

PARP Inhibitor Inhibits the Vasculogenic Mimicry through a NF-κB-PTX3 Axis Signaling in Breast Cancer Cells

 ,
, 
Abstract
:1. Introduction
2. Results
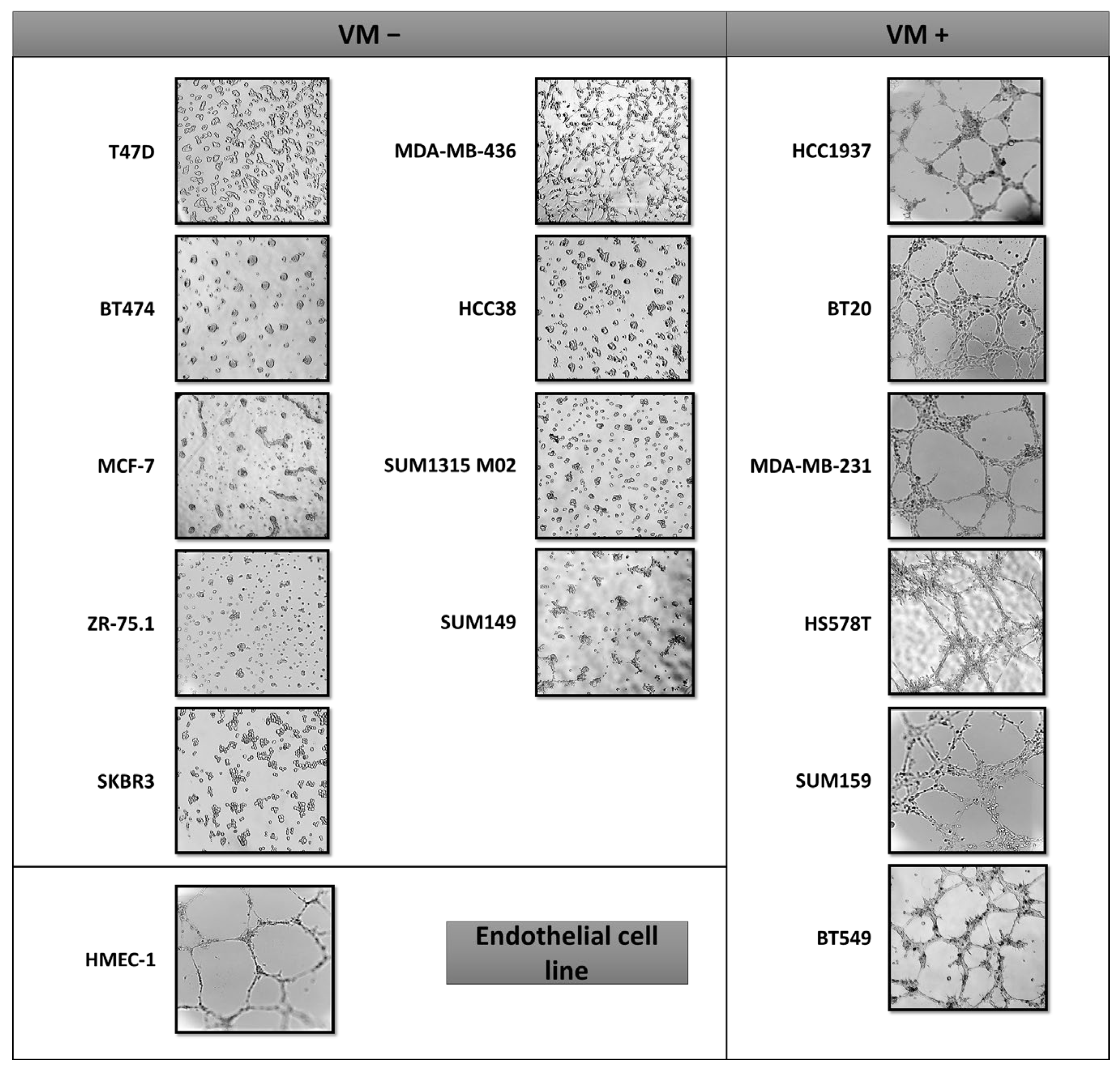
2.1. VM Formation in Triple Negative Breast Cancer Cells Is Independent of the BRCA Status
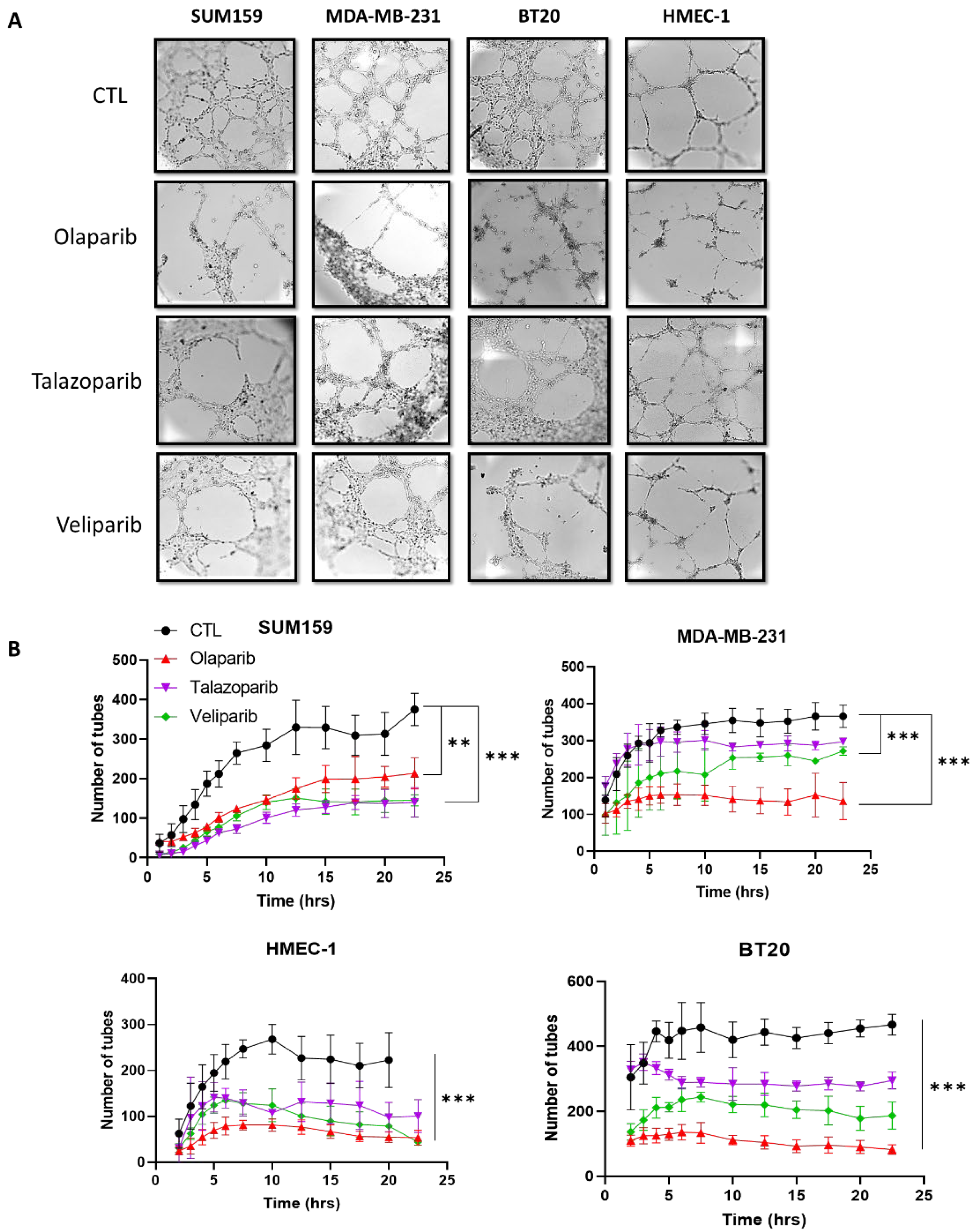
2.2. PARP Inhibition Suppresses the Vasculogenic Mimicry in Breast Cancer Cells
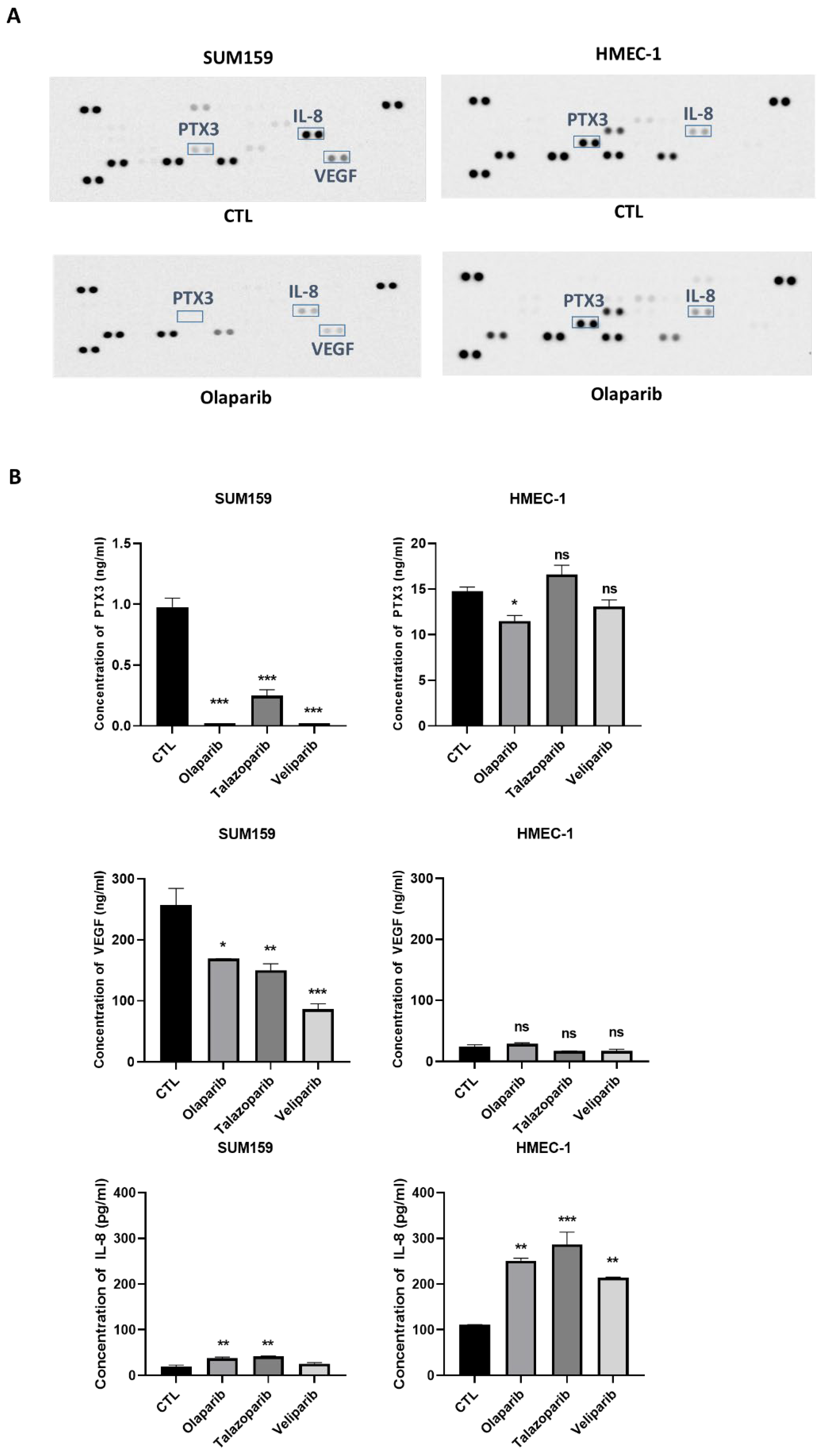
2.3. PARPi Treatment Caused the Decreased Expression of the Angiogenic Proteins
2.4. NF-κB Inactivation Is Required for the PARPi-Inhibited VM Formation
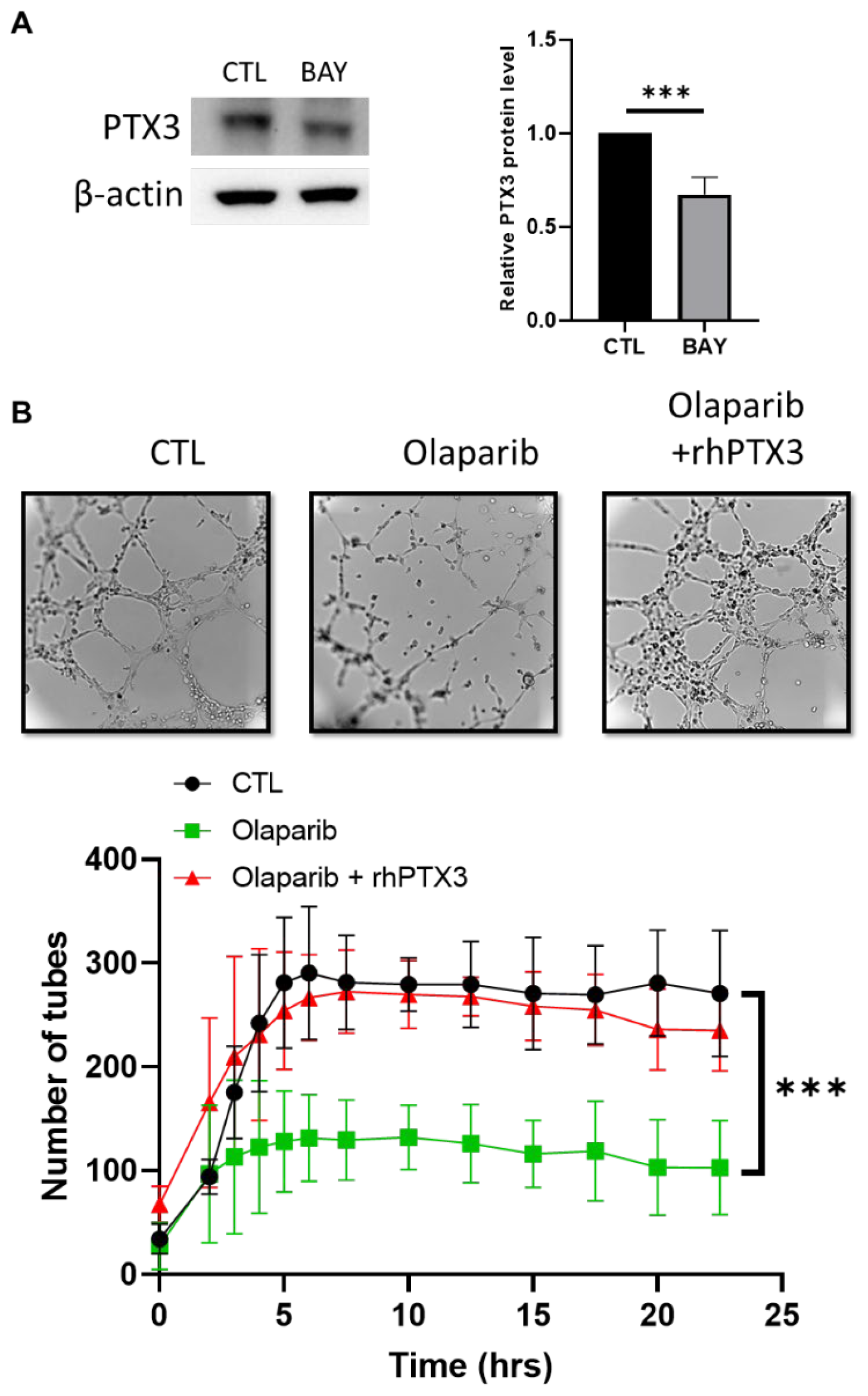
2.5. A NF-κB-PTX3 Axis Is Required for the VM Inhibition by PARPi
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Drug Treatment
4.2. Tube Formation Assay
4.3. Real-Time RT-PCR
4.4. Western Blot
4.5. ELISA
4.6. Luciferase Assay
4.7. Human Angiogenesis Proteome Array
4.8. Flow Cytometry and Cell Viability
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Global Burden of Disease Cancer Collaboration. The Global Burden of Cancer 2013. JAMA Oncol. 2015, 1, 505–527. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment. JAMA 2019, 321, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gligorov, J.; Doval, D.; Bines, J.; Alba, E.; Cortes, P.; Pierga, J.Y.; Gupta, V.; Costa, R.; Srock, S.; de Ducla, S.; et al. Maintenance capecitabine and bevacizumab versus bevacizumab alone after initial first-line bevacizumab and docetaxel for patients with HER2-negative metastatic breast cancer (IMELDA): A randomised, open-label, phase 3 trial. Lancet. Oncol. 2014, 15, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Kirschmann, D.A.; Seftor, E.A.; Hardy, K.M.; Seftor, R.E.; Hendrix, M.J. Molecular pathways: Vasculogenic mimicry in tumor cells: Diagnostic and therapeutic implications. Clin. Cancer Res. 2012, 18, 2726–2732. [Google Scholar] [CrossRef] [Green Version]
- Hendrix, M.J.; Seftor, E.A.; Meltzer, P.S.; Gardner, L.M.; Hess, A.R.; Kirschmann, D.A.; Schatteman, G.C.; Seftor, R.E. Expression and functional significance of VE-cadherin in aggressive human melanoma cells: Role in vasculogenic mimicry. Proc. Natl. Acad. Sci. USA 2001, 98, 8018–8023. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Zhang, D.; Yao, Z.; Lin, X.; Liu, J.; Gu, Q.; Dong, X.; Liu, F.; Wang, Y.; Yao, N.; et al. Anti-angiogenic treatment promotes triple-negative breast cancer invasion via vasculogenic mimicry. Cancer Biol. Ther. 2017, 18, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Wagenblast, E.; Soto, M.; Gutierrez-Angel, S.; Hartl, C.A.; Gable, A.L.; Maceli, A.R.; Erard, N.; Williams, A.M.; Kim, S.Y.; Dickopf, S.; et al. A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis. Nature 2015, 520, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Curtin, N.J.; Szabo, C. Poly(ADP-ribose) polymerase inhibition: Past, present and future. Nat. Rev. Drug. Discov. 2020, 19, 711–736. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef] [PubMed]
- Evans, K.W.; Yuca, E.; Akcakanat, A.; Scott, S.M.; Arango, N.P.; Zheng, X.; Chen, K.; Tapia, C.; Tarco, E.; Eterovic, A.K.; et al. A Population of Heterogeneous Breast Cancer Patient-Derived Xenografts Demonstrate Broad Activity of PARP Inhibitor in BRCA1/2 Wild-Type Tumors. Clin. Cancer Res. 2017, 23, 6468–6477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, S.; Esch, A.; Liby, T.; Gray, J.W.; Heiser, L.M. Pathway-Enriched Gene Signature Associated with 53BP1 Response to PARP Inhibition in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2017, 16, 2892–2901. [Google Scholar] [CrossRef] [Green Version]
- Keung, M.Y.; Wu, Y.; Badar, F.; Vadgama, J.V. Response of Breast Cancer Cells to PARP Inhibitors Is Independent of BRCA Status. J. Clin. Med. 2020, 9, 940. [Google Scholar] [CrossRef] [Green Version]
- Marti, J.M.; Fernandez-Cortes, M.; Serrano-Saenz, S.; Zamudio-Martinez, E.; Delgado-Bellido, D.; Garcia-Diaz, A.; Oliver, F.J. The Multifactorial Role of PARP-1 in Tumor Microenvironment. Cancers 2020, 12, 739. [Google Scholar] [CrossRef] [Green Version]
- Antolin, A.A.; Ameratunga, M.; Banerji, U.; Clarke, P.A.; Workman, P.; Al-Lazikani, B. The kinase polypharmacology landscape of clinical PARP inhibitors. Sci. Rep. 2020, 10, 2585. [Google Scholar] [CrossRef] [Green Version]
- Tentori, L.; Lacal, P.M.; Muzi, A.; Dorio, A.S.; Leonetti, C.; Scarsella, M.; Ruffini, F.; Xu, W.; Min, W.; Stoppacciaro, A.; et al. Poly(ADP-ribose) polymerase (PARP) inhibition or PARP-1 gene deletion reduces angiogenesis. Eur. J. Cancer 2007, 43, 2124–2133. [Google Scholar] [CrossRef] [PubMed]
