
Functional Roles of JNK and p38 MAPK Signaling in Nasopharyngeal Carcinoma

 , , , ,
, , , ,
Abstract
:1. Introduction
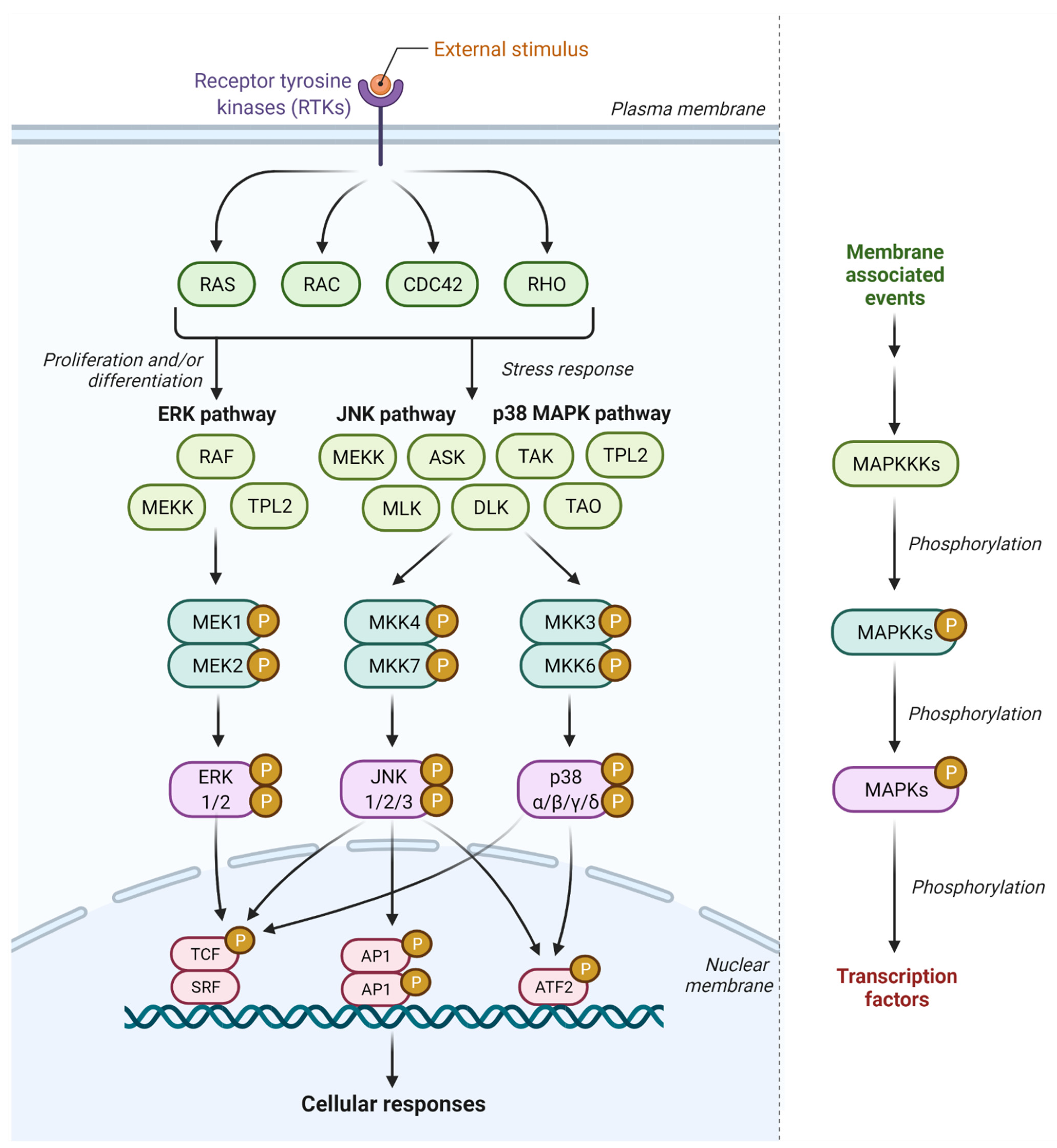
2. JNK Signaling Pathway
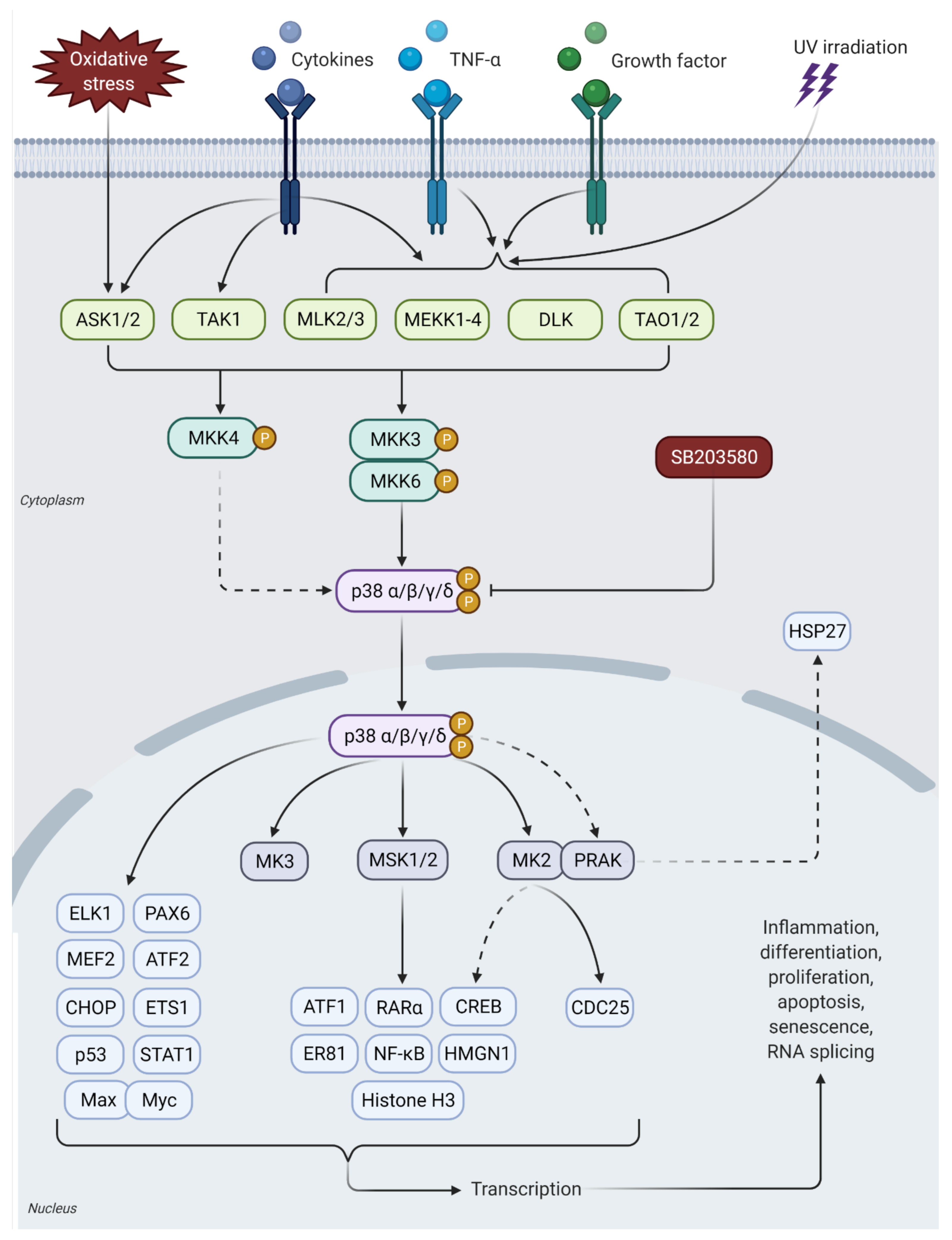
3. p38 MAPK Signaling Pathway
4. JNK and p38 MAPK Signaling in Human Cancers
5. Pro-Tumorigenic Functions of JNK and p38 MAPK Signaling in NPC
5.1. Activation of JNK Signaling Promotes NPC Cell Survival
5.2. JNK and p38 MAPK Activities Inhibit Pro-Apoptotic Signaling in NPC Cells
5.3. JNK and p38 MAPK Signaling Mediates LMP1 in EBV-Associated NPC
5.4. Activation of p38 MAPK Signaling Promotes Inflammatory Tumor Microenvironment
5.5. Activation of JNK and p38 MAPK Signaling Promotes NPC Cell Invasion and Metastasis
5.6. Activation of p38 MAPK Signaling Promotes Angiogenesis
6. Tumor Suppressive Functions of JNK and p38 MAPK Signaling in NPC
7. Targeting JNK and p38 MAPK in NPC
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bruce, J.P.; Yip, K.; Bratman, S.V.; Ito, E.; Liu, F. Nasopharyngeal cancer: Molecular landscape. J. Clin. Oncol. 2015, 33, 3346–3355. [Google Scholar] [CrossRef]
- Chen, Y.; Chan, A.T.; Le, Q.; Blanchard, P.; Sun, Y.; Ma, J. Nasopharyngeal carcinoma. Lancet 2019, 394, 64–80. [Google Scholar] [CrossRef]
- Zhao, L.; Fong, A.H.; Liu, N.; Cho, W.C. Molecular subtyping of nasopharyngeal carcinoma (NPC) and a microRNA-based prognostic model for distant metastasis. J. Biomed. Sci. 2018, 25, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Cancer Society. Key Statistics for Nasopharyngeal Cancer. 2017. Available online: https://www.cancer.org/cancer/nasopharyngeal-cancer/about/key-statistics.html (accessed on 7 December 2021).
