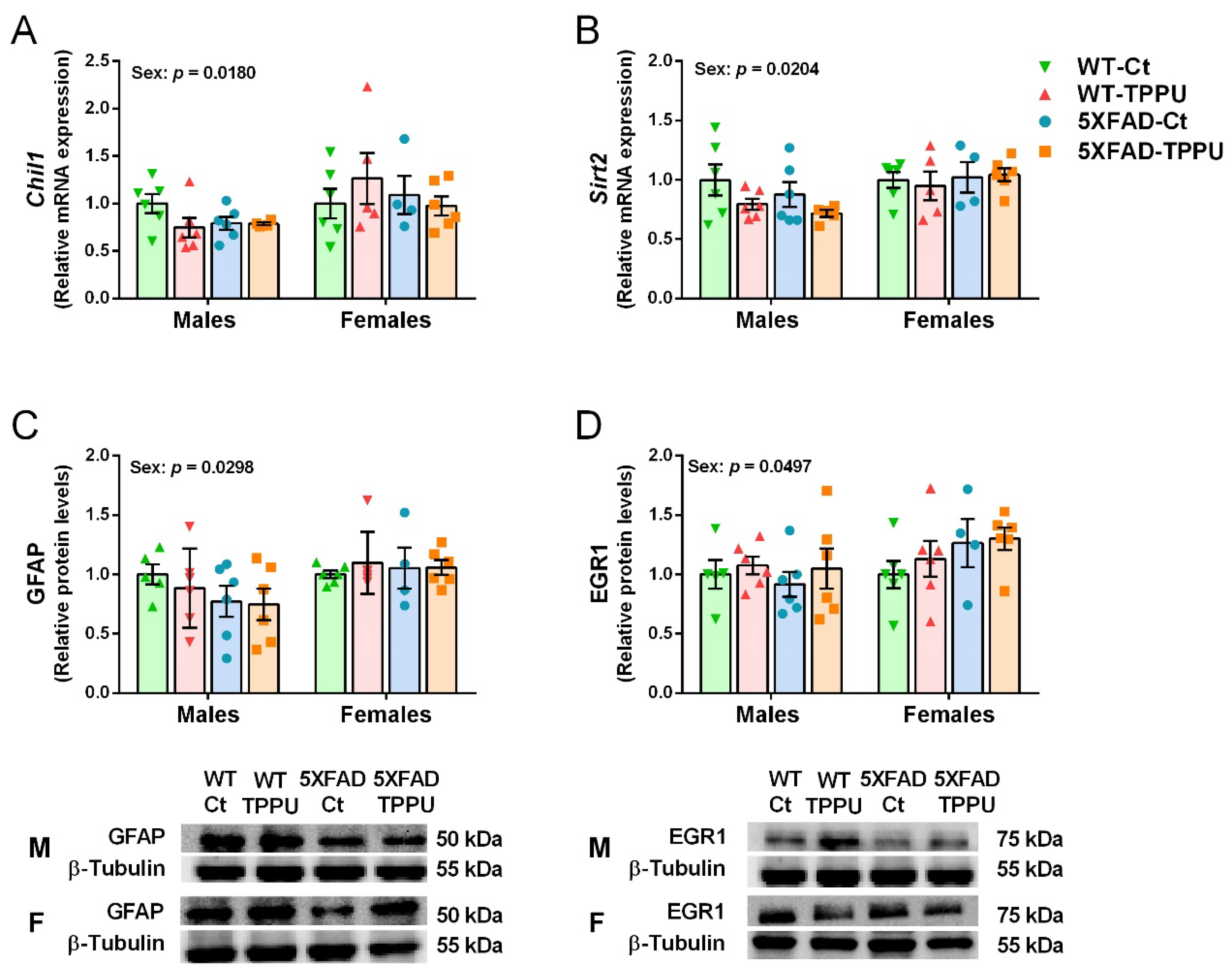
2.5. Female Mice Showed Higher Gliosis and Neurodegenerative Changes Than Male Mice
3. Discussion
4. Materials and Methods
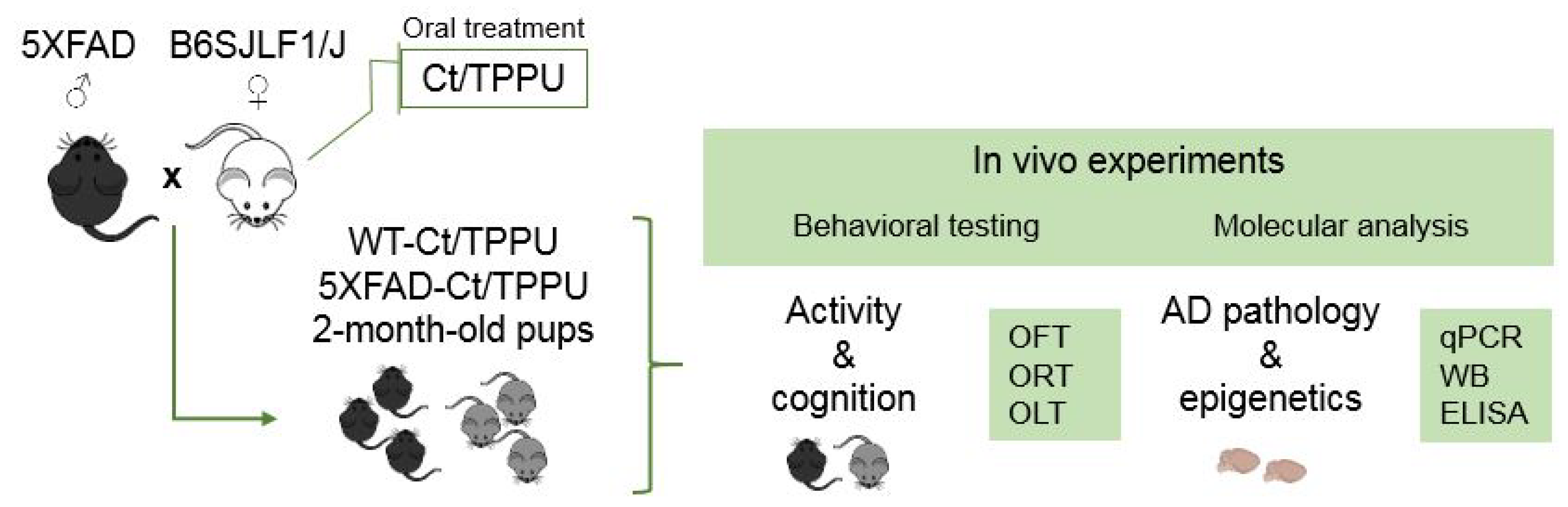
4.1. Animal Breeding and Experimental Design
4.2. Sample Collection
4.3. Behavioral Procedures
4.4. Real-Time Quantitative Polymerase Chain Reaction (qPCR)
4.5. Western Blotting (WB)
4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahimi, F. Alzheimer Disease: Controversies in Basic Science Research, Different Theories, and Reasons for Failed Trials. Biomedicines 2021, 9, 254. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s disease drug development pipeline: 2021. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2021, 7, e12179. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, T.; Patro, N.; Patro, I.K. Cumulative multiple early life hits- a potent threat leading to neurological disorders. Brain Res. Bull. 2019, 147, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Faa, G.; Marcialis, A.M.; Ravarino, A.; Piras, M.; Pintus, M.C.; Fanos, V. Fetal programming of the human brain: Is there a link with insurgence of neurodegenerative disorders in adulthood? Curr. Med. Chem. 2014, 21, 3854–3876. [Google Scholar] [CrossRef]
- Allegra, A.; Giarratana, R.M.; Scola, L.; Balistreri, C.R. The close link between the fetal programming imprinting and neurodegeneration in adulthood: The key role of “hemogenic endothelium” programming. Mech. Ageing Dev. 2021, 195, 111461. [Google Scholar] [CrossRef]
- Kubota, T.; Miyake, K.; Hirasawa, T. Epigenetic understanding of gene-environment interactions in psychiatric disorders: A new concept of clinical genetics. Specif. Gene Expr. Epigenetics Interplay Between Genome Its Environ. 2012, 4, 241–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maity, S.; Farrell, K.; Navabpour, S.; Narayanan, S.N.; Jarome, T.J. Epigenetic Mechanisms in Memory and Cognitive Decline Associated with Aging and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 12280. [Google Scholar] [CrossRef]
- Migliore, L.; Coppedè, F. Gene–environment interactions in Alzheimer disease: The emerging role of epigenetics. Nat. Rev. Neurol. 2022, 18, 643–660. [Google Scholar] [CrossRef]
- Bellver-Sanchis, A.; Pallàs, M.; Griñán-Ferré, C. The Contribution of Epigenetic Inheritance Processes on Age-Related Cognitive Decline and Alzheimer’s Disease. Epigenomes 2021, 5, 15. [Google Scholar] [CrossRef]
- Alldred, M.J.; Chao, H.M.; Lee, S.H.; Beilin, J.; Powers, B.E.; Petkova, E.; Strupp, B.J.; Ginsberg, S.D. Long-term effects of maternal choline supplementation on CA1 pyramidal neuron gene expression in the Ts65Dn mouse model of Down syndrome and Alzheimer’s disease. FASEB J. 2019, 33, 9871–9884. [Google Scholar] [CrossRef]
