
The Role of Calcium Signaling in Melanoma




{kind=link}
{kind=link}
Abstract
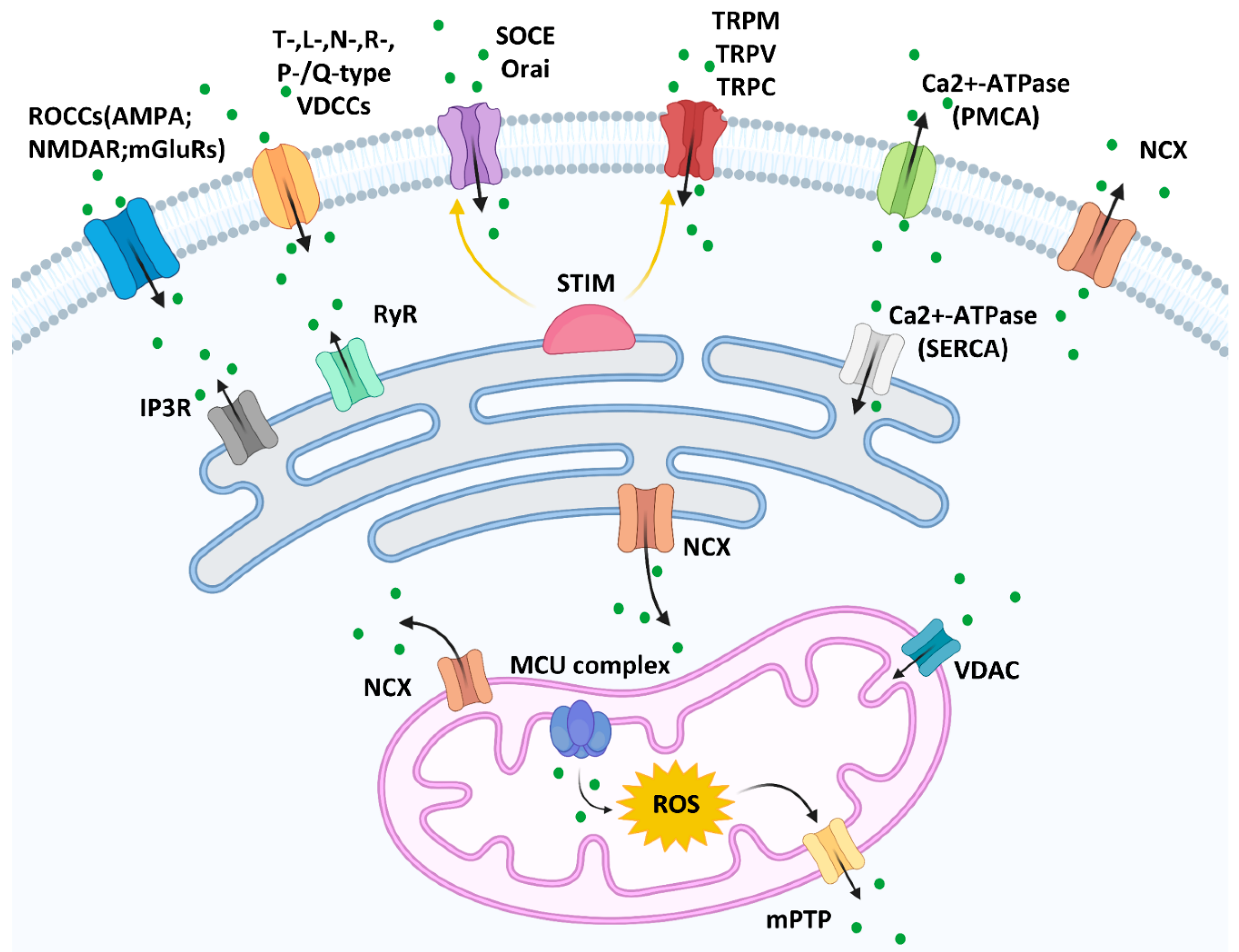
:1. Introduction
2. Calcium Signaling in Melanogenesis and Melanoma Tumorigenesis
2.1. Calcium Signaling in Melanogenesis
2.2. Calcium Signaling in Melanoma Tumorigenesis
3. Calcium Signaling in Melanoma Progression
3.1. [Ca2+]i Oscillation Influences Melanoma Progression
3.2. Calcium Channels Are Involved in Melanoma Progression
3.3. Ca2+ Signaling Influences Melanoma Progression through the Change of Morphological and Phenotypical Changes
3.4. Calcium-Related Pathways Participate in Melanoma Progression
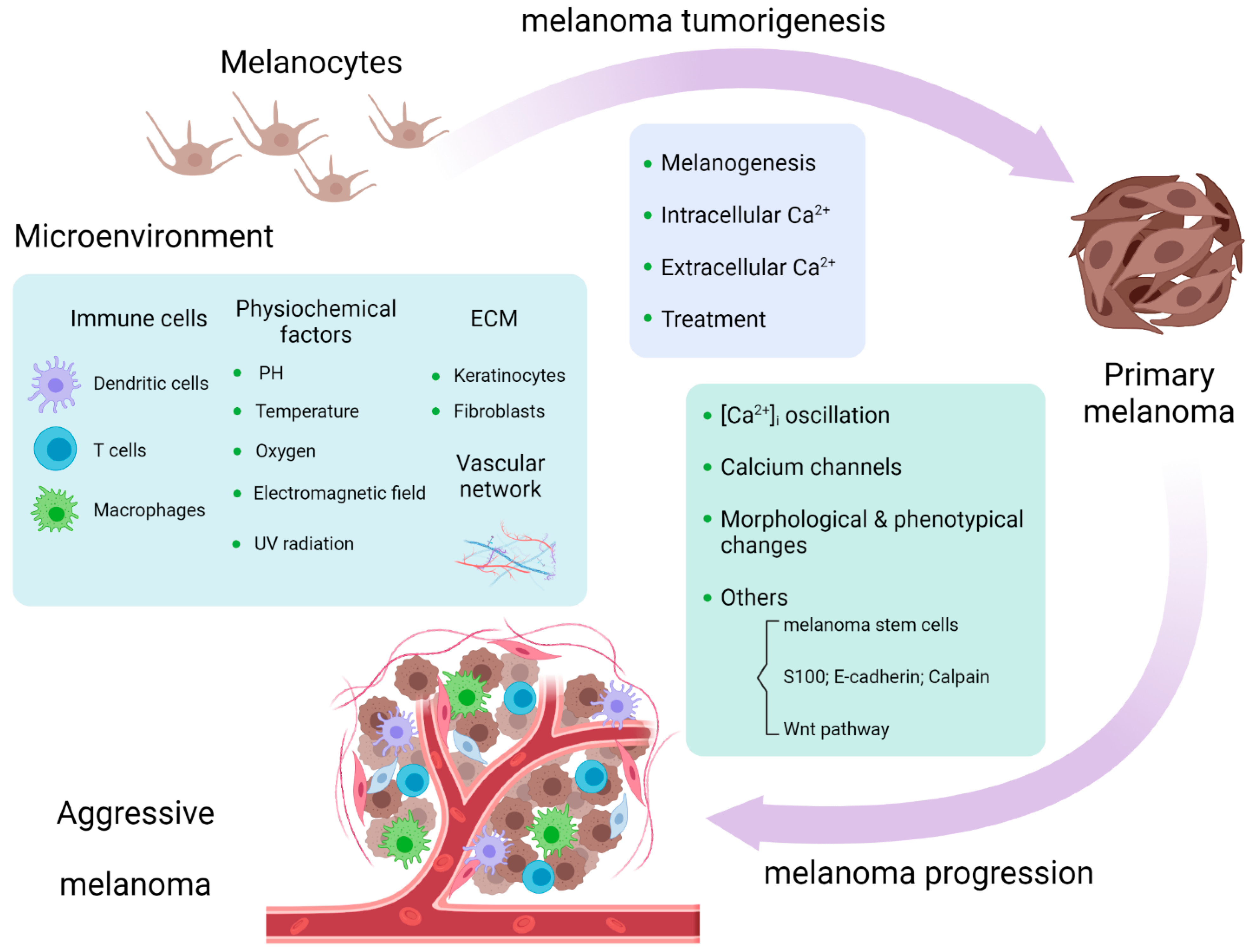
4. Calcium Signaling in Melanoma Microenvironment
4.1. Immune Cells
4.2. ECM and Vascular Network
4.3. Physical and Chemical Surroundings
5. Calcium Signaling and Other Ionic Channels in Melanoma
5.1. Sodium Channels
5.2. Potassium Channels
6. Calcium Signaling in Melanoma Treatment
6.1. Targeting Calcium, Mitochondria, and ER Stress in Melanoma
6.2. Drug Resistance and Combination Treatment
6.3. S100 Protein Family in Melanoma Prediction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allgower, C.; Kretz, A.L.; von Karstedt, S.; Wittau, M.; Henne-Bruns, D.; Lemke, J. Friend or Foe: S100 Proteins in Cancer. Cancers 2020, 12, 2037. [Google Scholar] [CrossRef]
- Brini, M.; Cali, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef]
- Tan, Y.; Deng, Y.L.; Qing, H. Calcium channel blockers and Alzheimer’s disease. Neural Regen. Res. 2012, 7, 137–140. [Google Scholar] [CrossRef]
- Ribeiro, M.P.C.; Nunes-Correia, I.; Santos, A.E.; Custodio, J.B.A. The combination of glutamate receptor antagonist MK-801 with tamoxifen and its active metabolites potentiates their antiproliferative activity in mouse melanoma K1735-M2 cells. Exp. Cell Res. 2014, 321, 288–296. [Google Scholar] [CrossRef]