- Tentori, L.; Muzi, A.; Dorio, A.S.; Bultrini, S.; Mazzon, E.; Lacal, P.M.; Shah, G.M.; Zhang, J.; Navarra, P.; Nocentini, G.; et al. Stable depletion of poly (ADP-ribose) polymerase-1 reduces in vivo melanoma growth and increases chemosensitivity. Eur. J. Cancer 2008, 44, 1302–1314. [Google Scholar] [CrossRef]
- Rodriguez, M.I.; Peralta-Leal, A.; O’Valle, F.; Rodriguez-Vargas, J.M.; Gonzalez-Flores, A.; Majuelos-Melguizo, J.; Lopez, L.; Serrano, S.; de Herreros, A.G.; Rodriguez-Manzaneque, J.C.; et al. PARP-1 regulates metastatic melanoma through modulation of vimentin-induced malignant transformation. PLoS Genet. 2013, 9, e1003531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacal, P.M.; Tentori, L.; Muzi, A.; Ruffini, F.; Dorio, A.S.; Xu, W.; Arcelli, D.; Zhang, J.; Graziani, G. Pharmacological inhibition of poly(ADP-ribose) polymerase activity down-regulates the expression of syndecan-4 and Id-1 in endothelial cells. Int. J. Oncol. 2009, 34, 861–872. [Google Scholar]
- Martin-Oliva, D.; Aguilar-Quesada, R.; O’Valle, F.; Munoz-Gamez, J.A.; Martinez-Romero, R.; Garcia Del Moral, R.; Ruiz de Almodovar, J.M.; Villuendas, R.; Piris, M.A.; Oliver, F.J. Inhibition of poly(ADP-ribose) polymerase modulates tumor-related gene expression, including hypoxia-inducible factor-1 activation, during skin carcinogenesis. Cancer Res. 2006, 66, 5744–5756. [Google Scholar] [CrossRef] [Green Version]
- Rajesh, M.; Mukhopadhyay, P.; Godlewski, G.; Batkai, S.; Hasko, G.; Liaudet, L.; Pacher, P. Poly(ADP-ribose)polymerase inhibition decreases angiogenesis. Biochem. Biophys. Res. Commun. 2006, 350, 1056–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorusso, D.; Guy, H.; Samyshkin, Y.; Hawkes, C.; Estenson, K.; Coleman, R.L. Feasibility Study of a Network Meta-Analysis and Unanchored Population-Adjusted Indirect Treatment Comparison of Niraparib, Olaparib, and Bevacizumab as Maintenance Therapies in Patients with Newly Diagnosed Advanced Ovarian Cancer. Cancers 2022, 14, 1285. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, A.; Ghedini, G.C.; Presta, M.; Ronca, R. Long pentraxin 3: A novel multifaceted player in cancer. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Presta, M.; Foglio, E.; Churruca Schuind, A.; Ronca, R. Long Pentraxin-3 Modulates the Angiogenic Activity of Fibroblast Growth Factor-2. Front. Immunol. 2018, 9, 2327. [Google Scholar] [CrossRef] [Green Version]