- Young, L.S.; Dawson, C.W. Epstein-Barr virus and nasopharyngeal carcinoma. Chin. J. Cancer 2014, 33, 581–590. [Google Scholar] [CrossRef]
- Huang, T.; Ploner, A.; Chang, E.T.; Liu, Q.; Cai, Y.; Zhang, Z.; Chen, G.; Huang, Q.; Xie, S.; Cao, S. Dietary patterns and risk of nasopharyngeal carcinoma: A population-based case-control study in southern China. Am. J. Clin. Nutr. 2021, 114, 462–471. [Google Scholar] [CrossRef]
- Lin, Z.; Khong, B.; Kwok, S.; Cao, H.; West, R.B.; Le, Q.; Kong, C.S. Human papillomavirus 16 detected in nasopharyngeal carcinomas in white Americans but not in endemic Southern Chinese patients. Head Neck 2014, 36, 709–714. [Google Scholar] [CrossRef]
- Tsao, S.W.; Yip, Y.L.; Tsang, C.M.; Pang, P.S.; Lau, V.M.Y.; Zhang, G.; Lo, K.W. Etiological factors of nasopharyngeal carcinoma. Oral Oncol. 2014, 50, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lu, T.; Huang, Y.; Han, F.; Chen, C.; Xiao, W. Clinical features of 337 patients with recurrent nasopharyngeal carcinoma. Chin. J. Cancer 2010, 29, 82–86. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.W.; Ng, W.T.; Chan, J.Y.; Corry, J.; Mäkitie, A.; Mendenhall, W.M.; Rinaldo, A.; Rodrigo, J.P.; Saba, N.F.; Strojan, P. Management of locally recurrent nasopharyngeal carcinoma. Cancer Treat. Rev. 2019, 79, 101890. [Google Scholar] [CrossRef]
- Abdullah, B.; Alias, A.; Hassan, S. Challenges in the management of nasopharyngeal carcinoma: A review. Malays J. Med. Sci. 2009, 16, 50–54. [Google Scholar] [PubMed]
- Plotnikov, A.; Flores, K.; Maik-Rachline, G.; Zehorai, E.; Kapri-Pardes, E.; Berti, D.A.; Hanoch, T.; Besser, M.J.; Seger, R. The nuclear translocation of ERK1/2 as an anticancer target. Nat. Commun. 2015, 6, 6685. [Google Scholar] [CrossRef] [PubMed]
- Koul, H.K.; Pal, M.; Koul, S. Role of p38 MAP kinase signal transduction in solid tumors. Genes Cancer 2013, 4, 342–359. [Google Scholar] [CrossRef] [PubMed]
- Burotto, M.; Chiou, V.L.; Lee, J.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Paton, E.L.; Turner, J.A.; Schlaepfer, I.R. Overcoming resistance to therapies targeting the MAPK pathway in BRAF-mutated tumours. J. Oncol. 2020, 2020, 1079827. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.; Zhang, X.; Liu, J.; Estrem, S.; Li, S.; Gong, X.Q.; Buchanan, S.; Henry, J.R.; Starling, J.J.; Peng, S.B. Reactivation of mitogen-activated protein kinase (MAPK) pathway by FGF receptor 3 (FGFR3)/Ras mediates resistance to vemurafenib in human BRAF V600E mutant melanoma. J. Biol. Chem. 2012, 287, 28087–28098. [Google Scholar] [CrossRef] [Green Version]
- Vo, U.; Vajpai, N.; Flavell, L.; Bobby, R.; Breeze, A.L.; Embrey, K.J.; Golovanov, A.P. Monitoring Ras interactions with the nucleotide exchange factor Son of Sevenless (SOS) using site-specific NMR reporter signals and intrinsic fluorescence. J. Biol. Chem. 2016, 291, 1703–1718. [Google Scholar] [CrossRef] [Green Version]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Richardson, D.R. Cellular iron depletion stimulates the JNK and p38 MAPK signaling transduction pathways, dissociation of ASK1-thioredoxin, and activation of ASK. J. Biol. Chem. 2011, 286, 15413–15427. [Google Scholar] [CrossRef] [Green Version]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O. A comprehensive review on MAPK: A promising therapeutic target in cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef] [Green Version]