- Choi, K.Y.; Chang, K.; Pickel, J.M.; Badger, J.D., 2nd; Roche, K.W. Expression of the metabotropic glutamate receptor 5 (mGluR5) induces melanoma in transgenic mice. Proc. Natl. Acad. Sci. USA 2011, 108, 15219–15224. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.J.; Wall, B.A.; Wangari-Talbot, J.; Chen, S. Metabotropic glutamate receptors in cancer. Neuropharmacology 2017, 115, 193–202. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Pushparaj, C.; Bahi, N.; Sorolla, A.; Herreros, J.; Pamplona, R.; Vilella, R.; Matias-Guiu, X.; Marti, R.M.; Canti, C. Functional expression of voltage-gated calcium channels in human melanoma. Pigment Cell Melanoma Res. 2012, 25, 200–212. [Google Scholar] [CrossRef]
- Serwach, K.; Gruszczynska-Biegala, J. Target Molecules of STIM Proteins in the Central Nervous System. Front. Mol. Neurosci. 2020, 13, 617422. [Google Scholar] [CrossRef]
- Martinsen, A.; Dessy, C.; Morel, N. Regulation of calcium channels in smooth muscle: New insights into the role of myosin light chain kinase. Channels (Austin) 2014, 8, 402–413. [Google Scholar] [CrossRef] [Green Version]
- Ushioda, R.; Miyamoto, A.; Inoue, M.; Watanabe, S.; Okumura, M.; Maegawa, K.I.; Uegaki, K.; Fujii, S.; Fukuda, Y.; Umitsu, M.; et al. Redox-assisted regulation of Ca2+ homeostasis in the endoplasmic reticulum by disulfide reductase ERdj5. Proc. Natl. Acad. Sci. USA 2016, 113, E6055–E6063. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Hao, Y.; Chen, H.; He, Q.; Yuan, Z.; Cheng, J. Mitochondrial calcium uniporter protein MCU is involved in oxidative stress-induced cell death. Protein Cell 2015, 6, 434–442. [Google Scholar] [CrossRef] [Green Version]
- Feno, S.; Butera, G.; Vecellio Reane, D.; Rizzuto, R.; Raffaello, A. Crosstalk between Calcium and ROS in Pathophysiological Conditions. Oxid. Med. Cell. Longev. 2019, 2019, 9324018. [Google Scholar] [CrossRef] [Green Version]
- Briston, T.; Roberts, M.; Lewis, S.; Powney, B.; Staddon, J.M.; Szabadkai, G.; Duchen, M.R. Mitochondrial permeability transition pore: Sensitivity to opening and mechanistic dependence on substrate availability. Sci. Rep. 2017, 7, 10492. [Google Scholar] [CrossRef]
- Adapted from “TGFb Signaling Pathway”, by BioRender.com. Available online: https://app.biorender.com/biorender-templates (accessed on 19 November 2021).