- Wesley, U.V.; Sutton, I.; Clark, P.A.; Cunningham, K.; Larrain, C.; Kuo, J.S.; Dempsey, R.J. Enhanced expression of pentraxin-3 in glioblastoma cells correlates with increased invasion and IL8-VEGF signaling axis. Brain. Res. 2022, 1776, 147752. [Google Scholar] [CrossRef]
- Tabruyn, S.P.; Griffioen, A.W. NF-kappa B: A new player in angiostatic therapy. Angiogenesis 2008, 11, 101–106. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.; Jang, H.; Kim, E.H.; Roh, J.L. Efficacy of poly (ADP-ribose) polymerase inhibitor olaparib against head and neck cancer cells: Predictions of drug sensitivity based on PAR-p53-NF-kappaB interactions. Cell Cycle 2016, 15, 3105–3114. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Sonnenblick, A.; de Azambuja, E.; Azim, H.A., Jr.; Piccart, M. An update on PARP inhibitors--moving to the adjuvant setting. Nat. Rev. Clin. Oncol. 2015, 12, 27–41. [Google Scholar]
- Mangerich, A.; Burkkle, A. Pleiotropic cellular functions of PARP1 in longevity and aging: Genome maintenance meets inflammation. Oxid. Med. Cell Longev. 2012, 2012, 321653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinatizadeh, M.R.; Schock, B.; Chalbatani, G.M.; Zarandi, P.K.; Jalali, S.A.; Miri, S. R The Nuclear Factor Kappa B (NF-kB) signaling in cancer development and immune diseases. Genes Dis. 2021, 8, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Good, D.J.; Polverini, P.J.; Rastinejad, F.; Le Beau, M.M.; Lemons, R.S.; Frazier, W.A.; Bouck, N.P. A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc. Natl. Acad. Sci. USA 1990, 87, 6624–6628. [Google Scholar] [CrossRef] [Green Version]
- Sabbah, M.; Prunier, C.; Ferrand, N.; Megalophonos, V.; Lambein, K.; De Wever, O.; Nazaret, N.; Lachuer, J.; Dumont, S.; Redeuilh, G. CCN5, a novel transcriptional repressor of the transforming growth factor beta signaling pathway. Mol. Cell Biol. 2011, 31, 1459–1469. [Google Scholar] [CrossRef] [Green Version]
- Inbar-Rozensal, D.; Castiel, A.; Visochek, L.; Castel, D.; Dantzer, F.; Izraeli, S.; Cohen-Armon, M. A selective eradication of human nonhereditary breast cancer cells by phenanthridine-derived polyADP-ribose polymerase inhibitors. Breast Cancer Res. 2009, 11, R78. [Google Scholar] [CrossRef] [Green Version]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, P.; Dey, P.; Ghosh, S.; Sarma, A.; Ghosh, U. Reduction of metastatic potential by inhibiting EGFR/Akt/p38/ERK signaling pathway and epithelial-mesenchymal transition after carbon ion exposure is potentiated by PARP-1 inhibition in non-small-cell lung cancer. BMC Cancer 2019, 19, 829. [Google Scholar] [CrossRef] [Green Version]