- Tulalamba, W.; Janvilisri, T. Nasopharyngeal carcinoma signaling pathway: An update on molecular biomarkers. Int. J. Cell Biol. 2012, 2012, 594681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabapathy, K. Role of the JNK pathway in human diseases. Prog. Mol. Biol. Transl. Sci. 2012, 106, 145–169. [Google Scholar] [PubMed]
- Yarza, R.; Vela, S.; Solas, M.; Ramirez, M.J. c-Jun N-terminal kinase (JNK) signaling as a therapeutic target for Alzheimer’s disease. Front. Pharmacol. 2016, 6, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogoyevitch, M.A.; Kobe, B. Uses for JNK: The many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 2006, 70, 1061–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gkouveris, I.; Nikitakis, N.G. Role of JNK signaling in oral cancer: A mini review. Tumor. Biol. 2017, 39, 1010428317711659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bubici, C.; Papa, S. JNK signalling in cancer: In need of new, smarter therapeutic targets. Br. J. Pharm. 2014, 171, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Haeusgen, W.; Boehm, R.; Zhao, Y.; Herdegen, T.; Waetzig, V. Specific activities of individual c-Jun N-terminal kinases in the brain. Neuroscience 2009, 161, 951–959. [Google Scholar] [CrossRef]
- New, L.; Han, J. The p38 MAP kinase pathway and its biological function. Trends Cardiovasc. Med. 1998, 8, 220–228. [Google Scholar] [CrossRef]
- Roy, S.; Roy, S.; Rana, A.; Akhter, Y.; Hande, M.P.; Banerjee, B. The role of p38 MAPK pathway in p53 compromised state and telomere mediated DNA damage response. Mutat. Res. Genet. Toxicol. Environ. Mutagenesis 2018, 836, 89–97. [Google Scholar] [CrossRef]
- Yang, Y.; Kim, S.C.; Yu, T.; Yi, Y.S.; Rhee, M.H.; Sung, G.H.; Yoo, B.C.; Cho, J.Y. Functional roles of p38 mitogen-activated protein kinase in macrophage-mediated inflammatory responses. Mediat. Inflamm. 2014, 2014, 352371. [Google Scholar] [CrossRef] [Green Version]
- Whitmarsh, A.; Davis, R. Role of mitogen-activated protein kinase kinase 4 in cancer. Oncogene 2007, 26, 3172–3184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiryaev, A.; Dumitriu, G.; Moens, U. Distinct roles of MK2 and MK5 in cAMP/PKA-and stress/p38 MAPK-induced heat shock protein 27 phosphorylation. J. Mol. Signal. 2011, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katopodis, P.; Kerslake, R.; Zikopoulos, A.; Beri, N.E.; Anikin, V. p38β-MAPK11 and its role in female cancers. J. Ovarian Res. 2020, 14, 84. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Liu, R.; Dai, Y.; Hong, S.; Dong, H.; Wang, H. The role of p38γ in cancer: From review to outlook. Int. J. Biol. Sci. 2021, 17, 4036. [Google Scholar] [CrossRef]
- O’Callaghan, C.; Fanning, L.J.; Barry, O.P. p38δ MAPK: Emerging roles of a neglected isoform. Int. J. Cell Biol. 2014, 2014, 272689. [Google Scholar] [CrossRef] [Green Version]
- Jing, L.; Anning, L. Role of JNK activation in apoptosis: A double-edged sword. Cell Res. 2005, 15, 36–42. [Google Scholar]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Tai, G. Role of c-Jun N-terminal kinase in hepatocellular carcinoma development. Target. Oncol. 2016, 11, 723–738. [Google Scholar] [CrossRef]
- Feng, C.; He, K.; Zhang, C.; Su, S.; Li, B.; Li, Y.; Duan, C.Y.; Chen, S.; Chen, R.; Liu, Y.; et al. JNK contributes to the tumorigenic potential of human cholangiocarcinoma cells through the mTOR pathway regulated GRP78 induction. PLoS ONE 2014, 9, e90388. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Zent, R.; Ghiassi, M.; McDonnell, M.; Moses, H.L. Integrin beta 1 signaling is necessary for transforming growth factor-beta activation of p38 MAPK and epithelial plasticity. J. Biol. Chem. 2001, 276, 46707–46713. [Google Scholar] [CrossRef]