- Davis, L.; Tarduno, A.; Lu, Y.C. Neoantigen-Reactive T Cells: The Driving Force behind Successful Melanoma Immunotherapy. Cancers 2021, 13, 6061. [Google Scholar] [CrossRef]
- Bellono, N.W.; Oancea, E.V. Ion transport in pigmentation. Arch. Biochem. Biophys. 2014, 563, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Devi, S.; Kedlaya, R.; Maddodi, N.; Bhat, K.M.; Weber, C.S.; Valdivia, H.; Setaluri, V. Calcium homeostasis in human melanocytes: Role of transient receptor potential melastatin 1 (TRPM1) and its regulation by ultraviolet light. Am. J. Physiol. Cell Physiol. 2009, 297, C679–C687. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Lu, F.; He, H.; Shen, J.; Messina, J.; Mathew, R.; Wang, D.; Sarnaik, A.A.; Chang, W.C.; Kim, M.; et al. STIM1- and Orai1-mediated Ca(2+) oscillation orchestrates invadopodium formation and melanoma invasion. J. Cell. Biol. 2014, 207, 535–548. [Google Scholar] [CrossRef] [Green Version]
- Stanisz, H.; Stark, A.; Kilch, T.; Schwarz, E.C.; Muller, C.S.; Peinelt, C.; Hoth, M.; Niemeyer, B.A.; Vogt, T.; Bogeski, I. ORAI1 Ca(2+) channels control endothelin-1-induced mitogenesis and melanogenesis in primary human melanocytes. J. Investig. Dermatol. 2012, 132, 1443–1451. [Google Scholar] [CrossRef] [Green Version]
- Alharbi, A.F.; Parrington, J. Endolysosomal Ca(2+) Signaling in Cancer: The Role of TPC2, From Tumorigenesis to Metastasis. Front. Cell Dev. Biol. 2019, 7, 302. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Liu, H.; Xu, Y.; Xie, J.; Zhu, D.; Amos, C.I.; Fang, S.; Lee, J.E.; Li, X.; Nan, H.; et al. Genetic variants in the calcium signaling pathway genes are associated with cutaneous melanoma-specific survival. Carcinogenesis 2019, 40, 279–288. [Google Scholar] [CrossRef]
- Dissanayake, S.K.; Weeraratna, A.T. Detecting PKC phosphorylation as part of the Wnt/calcium pathway in cutaneous melanoma. Methods Mol. Biol. 2008, 468, 157–172. [Google Scholar] [CrossRef] [Green Version]
- Oka, M.; Kikkawa, U. Protein kinase C in melanoma. Cancer Metastasis Rev. 2005, 24, 287–300. [Google Scholar] [CrossRef]
- Krenzer, S.; Peterziel, H.; Mauch, C.; Blaber, S.I.; Blaber, M.; Angel, P.; Hess, J. Expression and function of the kallikrein-related peptidase 6 in the human melanoma microenvironment. J. Investig. Dermatol. 2011, 131, 2281–2288. [Google Scholar] [CrossRef] [Green Version]
- Robert, G.; Gaggioli, C.; Bailet, O.; Chavey, C.; Abbe, P.; Aberdam, E.; Sabatie, E.; Cano, A.; Garcia de Herreros, A.; Ballotti, R.; et al. SPARC represses E-cadherin and induces mesenchymal transition during melanoma development. Cancer Res. 2006, 66, 7516–7523. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.B.; Islam, S.U.; Lee, Y.S. PRP4 Promotes Skin Cancer by Inhibiting Production of Melanin, Blocking Influx of Extracellular Calcium, and Remodeling Cell Actin Cytoskeleton. Int. J. Mol. Sci. 2021, 22, 6992. [Google Scholar] [CrossRef]
- Fedida-Metula, S.; Elhyany, S.; Tsory, S.; Segal, S.; Hershfinkel, M.; Sekler, I.; Fishman, D. Targeting lipid rafts inhibits protein kinase B by disrupting calcium homeostasis and attenuates malignant properties of melanoma cells. Carcinogenesis 2008, 29, 1546–1554. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Zaguilan, R.; Martinez, G.M.; Gomez, A.; Hendrix, M.J.C.; Gillies, R.J. Distinct regulation of pH(in) and [Ca2+](in) in human melanoma cells with different metastatic potential. J. Cell Physiol. 1998, 176, 196–205. [Google Scholar] [CrossRef]
- Baljinnyam, E.; Umemura, M.; De Lorenzo, M.S.; Xie, L.H.; Nowycky, M.; Iwatsubo, M.; Chen, S.; Goydos, J.S.; Iwatsubo, K. Gbetagamma subunits inhibit Epac-induced melanoma cell migration. BMC Cancer 2011, 11, 256. [Google Scholar] [CrossRef] [Green Version]
- Baljinnyam, E.; De Lorenzo, M.S.; Xie, L.H.; Iwatsubo, M.; Chen, S.; Goydos, J.S.; Nowycky, M.C.; Iwatsubo, K. Exchange protein directly activated by cyclic AMP increases melanoma cell migration by a Ca2+-dependent mechanism. Cancer Res. 2010, 70, 5607–5617. [Google Scholar] [CrossRef] [Green Version]
- Arozarena, I.; Sanchez-Laorden, B.; Packer, L.; Hidalgo-Carcedo, C.; Hayward, R.; Viros, A.; Sahai, E.; Marais, R. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5A. Cancer Cell 2011, 19, 45–57. [Google Scholar] [CrossRef]
- Kosnopfel, C.; Sinnberg, T.; Sauer, B.; Niessner, H.; Muenchow, A.; Fehrenbacher, B.; Schaller, M.; Mertens, P.R.; Garbe, C.; Thakur, B.K.; et al. Tumour Progression Stage-Dependent Secretion of YB-1 Stimulates Melanoma Cell Migration and Invasion. Cancers 2020, 12, 2328. [Google Scholar] [CrossRef]
- Gelis, L.; Jovancevic, N.; Bechara, F.G.; Neuhaus, E.M.; Hatt, H. Functional expression of olfactory receptors in human primary melanoma and melanoma metastasis. Exp. Dermatol. 2017, 26, 569–576. [Google Scholar] [CrossRef]
- D’Mello, S.A.; Joseph, W.R.; Green, T.N.; Leung, E.Y.; During, M.J.; Finlay, G.J.; Baguley, B.C.; Kalev-Zylinska, M.L. Selected GRIN2A mutations in melanoma cause oncogenic effects that can be modulated by extracellular glutamate. Cell Calcium 2016, 60, 384–395. [Google Scholar] [CrossRef]
- Umemura, M.; Baljinnyam, E.; Feske, S.; De Lorenzo, M.S.; Xie, L.H.; Feng, X.; Oda, K.; Makino, A.; Fujita, T.; Yokoyama, U.; et al. Store-operated Ca2+ entry (SOCE) regulates melanoma proliferation and cell migration. PLoS ONE 2014, 9, e89292. [Google Scholar] [CrossRef]
- D’Amore, A.; Hanbashi, A.A.; Di Agostino, S.; Palombi, F.; Sacconi, A.; Voruganti, A.; Taggi, M.; Canipari, R.; Blandino, G.; Parrington, J.; et al. Loss of Two-Pore Channel 2 (TPC2) Expression Increases the Metastatic Traits of Melanoma Cells by a Mechanism Involving the Hippo Signalling Pathway and Store-Operated Calcium Entry. Cancers 2020, 12, 2391. [Google Scholar] [CrossRef]