- Faraoni, I.; Aloisio, F.; De Gabrieli, A.; Consalvo, M.I.; Lavorgna, S.; Voso, M.T.; Lo-Coco, F.; Graziani, G. The poly(ADP-ribose) polymerase inhibitor olaparib induces up-regulation of death receptors in primary acute myeloid leukemia blasts by NF-kappaB activation. Cancer Lett. 2018, 423, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Nowsheen, S.; Cooper, T.; Bonner, J.A.; LoBuglio, A.F.; Yang, E.S. HER2 overexpression renders human breast cancers sensitive to PARP inhibition independently of any defect in homologous recombination DNA repair. Cancer Res. 2012, 72, 4796–4806. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.S. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. J. Clin. Investig. 2001, 107, 241–246. [Google Scholar] [CrossRef]
- Alles, V.V.; Bottazzi, B.; Peri, G.; Golay, J.; Introna, M.; Mantovani, A. Inducible expression of PTX3, a new member of the pentraxin family, in human mononuclear phagocytes. Blood 1994, 84, 3483–3493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmo, R.F.; Aroucha, D.; Vasconcelos, L.R.; Pereira, L.M.; Moura, P.; Cavalcanti, M.S. Genetic variation in PTX3 and plasma levels associated with hepatocellular carcinoma in patients with HCV. J. Viral Hepat. 2016, 23, 116–122. [Google Scholar] [CrossRef]
- Diamandis, E.P.; Goodglick, L.; Planque, C.; Thornquist, M.D. Pentraxin-3 is a novel biomarker of lung carcinoma. Clin. Cancer Res. 2011, 17, 2395–2399. [Google Scholar] [CrossRef] [Green Version]
- Kondo, S.; Ueno, H.; Hosoi, H.; Hashimoto, J.; Morizane, C.; Koizumi, F.; Tamura, K.; Okusaka, T. Clinical impact of pentraxin family expression on prognosis of pancreatic carcinoma. Br. J. Cancer 2013, 109, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Stallone, G.; Cormio, L.; Netti, G.S.; Infante, B.; Selvaggio, O.; Fino, G.D.; Ranieri, E.; Bruno, F.; Prattichizzo, C.; Sanguedolce, F.; et al. Pentraxin 3: A novel biomarker for predicting progression from prostatic inflammation to prostate cancer. Cancer Res. 2014, 74, 4230–4238. [Google Scholar] [CrossRef] [Green Version]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [Green Version]