- Izquierdo, V.; Palomera-Ávalos, V.; López-Ruiz, S.; Canudas, A.-M.; Pallàs, M.; Griñán-Ferré, C. Maternal Resveratrol Supplementation Prevents Cognitive Decline in Senescent Mice Offspring. Int. J. Mol. Sci. 2019, 20, 1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokor, S.; Vass, R.A.; Funke, S.; Ertl, T.; Molnár, D. Epigenetic Effect of Maternal Methyl-Group Donor Intake on Offspring’s Health and Disease. Life 2022, 12, 609. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, G.F.S.; Silva, G.D.B.; Pavan, A.R.; Chiba, D.E.; Chin, C.M.; Dos Santos, J.L. Epigenetic Regulatory Mechanisms Induced by Resveratrol. Nutrients 2017, 9, 1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarroca, S.; Molina-Martínez, P.; Aresté, C.; Etzrodt, M.; de Frutos, P.G.; Gasa, R.; Antonell, A.; Molinuevo, J.L.; Sánchez-Valle, R.; Saura, C.A.; et al. Preservation of cell-survival mechanisms by the presenilin-1 K239N mutation may cause its milder clinical phenotype. Neurobiol. Aging 2016, 46, 169–179. [Google Scholar] [CrossRef]
- Minaz, N.; Razdan, R.; Hammock, B.D.; Goswami, S.K. An inhibitor of soluble epoxide hydrolase ameliorates diabetes-induced learning and memory impairment in rats. Prostaglandins Other Lipid Mediat. 2018, 136, 84–89. [Google Scholar] [CrossRef]
- Ghosh, A.; Comerota, M.M.; Wan, D.; Chen, F.; Propson, N.E.; Hwang, S.H.; Hammock, B.D.; Zheng, H. An epoxide hydrolase inhibitor reduces neuroinflammation in a mouse model of Alzheimer’s disease. Sci. Transl. Med. 2020, 12, 1206. [Google Scholar] [CrossRef] [PubMed]
- García-Lara, E.; Aguirre, S.; Clotet, N.; Sawkulycz, X.; Bartra, C.; Almenara-Fuentes, L.; Suñol, C.; Corpas, R.; Olah, P.; Tripon, F.; et al. Antibody Protection against Long-Term Memory Loss Induced by Monomeric C-Reactive Protein in a Mouse Model of Dementia. Biomedicines 2021, 9, 828. [Google Scholar] [CrossRef]
- Matin, N.; Fisher, C.; Lansdell, T.A.; Hammock, B.D.; Yang, J.; Jackson, W.F.; Dorrance, A.M. Soluble epoxide hydrolase inhibition improves cognitive function and parenchymal artery dilation in a hypertensive model of chronic cerebral hypoperfusion. Microcirculation 2021, 28, 12653. [Google Scholar] [CrossRef]
- Sun, C.-P.; Zhang, X.-Y.; Zhou, J.-J.; Huo, X.-K.; Yu, Z.-L.; Morisseau, C.; Hammock, B.D.; Ma, X.-C. Inhibition of sEH via stabilizing the level of EETs alleviated Alzheimer’s disease through GSK3β signaling pathway. Food Chem. Toxicol. 2021, 156, 112516. [Google Scholar] [CrossRef]