- Hegedus, L.; Garay, T.; Molnar, E.; Varga, K.; Bilecz, A.; Torok, S.; Padanyi, R.; Paszty, K.; Wolf, M.; Grusch, M.; et al. The plasma membrane Ca(2+) pump PMCA4b inhibits the migratory and metastatic activity of BRAF mutant melanoma cells. Int. J. Cancer 2017, 140, 2758–2770. [Google Scholar] [CrossRef] [Green Version]
- Naffa, R.; Vogel, L.; Hegedus, L.; Paszty, K.; Toth, S.; Kelemen, K.; Singh, N.; Remenyi, A.; Kallay, E.; Cserepes, M.; et al. P38 MAPK Promotes Migration and Metastatic Activity of BRAF Mutant Melanoma Cells by Inducing Degradation of PMCA4b. Cells 2020, 9, 1209. [Google Scholar] [CrossRef]
- Long, T.; Su, J.; Tang, W.; Luo, Z.; Liu, S.; Liu, Z.; Zhou, H.; Qi, M.; Zeng, W.; Zhang, J.; et al. A novel interaction between calcium-modulating cyclophilin ligand and Basigin regulates calcium signaling and matrix metalloproteinase activities in human melanoma cells. Cancer Lett. 2013, 339, 93–101. [Google Scholar] [CrossRef]
- Maiques, O.; Barcelo, C.; Panosa, A.; Pijuan, J.; Orgaz, J.L.; Rodriguez-Hernandez, I.; Matas-Nadal, C.; Tell, G.; Vilella, R.; Fabra, A.; et al. T-type calcium channels drive migration/invasion in BRAFV600E melanoma cells through Snail1. Pigment Cell Melanoma Res. 2018, 31, 484–495. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Luo, Z.; Hao, Y.; Ba, W.; Wang, R.; Wang, W.; Ding, X.; Li, C. mTOR-mediated Na(+)/Ca(2+) exchange affects cell proliferation and metastasis of melanoma cells. Biomed. Pharmacother. 2017, 92, 744–749. [Google Scholar] [CrossRef]
- Schmidt, J.; Friebel, K.; Schonherr, R.; Coppolino, M.G.; Bosserhoff, A.K. Migration-associated secretion of melanoma inhibitory activity at the cell rear is supported by KCa3.1 potassium channels. Cell Res. 2010, 20, 1224–1238. [Google Scholar] [CrossRef] [Green Version]
- Jia, Q.; Hu, S.; Jiao, D.; Li, X.; Qi, S.; Fan, R. Synaptotagmin-4 promotes dendrite extension and melanogenesis in alpaca melanocytes by regulating Ca(2+) influx via TRPM1 channels. Cell Biochem. Funct. 2020, 38, 275–282. [Google Scholar] [CrossRef]
- Sun, J.; Lin, S.; Keeley, T.; Yang, S. Disseminating Melanoma Cells Surf on Calcium Waves. Mol. Cell. Oncol. 2015, 2, e1002714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.H.; Gill, N.K.; Nyberg, K.D.; Nguyen, A.V.; Hohlbauch, S.V.; Geisse, N.A.; Nowell, C.J.; Sloan, E.K.; Rowat, A.C. Cancer cells become less deformable and more invasive with activation of beta-adrenergic signaling. J. Cell Sci. 2016, 129, 4563–4575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meghnani, V.; Vetter, S.W.; Leclerc, E. RAGE overexpression confers a metastatic phenotype to the WM115 human primary melanoma cell line. Biochim. Biophys. Acta 2014, 1842, 1017–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terrie, E.; Coronas, V.; Constantin, B. Role of the calcium toolkit in cancer stem cells. Cell Calcium 2019, 80, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Neves de Oliveira, B.H.; Dalmaz, C.; Zeidan-Chulia, F. Network-Based Identification of Altered Stem Cell Pluripotency and Calcium Signaling Pathways in Metastatic Melanoma. Med. Sci. 2018, 6, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettum, I.J.; Gorad, S.S.; Barkovskaya, A.; Pettersen, S.; Moestue, S.A.; Vasiliauskaite, K.; Tenstad, E.; Oyjord, T.; Risa, O.; Nygaard, V.; et al. Metabolic reprogramming supports the invasive phenotype in malignant melanoma. Cancer Lett. 2015, 366, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Ito, T.; Nakahara, T.; Nagae, K.; Fuyuno, Y.; Nakao, M.; Akahoshi, M.; Nakagawa, R.; Tu, Y.; Uchi, H.; et al. Upregulation of S100P, receptor for advanced glycation end products and ezrin in malignant melanoma. J. Dermatol. 2013, 40, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Venza, M.; Visalli, M.; Catalano, T.; Biondo, C.; Beninati, C.; Teti, D.; Venza, I. DNA methylation-induced E-cadherin silencing is correlated with the clinicopathological features of melanoma. Oncol. Rep. 2016, 35, 2451–2460. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Zhu, L.; Li, Y.; Zheng, Z.; Lin, X.; Yang, C. LncRNA MEG3 promotes melanoma growth, metastasis and formation through modulating miR-21/E-cadherin axis. Cancer Cell Int. 2020, 20, 12. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Aladowicz, E.; Lanfrancone, L.; Goding, C.R. Tbx3 represses E-cadherin expression and enhances melanoma invasiveness. Cancer Res. 2008, 68, 7872–7881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimbourg, Q.; Perez, J.; Vandermeersch, S.; Prignon, A.; Hanouna, G.; Haymann, J.P.; Baud, L.; Letavernier, E. The calpain/calpastatin system has opposing roles in growth and metastatic dissemination of melanoma. PLoS ONE 2013, 8, e60469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weeraratna, A.T. A wnt-er wonderland—The complexity of wnt signaling in melanoma. Cancer Metast. Rev. 2005, 24, 237–250. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.P.; Fiori, J.L.; Baugher, K.M.; Indig, F.E.; French, A.D.; Camilli, T.C.; Frank, B.P.; Earley, R.; Hoek, K.S.; Hasskamp, J.H.; et al. Wnt5A activates the calpain-mediated cleavage of filamin A. J. Investig. Dermatol. 2009, 129, 1782–1789. [Google Scholar] [CrossRef] [Green Version]
- Witze, E.S.; Connacher, M.K.; Houel, S.; Schwartz, M.P.; Morphew, M.K.; Reid, L.; Sacks, D.B.; Anseth, K.S.; Ahn, N.G. Wnt5a directs polarized calcium gradients by recruiting cortical endoplasmic reticulum to the cell trailing edge. Dev. Cell 2013, 26, 645–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adapted from “Tumor Microenvironment”, by BioRender.com. Available online: https://app.biorender.com/biorender-templates (accessed on 19 December 2021).