- Scimeca, M.; Antonacci, C.; Colombo, D.; Bonfiglio, R.; Buonomo, O.C.; Bonanno, E. Emerging prognostic markers related to mesenchymal characteristics of poorly differentiated breast cancers. Tumour. Biol. 2016, 37, 5427–5435. [Google Scholar] [CrossRef]
- Thomas, C.; Henry, W.; Cuiffo, B.G.; Collmann, A.Y.; Marangoni, E.; Benhamo, V.; Bhasin, M.K.; Fan, C.; Fuhrmann, L.; Baldwin, A.S.; et al. Pentraxin-3 is a PI3K signaling target that promotes stem cell-like traits in basal-like breast cancers. Sci. Signal 2017, 10, eaah4674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathore, M.; Girard, C.; Ohanna, M.; Tichet, M.; Ben Jouira, R.; Garcia, E.; Larbret, F.; Gesson, M.; Audebert, S.; Lacour, J.P.; et al. Cancer cell-derived long pentraxin 3 (PTX3) promotes melanoma migration through a toll-like receptor 4 (TLR4)/NF-kappaB signaling pathway. Oncogene 2019, 38, 5873–5889. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.; Lee, E.J.; Song, D.H.; Yoon, S.C.; Chung, Y.H.; Jang, Y.; Kim, S.M.; Song, Y.; Kang, S.W.; Yoon, S.Y.; et al. Elevated Pentraxin 3 in bone metastatic breast cancer is correlated with osteolytic function. Oncotarget 2014, 5, 481–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravenna, L.; Sale, P.; Di Vito, M.; Russo, A.; Salvatori, L.; Tafani, M.; Mari, E.; Sentinelli, S.; Petrangeli, E.; Gallucci, M.; et al. Up-regulation of the inflammatory-reparative phenotype in human prostate carcinoma. Prostate 2009, 69, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Yeni, M.; Korkut, E.; Aksungur, N.; Kara, S.; Askin, S.; Kartal, M. Determination of Pentraxin-3, Interleukin-8 and Vascular Endothelial Growth Factor Levels in Patients with Gastric Adenocarcinoma. Asian Pac. J. Cancer Prev. 2021, 22, 1507–1512. [Google Scholar] [CrossRef]
- Hida, K.; Maishi, N.; Kawamoto, T.; Akiyama, K.; Ohga, N.; Hida, Y.; Yamada, K.; Hojo, T.; Kikuchi, H.; Sato, M.; et al. Tumor endothelial cells express high pentraxin 3 levels. Pathol. Int. 2016, 66, 687–694. [Google Scholar] [CrossRef]
- Aguilera-Aguirre, L.; Bacsi, A.; Radak, Z.; Hazra, T.K.; Mitra, S.; Sur, S.; Brasier, A.R.; Ba, X.; Boldogh, I. Innate inflammation induced by the 8-oxoguanine DNA glycosylase-1-KRAS-NF-kappaB pathway. J. Immunol. 2014, 193, 4643–4653. [Google Scholar] [CrossRef] [Green Version]