- Griñán-Ferré, C.; Codony, S.; Pujol, E.; Yang, J.; Leiva, R.; Escolano, C.; Puigoriol-Illamola, D.; Companys-Alemany, J.; Corpas, R.; Sanfeliu, C.; et al. Pharmacological Inhibition of Soluble Epoxide Hydrolase as a New Therapy for Alzheimer’s Disease. Neurotherapeutics 2020, 17, 1825–1835. [Google Scholar] [CrossRef]
- Waldman, M.; Bellner, L.; Vanella, L.; Schragenheim, J.; Sodhi, K.; Singh, S.P.; Lin, D.; Lakhkar, A.; Li, J.; Hochhauser, E.; et al. Epoxyeicosatrienoic Acids Regulate Adipocyte Differentiation of Mouse 3T3 Cells, Via PGC-1α Activation, Which Is Required for HO-1 Expression and Increased Mitochondrial Function. Stem Cells Dev. 2016, 25, 1084–1094. [Google Scholar] [CrossRef] [Green Version]
- Spector, A.A.; Norris, A. Action of epoxyeicosatrienoic acids on cellular function. Am. J. Physiol. Cell Physiol. 2007, 292, C996–C1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliff, J.J.; Jia, J.; Nelson, J.; Goyagi, T.; Klaus, J.; Alkayed, N.J. Epoxyeicosanoid signaling in CNS function and disease. Prostaglandins Other Lipid Mediat. 2010, 91, 68–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, K.M.; McReynolds, C.B.; Schmidt, W.K.; Hammock, B.D. Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases. Pharmacol. Ther. 2017, 180, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Zarriello, S.; Tuazon, J.P.; Corey, S.; Schimmel, S.; Rajani, M.; Gorsky, A.; Incontri, D.; Hammock, B.D.; Borlongan, C.V. Humble beginnings with big goals: Small molecule soluble epoxide hydrolase inhibitors for treating CNS disorders. Prog. Neurobiol. 2019, 172, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Gabbs, M.; Leng, S.; Devassy, J.G.; Monirujjaman, M.; Aukema, H.M. Advances in Our Understanding of Oxylipins Derived from Dietary PUFAs. Adv. Nutr. 2015, 6, 513–540. [Google Scholar] [CrossRef] [Green Version]
- González-Domínguez, R.; González-Domínguez, Á.; Sayago, A.; González-Sanz, J.; Lechuga-Sancho, A.; Fernández-Recamales, Á. Mechanistic Insights into Alzheimer’s Disease Unveiled through the Investigation of Disturbances in Central Metabolites and Metabolic Pathways. Biomedicines 2021, 9, 298. [Google Scholar] [CrossRef]
- Peña-Bautista, C.; Álvarez-Sánchez, L.; Cañada-Martínez, A.J.; Baquero, M.; Cháfer-Pericás, C. Epigenomics and Lipidomics Integration in Alzheimer Disease: Pathways Involved in Early Stages. Biomedicines 2021, 9, 1812. [Google Scholar] [CrossRef]
- Jang, H.S.; Shin, W.J.; Lee, J.E.; Do, J.T. CpG and Non-CpG Methylation in Epigenetic Gene Regulation and Brain Function. Genes 2017, 8, 148. [Google Scholar] [CrossRef] [Green Version]
- Poon, C.H.; Tse, L.S.R.; Lim, L.W. DNA methylation in the pathology of Alzheimer’s disease: From gene to cognition. Ann. New York Acad. Sci. 2020, 1475, 15–33. [Google Scholar] [CrossRef]