- Singh, K.; Rosenberg, P. Anti-tumour activity and store operated calcium entry: New roles in immunology. EMBO Mol. Med. 2013, 5, 1297–1299. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.D.; Bae, S.; Capece, T.; Nedelkovska, H.; de Rubio, R.G.; Smrcka, A.V.; Jun, C.D.; Jung, W.; Park, B.; Kim, T.I.; et al. Targeted calcium influx boosts cytotoxic T lymphocyte function in the tumour microenvironment. Nat. Commun. 2017, 8, 15365. [Google Scholar] [CrossRef]
- Mookerjee-Basu, J.; Hooper, R.; Gross, S.; Schultz, B.; Go, C.K.; Samakai, E.; Ladner, J.; Nicolas, E.; Tian, Y.; Zhou, B.; et al. Suppression of Ca(2+) signals by EGR4 controls Th1 differentiation and anti-cancer immunity in vivo. EMBO Rep. 2020, 21, e48904. [Google Scholar] [CrossRef]
- Truta-Feles, K.; Lagadari, M.; Lehmann, K.; Berod, L.; Cubillos, S.; Piehler, S.; Herouy, Y.; Barz, D.; Kamradt, T.; Maghazachi, A.; et al. Histamine modulates gammadelta-T lymphocyte migration and cytotoxicity, via Gi and Gs protein-coupled signalling pathways. Br. J. Pharmacol. 2010, 161, 1291–1300. [Google Scholar] [CrossRef] [Green Version]
- Key, P.N.; Germino, J.; Yang, L.; Piersma, S.J.; Tripathy, S.K. Chronic Ly49H Receptor Engagement in vivo Decreases NK Cell Response to Stimulation Through ITAM-Dependent and Independent Pathways Both in vitro and in vivo. Front. Immunol. 2019, 10, 1692. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Hollander, L.; Guo, X.; Velazquez, H.; Chang, J.; Safirstein, R.; Kluger, H.; Cha, C.; Desir, G.V. Renalase Expression by Melanoma and Tumor-Associated Macrophages Promotes Tumor Growth through a STAT3-Mediated Mechanism. Cancer Res. 2016, 76, 3884–3894. [Google Scholar] [CrossRef] [Green Version]
- Hakonen, E.; Chandra, V.; Fogarty, C.L.; Yu, N.Y.; Ustinov, J.; Katayama, S.; Galli, E.; Danilova, T.; Lindholm, P.; Vartiainen, A.; et al. MANF protects human pancreatic beta cells against stress-induced cell death. Diabetologia 2018, 61, 2202–2214. [Google Scholar] [CrossRef] [Green Version]
- Peled, M.; Bar-Lev, T.H.; Talalai, E.; Aspitz, H.Z.; Daniel-Meshulam, I.; Bar, J.; Kamer, I.; Ofek, E.; Mor, A.; Onn, A. Mesencephalic astrocyte-derived neurotrophic factor is secreted from interferon-gamma-activated tumor cells through ER calcium depletion. PLoS ONE 2021, 16, e0250178. [Google Scholar] [CrossRef]
- Tremble, L.F.; Heffron, C.; Forde, P.F. The effect of calcium electroporation on viability, phenotype and function of melanoma conditioned macrophages. Sci. Rep. 2020, 10, 20645. [Google Scholar] [CrossRef]
- Falk, H.; Matthiessen, L.W.; Wooler, G.; Gehl, J. Calcium electroporation for treatment of cutaneous metastases; a randomized double-blinded phase II study, comparing the effect of calcium electroporation with electrochemotherapy. Acta Oncol. 2018, 57, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Zhang, Y.; Ma, G.; Tan, P.; Li, Z.; Zang, S.; Wu, X.; Jing, J.; Fang, S.; Zhou, L.; et al. Near-infrared photoactivatable control of Ca(2+) signaling and optogenetic immunomodulation. Elife 2015, 4, e10024. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Moreno, I.G.; Ibarra-Sanchez, A.; Castillo-Arellano, J.I.; Blank, U.; Gonzalez-Espinosa, C. Mast Cells Localize in Hypoxic Zones of Tumors and Secrete CCL-2 under Hypoxia through Activation of L-Type Calcium Channels. J. Immunol. 2020, 204, 1056–1068. [Google Scholar] [CrossRef] [PubMed]
- Shahan, T.A.; Fawzi, A.; Bellon, G.; Monboisse, J.C.; Kefalides, N.A. Regulation of tumor cell chemotaxis by type IV collagen is mediated by a Ca(2+)-dependent mechanism requiring CD47 and the integrin alpha(V)beta(3). J. Biol. Chem. 2000, 275, 4796–4802. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, L.; Dong, C. [Ca2+](i) as a potential downregulator of alpha(2)beta(1)-integrin-mediated A2058 tumor cell migration to type IV collagen. Am. J. Physiol.-Cell Physiol. 2001, 281, C106–C113. [Google Scholar] [CrossRef]
- Huang, H.C.; Shi, G.Y.; Jiang, S.J.; Shi, C.S.; Wu, C.M.; Yang, H.Y.; Wu, H.L. Thrombomodulin-mediated cell adhesion: Involvement of its lectin-like domain. J. Biol. Chem. 2003, 278, 46750–46759. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.; Jung, H.; Jho, E.H.; Multhaupt, H.A.B.; Couchman, J.R.; Oh, E.S. Keratinocytes negatively regulate the N-cadherin levels of melanoma cells via contact-mediated calcium regulation. Biochem. Biophys. Res. Commun. 2018, 503, 615–620. [Google Scholar] [CrossRef]
- Slater, M.; Scolyer, R.A.; Gidley-Baird, A.; Thompson, J.F.; Barden, J.A. Increased expression of apoptotic markers in melanoma. Melanoma. Res. 2003, 13, 137–145. [Google Scholar] [CrossRef]
- Li, G.; Satyamoorthy, K.; Meier, F.; Berking, C.; Bogenrieder, T.; Herlyn, M. Function and regulation of melanoma-stromal fibroblast interactions: When seeds meet soil. Oncogene 2003, 22, 3162–3171. [Google Scholar] [CrossRef] [Green Version]
- Ekstrom, E.J.; Bergenfelz, C.; von Bulow, V.; Serifler, F.; Carlemalm, E.; Jonsson, G.; Andersson, T.; Leandersson, K. WNT5A induces release of exosomes containing pro-angiogenic and immunosuppressive factors from malignant melanoma cells. Mol. Cancer 2014, 13, 88. [Google Scholar] [CrossRef] [Green Version]
- Favia, A.; Pafumi, I.; Desideri, M.; Padula, F.; Montesano, C.; Passeri, D.; Nicoletti, C.; Orlandi, A.; Del Bufalo, D.; Sergi, M.; et al. NAADP-Dependent Ca(2+) Signaling Controls Melanoma Progression, Metastatic Dissemination and Neoangiogenesis. Sci. Rep. 2016, 6, 18925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Zhou, P.; Meng, A.; Zhang, R.; Zhou, Y. Down-regulating Myoferlin inhibits the vasculogenic mimicry of melanoma via decreasing MMP-2 and inducing mesenchymal-to-epithelial transition. J. Cell. Mol. Med. 2018, 22, 1743–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vartanian, A.; Stepanova, E.; Grigorieva, I.; Solomko, E.; Belkin, V.; Baryshnikov, A.; Lichinitser, M. Melanoma vasculogenic mimicry capillary-like structure formation depends on integrin and calcium signaling. Microcirculation 2011, 18, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Oliver, V.K.; Patton, A.M.; Desai, S.; Lorang, D.; Libutti, S.K.; Kohn, E.C. Regulation of the pro-angiogenic microenvironment by carboxyamido-triazole. J. Cell. Physiol. 2003, 197, 139–148. [Google Scholar] [CrossRef]