- Ba, X.; Bacsi, A.; Luo, J.; Aguilera-Aguirre, L.; Zeng, X.; Radak, Z.; Brasier, A.R.; Boldogh, I. 8-oxoguanine DNA glycosylase-1 augments proinflammatory gene expression by facilitating the recruitment of site-specific transcription factors. J. Immunol. 2014, 192, 2384–2394. [Google Scholar] [CrossRef] [Green Version]
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Chen, X.; Xu, X.; Qian, Y.; Liang, G.; Yao, F.; Yao, Z.; Wu, H.; Zhang, J.; He, Q.; et al. PARP1 Suppresses the Transcription of PD-L1 by Poly(ADP-Ribosyl)ating STAT3. Cancer Immunol. Res. 2019, 7, 136–149. [Google Scholar] [CrossRef] [Green Version]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrand, N.; Fert, A.; Morichon, R.; Radosevic-Robin, N.; Zaoui, M.; Sabbah, M. WISP2/CCN5 Suppresses Vasculogenic Mimicry through Inhibition of YAP/TAZ Signaling in Breast Cancer Cells. Cancers 2022, 14, 1487. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhou, L.; Ling, L.; Meng, X.; Chu, F.; Zhang, S.; Zhou, F. The Crosstalk Between Hippo-YAP Pathway and Innate Immunity. Front. Immunol. 2020, 11, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanisavljevic, J.; Porta-de-la-Riva, M.; Batlle, R.; de Herreros, A.G.; Baulida, J. The p65 subunit of NF-kappaB and PARP1 assist Snail1 in activating fibronectin transcription. J. Cell Sci. 2011, 124, 4161–4171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Zhang, Y.; Wang, Z.; Dong, H.; Fu, N.; Han, X. The roles and signaling pathways of prolyl-4-hydroxylase 2 in the tumor microenvironment. Chem. Biol. Interact. 2019, 303, 40–49. [Google Scholar] [CrossRef]
- Carpentier, G.; Berndt, S.; Ferratge, S.; Rasband, W.; Cuendet, M.; Uzan, G.; Albanese, P. Angiogenesis Analyzer for ImageJ—A comparative morphometric analysis of “Endothelial Tube Formation Assay” and “Fibrin Bead Assay”. Sci. Rep. 2020, 10, 11568. [Google Scholar] [CrossRef]
- Ferrand, N.; Gnanapragasam, A.; Dorothee, G.; Redeuilh, G.; Larsen, A.K.; Sabbah, M. Loss of WISP2/CCN5 in estrogen-dependent MCF7 human breast cancer cells promotes a stem-like cell phenotype. PLoS ONE 2014, 9, e87878. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Classification | ER | PR | HER2 | Tumor | BRCA Status |
|---|---|---|---|---|---|---|
| T47D | Lum A | + | + | − | IDC | WT |
| BT474 | Lum B | + | + | + | IDC | BRCA2 |
| MCF-7 | Lum A | + | + | − | IDC | WT |
| ZR-75.1 | Lum A | + | + − | − | IDC | WT |
| SKBR3 | HER2 | − | − | + | AC | WT |