- Penney, J.; Tsai, L.-H. Histone deacetylases in memory and cognition. Sci. Signal. 2014, 7, re12. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griñán-Ferré, C.; Sarroca, S.; Ivanova, A.; Puigoriol-Illamola, D.; Aguado, F.; Camins, A.; Sanfeliu, C.; Pallàs, M. Epigenetic mechanisms underlying cognitive impairment and Alzheimer disease hallmarks in 5XFAD mice. Aging 2016, 8, 664–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Mesa, Y.; López-Ramos, J.C.; Giménez-Llort, L.; Revilla, S.; Guerra, R.; Gruart, A.; Laferla, F.M.; Cristòfol, R.; Delgado-García, J.M.; Sanfeliu, C. Physical exercise protects against Alzheimer’s disease in 3xTg-AD mice. J. Alzheimer’s. Dis. 2011, 24, 421–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumati, S.; Herrmann, N.; Marotta, G.; Li, A.; Lanctôt, K.L. Blood-based biomarkers of agitation in Alzheimer’s disease: Advances and future prospects. Neurochem. Int. 2022, 152, 105250. [Google Scholar] [CrossRef] [PubMed]
- Ulu, A.; Inceoglu, B.; Yang, J.; Singh, V.; Vito, S.; Wulff, H.; Hammock, B.D. Inhibition of Soluble Epoxide Hydrolase as a Novel Approach to High Dose Diazepam Induced Hypotension. J. Clin. Toxicol. 2016, 6, 1000300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, E.; Hor, K.; Drake, A.J. Maternal influences on fetal brain development: The role of nutrition, infection and stress, and the potential for intergenerational consequences. Early Hum. Dev. 2020, 150, 105190. [Google Scholar] [CrossRef]
- Queiroz, M.P.; Lima, M.D.S.; Barbosa, M.Q.; De Melo, M.F.F.T.; Bertozzo, C.C.D.M.S.; De Oliveira, M.E.G.; Bessa, R.J.B.; Alves, S.P.A.; Souza, M.I.A.; Queiroga, R.D.C.R.D.E.; et al. Effect of Conjugated Linoleic Acid on Memory and Reflex Maturation in Rats Treated During Early Life. Front. Neurosci. 2019, 13, 370. [Google Scholar] [CrossRef]
- Ma, M.; Ren, Q.; Yang, J.; Zhang, K.; Xiong, Z.; Ishima, T.; Pu, Y.; Hwang, S.H.; Toyoshima, M.; Iwayama, Y.; et al. Key role of soluble epoxide hydrolase in the neurodevelopmental disorders of offspring after maternal immune activation. Proc. Natl. Acad. Sci. USA 2019, 116, 7083–7088. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Xu, S.; Mo, M.; Yang, Q.; Li, J.; Zhong, Y.; Zhang, J.; Zhang, L.; Ye, X.; Liu, Z.; et al. Quantification of DNA methylation and hydroxymethylation in Alzheimer’s disease mouse model using LC-MS/MS. J. Mass Spectrom. 2018, 53, 590–594. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabaneda-Bueno, R.; Mena-Montes, B.; Torres-Castro, S.; Torres-Carrillo, N.; Torres-Carrillo, N.M. Advances in Genetics and Epigenetic Alterations in Alzheimer’s Disease: A Notion for Therapeutic Treatment. Genes 2021, 12, 1959. [Google Scholar] [CrossRef] [PubMed]