- Frandsen, S.K.; Gissel, H.; Hojman, P.; Tramm, T.; Eriksen, J.; Gehl, J. Direct therapeutic applications of calcium electroporation to effectively induce tumor necrosis. Cancer Res. 2012, 72, 1336–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staresinic, B.; Jesenko, T.; Kamensek, U.; Krog Frandsen, S.; Sersa, G.; Gehl, J.; Cemazar, M. Effect of calcium electroporation on tumour vasculature. Sci. Rep. 2018, 8, 9412. [Google Scholar] [CrossRef] [Green Version]
- Boda-Heggemann, J.; Regnier-Vigouroux, A.; Franke, W.W. Beyond vessels: Occurrence and regional clustering of vascular endothelial (VE-)cadherin-containing junctions in non-endothelial cells. Cell Tissue Res. 2009, 335, 49–65. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.H.; Hodgson, L.; Henderson, A.J.; Dong, C. Involvement of phospholipase C signaling in melanoma cell-induced endothelial junction disassembly. Front Biosci. 2005, 10, 1597–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.H.; Dong, C. Systemic Analysis of Tumor Cell-Induced Endothelial Calcium Signaling and Junction Disassembly. Cell. Mol. Bioeng. 2009, 2, 375–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, Y.; Ozawa, S.; Tsukuda, M.; Kubota, E.; Miyazaki, K.; St-Pierre, Y.; Hata, R. Acidic extracellular pH increases calcium influx-triggered phospholipase D activity along with acidic sphingomyelinase activation to induce matrix metalloproteinase-9 expression in mouse metastatic melanoma. FEBS J. 2007, 274, 3171–3183. [Google Scholar] [CrossRef]
- Noguchi, F.; Inui, S.; Fedele, C.; Shackleton, M.; Itami, S. Calcium-Dependent Enhancement by Extracellular Acidity of the Cytotoxicity of Mitochondrial Inhibitors against Melanoma. Mol. Cancer Ther. 2017, 16, 936–947. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.; Gebhardt, L.; Arndt, S.; Karrer, S.; Zimmermann, J.L.; Fischer, M.J.M.; Bosserhoff, A.K. Acidification is an Essential Process of Cold Atmospheric Plasma and Promotes the Anti-Cancer Effect on Malignant Melanoma Cells. Cancers 2019, 11, 671. [Google Scholar] [CrossRef] [Green Version]
- Hung, W.C.; Yang, J.R.; Yankaskas, C.L.; Wong, B.S.; Wu, P.H.; Pardo-Pastor, C.; Serra, S.A.; Chiang, M.J.; Gu, Z.; Wirtz, D.; et al. Confinement Sensing and Signal Optimization via Piezo1/PKA and Myosin II Pathways. Cell Rep. 2016, 15, 1430–1441. [Google Scholar] [CrossRef] [Green Version]
- Buckner, C.A.; Buckner, A.L.; Koren, S.A.; Persinger, M.A.; Lafrenie, R.M. Inhibition of cancer cell growth by exposure to a specific time-varying electromagnetic field involves T-type calcium channels. PLoS ONE 2015, 10, e0124136. [Google Scholar] [CrossRef]
- Yu, S.; Li, C.; Ding, Y.; Huang, S.; Wang, W.; Wu, Y.; Wang, F.; Wang, A.; Han, Y.; Sun, Z.; et al. Exploring the ‘cold/hot’ properties of traditional Chinese medicine by cell temperature measurement. Pharm. Biol. 2020, 58, 208–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, J.H.; Lee, D.U. Foeniculum vulgare extract and its constituent, trans-anethole, inhibit UV-induced melanogenesis via ORAI1 channel inhibition. J. Dermatol. Sci. 2016, 84, 305–313. [Google Scholar] [CrossRef]
- Slominski, A.T.; Brozyna, A.A.; Zmijewski, M.A.; Jozwicki, W.; Jetten, A.M.; Mason, R.S.; Tuckey, R.C.; Elmets, C.A. Vitamin D signaling and melanoma: Role of vitamin D and its receptors in melanoma progression and management. Lab. Investig. 2017, 97, 706–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleszczynski, K.; Bilska, B.; Stegemann, A.; Flis, D.J.; Ziolkowski, W.; Pyza, E.; Luger, T.A.; Reiter, R.J.; Bohm, M.; Slominski, A.T. Melatonin and Its Metabolites Ameliorate UVR-Induced Mitochondrial Oxidative Stress in Human MNT-1 Melanoma Cells. Int. J. Mol. Sci. 2018, 19, 3786. [Google Scholar] [CrossRef] [Green Version]
- Chovancova, B.; Liskova, V.; Babula, P.; Krizanova, O. Role of Sodium/Calcium Exchangers in Tumors. Biomolecules 2020, 10, 1257. [Google Scholar] [CrossRef] [PubMed]
- Carrithers, M.D.; Chatterjee, G.; Carrithers, L.M.; Offoha, R.; Iheagwara, U.; Rahner, C.; Graham, M.; Waxman, S.G. Regulation of podosome formation in macrophages by a splice variant of the sodium channel SCN8A. J. Biol. Chem. 2009, 284, 8114–8126. [Google Scholar] [CrossRef] [Green Version]
- Sennoune, S.R.; Santos, J.M.; Hussain, F.; Martinez-Zaguilan, R. Sodium calcium exchanger operates in the reverse mode in metastatic human melanoma cells. Cell Mol. Biol. 2015, 61, 40–49. [Google Scholar] [PubMed]
- Esteves, G.N.N.; Ferraz, L.S.; Alvarez, M.M.P.; Costa, C.A.D.; Lopes, R.M.; Tersariol, I.; Rodrigues, T. BRAF and NRAS mutated melanoma: Different Ca(2+) responses, Na(+)/Ca(2+) exchanger expression, and sensitivity to inhibitors. Cell Calcium 2020, 90, 102241. [Google Scholar] [CrossRef]
- Gueguinou, M.; Chantome, A.; Fromont, G.; Bougnoux, P.; Vandier, C.; Potier-Cartereau, M. KCa and Ca(2+) channels: The complex thought. Biochim. Biophys. Acta 2014, 1843, 2322–2333. [Google Scholar] [CrossRef] [Green Version]
- Tajima, N.; Schonherr, K.; Niedling, S.; Kaatz, M.; Kanno, H.; Schonherr, R.; Heinemann, S.H. Ca2+-activated K+ channels in human melanoma cells are up-regulated by hypoxia involving hypoxia-inducible factor-1alpha and the von Hippel-Lindau protein. J. Physiol. 2006, 571, 349–359. [Google Scholar] [CrossRef]