| MDA-MB-436 | Basal A | − | − | − | AC | BRCA1 |
| HCC38 | Basal B (Claudin Low) | − | − | − | DC | WT |
| SUM1315 M02 | Basal B | − | − | − | IDC | BRCA1 |
| BT549 | Basal B (Claudin Low) | − | − | − | DC | BRCA1 |
| SUM149 | Basal B (Claudin Low) | − | − | − | DC | BRCA1 |
| HCC1937 | Basal A | − | − | − | DC | BRCA1 |
| BT20 | Basal A | − | − | − | IDC | WT |
| MDA-MB-231 | Basal B | − | − | − | IDC | WT |
| HS578T | Basal B (Claudin Low) | − | − | − | IDC | WT |
| SUM159 | Basal B (Claudin Low) | − | − | − | AnC | WT |
| HMEC-1 | Endothéliale | WT |
| Target | Forward (5′ → 3′) | Reverse (5′ → 3′) |
|---|---|---|
| β-Actin | GGACTTCG AGCAAGAGATGG | AGCACTGTGTTGGCGTACAG |
| 36B4 | TCGACAATGGCAGCATCTAC | GCCTTGACCTTTTCAGCAAG |
| PTX3 | CGTCTCTCCAGCAATGCAT | AAGAGCTTGTCCCATTCCGA |
| VEGF | ATGACCCAGTTTGGGAACA | TCCTGAATCTTCCAGGCAGT |
| uPA | TGCCCTGAAGTCGTTAGTGT | ATCTCCTGTGCQTGGGTGQQ |
| PAI1 | CTCTCTCTGCCCTCACCAAC | GTGGAGAGGCTCTTGGTCTG |
| IL-8 | TAGCAAAATTGAGGCCAAGG | AAACAAGGCACAGTGGAAC |
| TIMP-1 | CTTCTGCAATTCCGACCTCGT | ACGCTGGTATAAGGTGGTCTG |
| THSB1 | TTGTCTTTGGAACCACACCA | CTGGACAGCTCATCACAGGA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chivot, J.; Ferrand, N.; Fert, A.; Van Dreden, P.; Morichon, R.; Sabbah, M. PARP Inhibitor Inhibits the Vasculogenic Mimicry through a NF-κB-PTX3 Axis Signaling in Breast Cancer Cells. Int. J. Mol. Sci. 2022, 23, 16171. https://doi.org/10.3390/ijms232416171
Chivot J, Ferrand N, Fert A, Van Dreden P, Morichon R, Sabbah M. PARP Inhibitor Inhibits the Vasculogenic Mimicry through a NF-κB-PTX3 Axis Signaling in Breast Cancer Cells. International Journal of Molecular Sciences. 2022; 23(24):16171. https://doi.org/10.3390/ijms232416171
Chicago/Turabian StyleChivot, Justine, Nathalie Ferrand, Aude Fert, Patrick Van Dreden, Romain Morichon, and Michèle Sabbah. 2022. "PARP Inhibitor Inhibits the Vasculogenic Mimicry through a NF-κB-PTX3 Axis Signaling in Breast Cancer Cells" International Journal of Molecular Sciences 23, no. 24: 16171. https://doi.org/10.3390/ijms232416171
APA StyleChivot, J., Ferrand, N., Fert, A., Van Dreden, P., Morichon, R., & Sabbah, M. (2022). PARP Inhibitor Inhibits the Vasculogenic Mimicry through a NF-κB-PTX3 Axis Signaling in Breast Cancer Cells. International Journal of Molecular Sciences, 23(24), 16171. https://doi.org/10.3390/ijms232416171