- Gräff, J.; Rei, D.; Guan, J.-S.; Wang, W.-Y.; Seo, J.; Hennig, K.M.; Nieland, T.J.F.; Fass, D.M.; Kao, P.F.; Kahn, M.; et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 2012, 483, 222–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schueller, E.; Paiva, I.; Blanc, F.; Wang, X.-L.; Cassel, J.-C.; Boutillier, A.-L.; Bousiges, O. Dysregulation of histone acetylation pathways in hippocampus and frontal cortex of Alzheimer’s disease patients. Eur. Neuropsychopharmacol. 2020, 33, 101–116. [Google Scholar] [CrossRef]
- D’Mello, S.R. Regulation of CNS Development by Class I HDACs. Physiol. Behav. 2016, 176, 100–106. [Google Scholar]
- Kumar, V.; Kundu, S.; Singh, A.; Singh, S. Understanding the role of histone deacetylase and their inhibitors in neurodegenerative disorders: Current targets and future perspective. Curr. Neuropharmacol. 2022, 20, 158–178. [Google Scholar] [CrossRef]
- Guan, J.-S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.-H.; Joseph, N.; Gao, J.; Nieland, T.J.F.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009, 459, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Bardai, F.H.; Price, V.; Zaayman, M.; Wang, L.; D’Mello, S.R. Histone Deacetylase-1 (HDAC1) Is a Molecular Switch between Neuronal Survival and Death. J. Biol. Chem. 2012, 287, 35444–35453. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Chen, L.; Guo, Y.; Wang, C.; Zhang, C.; Kong, L.; Ma, H. Class I HDAC Inhibitor Improves Synaptic Proteins and Repairs Cytoskeleton Through Regulating Synapse-Related Genes In vitro and In vivo. Front. Aging Neurosci. 2021, 12, 619866. [Google Scholar] [CrossRef]
- Xu, K.; Dai, X.-L.; Huang, H.-C.; Jiang, Z.-F. Targeting HDACs: A Promising Therapy for Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2011, 2011, 143269. [Google Scholar] [CrossRef] [Green Version]
- Barron, M.; Gartlon, J.; Dawson, L.A.; Atkinson, P.J.; Pardon, M.-C. A state of delirium: Deciphering the effect of inflammation on tau pathology in Alzheimer’s disease. Exp. Gerontol. 2017, 94, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R. Advances in Alzheimer’s disease research over the past two decades. Lancet Neurol. 2022, 21, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Onyango, I.G.; Jauregui, G.V.; Bennett, J.P., Jr.; Stokin, G.B. Neuroinflammation in Alzheimer’s Disease. Biomedicines 2021, 9, 524. [Google Scholar] [CrossRef]
- Al-Ghraiybah, N.F.; Wang, J.; Alkhalifa, A.E.; Roberts, A.B.; Raj, R.; Yang, E.; Kaddoumi, A. Glial Cell-Mediated Neuroinflammation in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 10572. [Google Scholar] [CrossRef] [PubMed]