- Tajima, N.; Itokazu, Y.; Korpi, E.R.; Somerharju, P.; Kakela, R. Activity of BK(Ca) channel is modulated by membrane cholesterol content and association with Na+/K+-ATPase in human melanoma IGR39 cells. J. Biol. Chem. 2011, 286, 5624–5638. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.Y.; Ridgway, L.D.; Dryer, S.E. Interactions with filamin A stimulate surface expression of large-conductance Ca2+-activated K+ channels in the absence of direct actin binding. Mol. Pharmacol. 2007, 72, 622–630. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.J.; Liu, L.Y.; Jiao, S.; Wei, S.M.; Mei, Y.A. Ca2+-inactivated K+ current is modulated by endothelin-1 in B-16 murine melanoma cells. Pigm. Cell Res. 2003, 16, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Bahar, E.; Kim, H.; Yoon, H. ER Stress-Mediated Signaling: Action Potential and Ca(2+) as Key Players. Int. J. Mol. Sci. 2016, 17, 1558. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jiang, Z.; Zhang, X.; Feng, J.; Ling, Y. Nacetyl-S-(p-chlorophenylcarbamoyl)cysteine induces mitochondrial-mediated apoptosis and suppresses migration in melanoma cells. Oncol. Rep. 2015, 34, 2547–2556. [Google Scholar] [CrossRef] [PubMed]
- Feldman, B.; Fedida-Metula, S.; Nita, J.; Sekler, I.; Fishman, D. Coupling of mitochondria to store-operated Ca(2+)-signaling sustains constitutive activation of protein kinase B/Akt and augments survival of malignant melanoma cells. Cell Calcium 2010, 47, 525–537. [Google Scholar] [CrossRef]
- Nakagawa, C.; Suzuki-Karasaki, M.; Suzuki-Karasaki, M.; Ochiai, T.; Suzuki-Karasaki, Y. The Mitochondrial Ca(2+) Overload via Voltage-Gated Ca(2+) Entry Contributes to an Anti-Melanoma Effect of Diallyl Trisulfide. Int. J. Mol. Sci. 2020, 21, 491. [Google Scholar] [CrossRef] [Green Version]
- Rouaud, F.; Boucher, J.L.; Slama-Schwok, A.; Rocchi, S. Mechanism of melanoma cells selective apoptosis induced by a photoactive NADPH analogue. Oncotarget 2016, 7, 82804–82819. [Google Scholar] [CrossRef] [Green Version]
- Marciel, M.P.; Hoffmann, P.R. Molecular Mechanisms by Which Selenoprotein K Regulates Immunity and Cancer. Biol. Trace Elem Res. 2019, 192, 60–68. [Google Scholar] [CrossRef]
- Marciel, M.P.; Khadka, V.S.; Deng, Y.; Kilicaslan, P.; Pham, A.; Bertino, P.; Lee, K.; Chen, S.; Glibetic, N.; Hoffmann, F.W.; et al. Selenoprotein K deficiency inhibits melanoma by reducing calcium flux required for tumor growth and metastasis. Oncotarget 2018, 9, 13407–13422. [Google Scholar] [CrossRef] [Green Version]
- Loubiere, C.; Clavel, S.; Gilleron, J.; Harisseh, R.; Fauconnier, J.; Ben-Sahra, I.; Kaminski, L.; Laurent, K.; Herkenne, S.; Lacas-Gervais, S.; et al. The energy disruptor metformin targets mitochondrial integrity via modification of calcium flux in cancer cells. Sci. Rep. 2017, 7, 5040. [Google Scholar] [CrossRef] [Green Version]
- Aranda-Souza, M.A.; Rossato, F.A.; Costa, R.A.; Figueira, T.R.; Castilho, R.F.; Guarniere, M.C.; Nunes, E.S.; Coelho, L.C.; Correia, M.T.; Vercesi, A.E. A lectin from Bothrops leucurus snake venom raises cytosolic calcium levels and promotes B16-F10 melanoma necrotic cell death via mitochondrial permeability transition. Toxicon 2014, 82, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Qin, L.J.; Qiu, H.J.; Shi, D.B.; Sun, R.; Li, W.B.; Liu, T.Z.; Wang, J.S.; Xu, T.T.; Guo, W.; et al. RPS3 regulates melanoma cell growth and apoptosis by targeting Cyto C/Ca2+/MICU1 dependent mitochondrial signaling. Oncotarget 2015, 6, 29614–29625. [Google Scholar] [CrossRef] [Green Version]
- Raimondi, M.; Fontana, F.; Marzagalli, M.; Audano, M.; Beretta, G.; Procacci, P.; Sartori, P.; Mitro, N.; Limonta, P. Ca(2+) overload- and ROS-associated mitochondrial dysfunction contributes to delta-tocotrienol-mediated paraptosis in melanoma cells. Apoptosis 2021, 26, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Kang, K.A.; Ryu, Y.S.; Piao, M.J.; Han, X.; Oh, M.C.; Boo, S.J.; Jeong, S.U.; Jeong, Y.J.; Chae, S.; et al. Induction of Endoplasmic Reticulum Stress via Reactive Oxygen Species Mediated by Luteolin in Melanoma Cells. Anticancer Res. 2016, 36, 2281–2289. [Google Scholar] [PubMed]
- Burgeiro, A.; Bento, A.C.; Gajate, C.; Oliveira, P.J.; Mollinedo, F. Rapid human melanoma cell death induced by sanguinarine through oxidative stress. Eur. J. Pharmacol. 2013, 705, 109–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forno, F.; Maatuf, Y.; Boukeileh, S.; Dipta, P.; Mahameed, M.; Darawshi, O.; Ferreira, V.; Rada, P.; Garcia-Martinez, I.; Gross, E.; et al. Aripiprazole Cytotoxicity Coincides with Activation of the Unfolded Protein Response in Human Hepatic Cells. J. Pharmacol. Exp. Ther. 2020, 374, 452–461. [Google Scholar] [CrossRef]
- Prieto, K.; Cao, Y.; Mohamed, E.; Trillo-Tinoco, J.; Sierra, R.A.; Uruena, C.; Sandoval, T.A.; Fiorentino, S.; Rodriguez, P.C.; Barreto, A. Polyphenol-rich extract induces apoptosis with immunogenic markers in melanoma cells through the ER stress-associated kinase PERK. Cell Death Discov. 2019, 5, 134. [Google Scholar] [CrossRef] [Green Version]
- Eskiocak, U.; Ramesh, V.; Gill, J.G.; Zhao, Z.; Yuan, S.W.; Wang, M.; Vandergriff, T.; Shackleton, M.; Quintana, E.; Johnson, T.M.; et al. Synergistic effects of ion transporter and MAP kinase pathway inhibitors in melanoma. Nat. Commun. 2016, 7, 12336. [Google Scholar] [CrossRef]
- El-Khattouti, A.; Selimovic, D.; Hannig, M.; Taylor, E.B.; Abd Elmageed, Z.Y.; Hassan, S.Y.; Haikel, Y.; Kandil, E.; Leverkus, M.; Brodell, R.T.; et al. Imiquimod-induced apoptosis of melanoma cells is mediated by ER stress-dependent Noxa induction and enhanced by NF-kappaB inhibition. J. Cell. Mol. Med. 2016, 20, 266–286. [Google Scholar] [CrossRef]
- Nyberg, W.A.; Espinosa, A. Imiquimod induces ER stress and Ca(2+) influx independently of TLR7 and TLR8. Biochem. Biophys. Res. Commun. 2016, 473, 789–794. [Google Scholar] [CrossRef] [Green Version]
- Onoe-Takahashi, A.; Suzuki-Karasaki, M.; Suzuki-Karasaki, M.; Ochiai, T.; Suzuki-Karasaki, Y. Autophagy inhibitors regulate TRAIL sensitivity in human malignant cells by targeting the mitochondrial network and calcium dynamics. Int J Oncol 2019, 54, 1734–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenblatt, R.B.; Frank, J.A.; Burks, S.R. Cytosolic Ca(2+) transients during pulsed focused ultrasound generate reactive oxygen species and cause DNA damage in tumor cells. Theranostics 2021, 11, 602–613. [Google Scholar] [CrossRef] [PubMed]
- Nardin, C.; Peres, C.; Mazzarda, F.; Ziraldo, G.; Salvatore, A.M.; Mammano, F. Photosensitizer Activation Drives Apoptosis by Interorganellar Ca(2+) Transfer and Superoxide Production in Bystander Cancer Cells. Cells 2019, 8, 1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granados, K.; Huser, L.; Federico, A.; Sachindra, S.; Wolff, G.; Hielscher, T.; Novak, D.; Madrigal-Gamboa, V.; Sun, Q.; Vierthaler, M.; et al. T-type calcium channel inhibition restores sensitivity to MAPK inhibitors in de-differentiated and adaptive melanoma cells. Br. J. Cancer 2020, 122, 1023–1036. [Google Scholar] [CrossRef] [Green Version]
- Barcelo, C.; Siso, P.; Maiques, O.; Garcia-Mulero, S.; Sanz-Pamplona, R.; Navaridas, R.; Megino, C.; Felip, I.; Urdanibia, I.; Eritja, N.; et al. T-Type Calcium Channels as Potential Therapeutic Targets in Vemurafenib-Resistant BRAF(V600E) Melanoma. J. Investig. Dermatol. 2020, 140, 1253–1265. [Google Scholar] [CrossRef]
- Alza, L.; Visa, A.; Herreros, J.; Canti, C. The rise of T-type channels in melanoma progression and chemotherapeutic resistance. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188364. [Google Scholar] [CrossRef]
- Takata, N.; Ohshima, Y.; Suzuki-Karasaki, M.; Yoshida, Y.; Tokuhashi, Y.; Suzuki-Karasaki, Y. Mitochondrial Ca2+ removal amplifies TRAIL cytotoxicity toward apoptosis-resistant tumor cells via promotion of multiple cell death modalities. Int. J. Oncol. 2017, 51, 193–203. [Google Scholar] [CrossRef]
- Ohshima, Y.; Takata, N.; Suzuki-Karasaki, M.; Yoshida, Y.; Tokuhashi, Y.; Suzuki-Karasaki, Y. Disrupting mitochondrial Ca2+ homeostasis causes tumor-selective TRAIL sensitization through mitochondrial network abnormalities. Int. J. Oncol. 2017, 51, 1146–1158. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, T.; Ando, T.; Suzuki-Karasaki, M.; Ito, T.; Onoe-Takahashi, A.; Ochiai, T.; Soma, M.; Suzuki-Karasaki, Y. Plasma-stimulated medium kills TRAIL-resistant human malignant cells by promoting caspase-independent cell death via membrane potential and calcium dynamics modulation. Int. J. Oncol. 2018, 52, 697–708. [Google Scholar] [CrossRef] [Green Version]
- Ivankovic, S.; Stojkovic, R.; Maksimovic, M.; Galic, B.; Milos, M. Impact of calcium ion on cytotoxic effect of the boroxine derivative, K2[B3O3F4OH]. J. Enzym. Inhib. Med. Chem. 2016, 31, 70–74. [Google Scholar] [CrossRef] [Green Version]
- Sedaghat, F.; Notopoulos, A. S100 protein family and its application in clinical practice. Hippokratia 2008, 12, 198–204. [Google Scholar]
- Deckers, E.A.; Kruijff, S.; Brouwers, A.H.; van der Steen, K.; Hoekstra, H.J.; Thompson, J.F.; Vallez Garcia, D.; Wevers, K.P. The association between active tumor volume, total lesion glycolysis and levels of S-100B and LDH in stage IV melanoma patients. Eur. J. Surg. Oncol. 2020, 46, 2147–2153. [Google Scholar] [CrossRef]
- Wagner, N.B.; Forschner, A.; Leiter, U.; Garbe, C.; Eigentler, T.K. S100B and LDH as early prognostic markers for response and overall survival in melanoma patients treated with anti-PD-1 or combined anti-PD-1 plus anti-CTLA-4 antibodies. Br. J. Cancer 2018, 119, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Gassenmaier, M.; Lenders, M.M.; Forschner, A.; Leiter, U.; Weide, B.; Garbe, C.; Eigentler, T.K.; Wagner, N.B. Serum S100B and LDH at Baseline and During Therapy Predict the Outcome of Metastatic Melanoma Patients Treated with BRAF Inhibitors. Target Oncol. 2021, 16, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Felix, J.; Cassinat, B.; Porcher, R.; Schlageter, M.H.; Maubec, E.; Pages, C.; Baroudjian, B.; Homyrda, L.; Boukouaci, W.; Tamouza, R.; et al. Relevance of serum biomarkers associated with melanoma during follow-up of anti-CTLA-4 immunotherapy. Int. Immunopharmacol. 2016, 40, 466–473. [Google Scholar] [CrossRef]
- Nordlinger, A.; Dror, S.; Elkahloun, A.; Del Rio, J.; Stubbs, E.; Golan, T.; Malcov, H.; Pricket, T.D.; Cronin, J.C.; Parikh, S.; et al. Mutated MITF-E87R in Melanoma Enhances Tumor Progression via S100A4. J. Investig. Dermatol. 2018, 138, 2216–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massi, D.; Landriscina, M.; Piscazzi, A.; Cosci, E.; Kirov, A.; Paglierani, M.; Di Serio, C.; Mourmouras, V.; Fumagalli, S.; Biagioli, M.; et al. S100A13 is a new angiogenic marker in human melanoma. Mod. Pathol. 2010, 23, 804–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, M.; Savage, P.; Lovato, J.; Schwartz, G.G. Serum calcium, albumin and tumor stage in cutaneous malignant melanoma. Future Oncol. 2016, 12, 2205–2214. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Chen, Z.; Zhang, A.; Gupte, A.A.; Hamilton, D.J. The Role of Calcium Signaling in Melanoma. Int. J. Mol. Sci. 2022, 23, 1010. https://doi.org/10.3390/ijms23031010
Zhang H, Chen Z, Zhang A, Gupte AA, Hamilton DJ. The Role of Calcium Signaling in Melanoma. International Journal of Molecular Sciences. 2022; 23(3):1010. https://doi.org/10.3390/ijms23031010
Chicago/Turabian StyleZhang, Haoran, Zhe Chen, Aijun Zhang, Anisha A. Gupte, and Dale J. Hamilton. 2022. "The Role of Calcium Signaling in Melanoma" International Journal of Molecular Sciences 23, no. 3: 1010. https://doi.org/10.3390/ijms23031010
APA StyleZhang, H., Chen, Z., Zhang, A., Gupte, A. A., & Hamilton, D. J. (2022). The Role of Calcium Signaling in Melanoma. International Journal of Molecular Sciences, 23(3), 1010. https://doi.org/10.3390/ijms23031010