- Molina-Martínez, P.; Corpas, R.; García-Lara, E.; Cosín-Tomás, M.; Cristòfol, R.; Kaliman, P.; Solà, C.; Molinuevo, J.L.; Sánchez-Valle, R.; Antonell, A.; et al. Microglial Hyperreactivity Evolved to Immunosuppression in the Hippocampus of a Mouse Model of Accelerated Aging and Alzheimer’s Disease Traits. Front. Aging Neurosci. 2021, 12, 622360. [Google Scholar] [CrossRef]
- Lee, J.-W.; Chun, W.; Lee, H.J.; Kim, S.-M.; Min, J.-H.; Kim, D.-Y.; Kim, M.-O.; Ryu, H.W.; Lee, S.U. The Role of Microglia in the Development of Neurodegenerative Diseases. Biomedicines 2021, 9, 1449. [Google Scholar] [CrossRef]
- Cheray, M.; Joseph, B. Epigenetics Control Microglia Plasticity. Front. Cell. Neurosci. 2018, 12, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Yu, C.-C.; Liu, X.-Y.; Deng, X.-N.; Tian, Q.; Du, Y.-J. Epigenetic Modulation of Microglia Function and Phenotypes in Neurodegenerative Diseases. Neural Plast. 2021, 2021, 9912686. [Google Scholar] [CrossRef]
- Yang, Y.-M.; Sun, D.; Kandhi, S.; Froogh, G.; Zhuge, J.; Huang, W.; Hammock, B.D.; Huang, A. Estrogen-dependent epigenetic regulation of soluble epoxide hydrolase via DNA methylation. Proc. Natl. Acad. Sci. USA 2018, 115, 613–618. [Google Scholar] [CrossRef] [Green Version]
- Luan, Y.-Y.; Yao, Y.-M. The Clinical Significance and Potential Role of C-Reactive Protein in Chronic Inflammatory and Neurodegenerative Diseases. Front. Immunol. 2018, 9, 1302. [Google Scholar] [CrossRef] [Green Version]
- Harting, K.; Knöll, B. SIRT2-mediated protein deacetylation: An emerging key regulator in brain physiology and pathology. Eur. J. Cell Biol. 2010, 89, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Carril, J.C.; Cacabelos, N.; Kazantsev, A.G.; Vostrov, A.V.; Corzo, L.; Cacabelos, P.; Goldgaber, D. Sirtuins in Alzheimer’s Disease: SIRT2-Related GenoPhenotypes and Implications for PharmacoEpiGenetics. Int. J. Mol. Sci. Artic. 2019, 20, 1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco-Palmero, V.A.; Rubio-Fernández, M.; Antequera, D.; Villarejo-Galende, A.; Molina, J.A.; Ferrer, I.; Bartolome, F.; Carro, E. Increased YKL-40 but Not C-Reactive Protein Levels in Patients with Alzheimer’s Disease. Biomedicines 2021, 27, 1094. [Google Scholar] [CrossRef] [PubMed]
- Hok-A-Hin, Y.S.; Hoozemans, J.J.M.; Hu, W.T.; Wouters, D.; Howell, J.C.; Rábano, A.; van der Flier, W.M.; Pijnenburg, Y.A.L.; Teunissen, C.E.; del Campo, M. YKL-40 changes are not detected in post-mortem brain of patients with Alzheimer’s disease and frontotemporal lobar degeneration. Alzheimer’s Res. Ther. 2022, 14, 100. [Google Scholar] [CrossRef]
- Ferrari-Souza, J.P.; Ferreira, P.C.L.; Bellaver, B.; Tissot, C.; Wang, Y.-T.; Leffa, D.T.; Brum, W.S.; Benedet, A.L.; Ashton, N.J.; De Bastiani, M.A.; et al. Astrocyte biomarker signatures of amyloid-β and tau pathologies in Alzheimer’s disease. Mol. Psychiatry 2022. [Google Scholar] [CrossRef]
- Bossers, K.; Wirz, K.T.; Meerhoff, G.F.; Essing, A.H.; van Dongen, J.W.; Houba, P.; Kruse, C.G.; Verhaagen, J.; Swaab, D.F. Concerted changes in transcripts in the prefrontal cortex precede neuropathology in Alzheimer’s disease. Brain 2010, 133, 3699–3723. [Google Scholar] [CrossRef] [Green Version]
- Mazure, C.M.; Swendsen, J. Sex differences in Alzheimer’s disease and other dementias. Lancet Neurol. 2016, 15, 451–452. [Google Scholar] [CrossRef] [Green Version]
- Mielke, M.M.; Vemuri, P.; Rocca, W.A. Clinical Epidemiology Dovepress Clinical epidemiology of Alzheimer’s disease: Assessing sex and gender differences. Clin. Epidemiol. 2014, 6, 37–48. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartra, C.; Irisarri, A.; Villoslada, A.; Corpas, R.; Aguirre, S.; García-Lara, E.; Suñol, C.; Pallàs, M.; Griñán-Ferré, C.; Sanfeliu, C. Neuroprotective Epigenetic Changes Induced by Maternal Treatment with an Inhibitor of Soluble Epoxide Hydrolase Prevents Early Alzheimer′s Disease Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 15151. https://doi.org/10.3390/ijms232315151
Bartra C, Irisarri A, Villoslada A, Corpas R, Aguirre S, García-Lara E, Suñol C, Pallàs M, Griñán-Ferré C, Sanfeliu C. Neuroprotective Epigenetic Changes Induced by Maternal Treatment with an Inhibitor of Soluble Epoxide Hydrolase Prevents Early Alzheimer′s Disease Neurodegeneration. International Journal of Molecular Sciences. 2022; 23(23):15151. https://doi.org/10.3390/ijms232315151
Chicago/Turabian StyleBartra, Clara, Alba Irisarri, Ainhoa Villoslada, Rubén Corpas, Samuel Aguirre, Elisa García-Lara, Cristina Suñol, Mercè Pallàs, Christian Griñán-Ferré, and Coral Sanfeliu. 2022. "Neuroprotective Epigenetic Changes Induced by Maternal Treatment with an Inhibitor of Soluble Epoxide Hydrolase Prevents Early Alzheimer′s Disease Neurodegeneration" International Journal of Molecular Sciences 23, no. 23: 15151. https://doi.org/10.3390/ijms232315151
APA StyleBartra, C., Irisarri, A., Villoslada, A., Corpas, R., Aguirre, S., García-Lara, E., Suñol, C., Pallàs, M., Griñán-Ferré, C., & Sanfeliu, C. (2022). Neuroprotective Epigenetic Changes Induced by Maternal Treatment with an Inhibitor of Soluble Epoxide Hydrolase Prevents Early Alzheimer′s Disease Neurodegeneration. International Journal of Molecular Sciences, 23(23), 15151. https://doi.org/10.3390/ijms232315151