- Cheng, T.; Symons, M.; Jou, T. Regulation of anoikis by Cdc42 and Rac. Exp. Cell Res. 2004, 295, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Limón, A.; Joaquin, M.; Caballero, M.; Posas, F.; De Nadal, E. The p38 pathway: From biology to cancer therapy. Int. J. Mol. Sci. 2020, 21, 1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Q.; Chen, J.; Beezhold, K.J.; Castranova, V.; Shi, X.; Chen, F. JNK1 activation predicts the prognostic outcome of the human hepatocellular carcinoma. Mol. Cancer 2009, 8, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.; Hu, J. The role of JNK in prostate cancer progression and therapeutic strategies. Biomed. Pharmacother. 2020, 121, 109679. [Google Scholar] [CrossRef]
- Royuela, M.; Arenas, M.I.; Bethencourt, F.R.; Sánchez-Chapado, M.; Fraile, B.; Paniagua, R. Regulation of proliferation/apoptosis equilibrium by mitogen-activated protein kinases in normal, hyperplastic, and carcinomatous human prostate. Hum. Pathol. 2002, 33, 299–306. [Google Scholar] [CrossRef]
- Yeh, Y.; Hou, M.; Chung, Y.; Chen, Y.; Yang, S.; Chen, D.; Su, J.; Yuan, S.F. Decreased expression of phosphorylated JNK in breast infiltrating ductal carcinoma is associated with a better overall survival. Int. J. Cancer 2006, 118, 2678–2684. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.W.; Liu, H.; Zhao, Y.; Qian, C.; Wang, L.; Qi, J. JNK2 downregulation promotes tumorigenesis and chemoresistance by decreasing p53 stability in bladder cancer. Oncotarget 2016, 7, 35119–35131. [Google Scholar] [CrossRef]
- Liu, J.; Wang, T.; Creighton, C.J.; Wu, S.; Ray, M.; Janardhan, K.S.; Willson, C.J.; Cho, S.; Castro, P.D.; Ittmann, M.M. JNK 1/2 represses Lkb 1-deficiency-induced lung squamous cell carcinoma progression. Nat. Commun. 2019, 10, 2148. [Google Scholar] [CrossRef]
- Wang, X.; Chao, L.; Zhen, J.; Chen, L.; Ma, G.; Li, X. Phosphorylated c-Jun NH2-terminal kinase is overexpressed in human papillary thyroid carcinomas and associates with lymph node metastasis. Cancer Lett. 2010, 293, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.M.; Tong, J.L.; Xu, Q.; Nie, F.; Xu, X.T.; Xiao, S.D.; Ran, Z.H. Increased JNK1 signaling pathway is responsible for ABCG2-mediated multidrug resistance in human colon cancer. PLoS ONE 2012, 7, 41763. [Google Scholar] [CrossRef] [PubMed]
- Gkouveris, I.; Nikitakis, N.; Karanikou, M.; Rassidakis, G.; Sklavounou, A. JNK1/2 expression and modulation of STAT3 signaling in oral cancer. Oncol. Lett. 2016, 12, 699–706. [Google Scholar] [CrossRef] [Green Version]
- Hammouda, M.B.; Ford, A.E.; Liu, Y.; Zhang, J.Y. The JNK signaling pathway in inflammatory skin disorders and cancer. Cells 2020, 9, 857. [Google Scholar] [CrossRef] [Green Version]
- Katagiri, C.; Nakanishi, J.; Kadoya, K.; Hibino, T. Serpin squamous cell carcinoma antigen inhibits UV-induced apoptosis via suppression of c-JUN NH2-terminal kinase. J. Cell Biol. 2006, 172, 983–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Aguayo, C.; Vivas-Mejia, P.; Benito, J.M.; Fernandez, A.; Francois-Xavier, C.; Chavez-Reyes, A.; Sood, A.K.; Lopez-Berestein, G. JNK-1 inhibition leads to antitumor activity in ovarian cancer. Clin. Cancer Res. 2010, 70, 5468. [Google Scholar]
- Seino, M.; Okada, M.; Sakaki, H.; Takeda, H.; Watarai, H.; Suzuki, S.; Seino, S.; Kuramoto, K.; Ohta, T.; Nagase, S. Time-staggered inhibition of JNK effectively sensitizes chemoresistant ovarian cancer cells to cisplatin and paclitaxel. Oncol. Rep. 2016, 35, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, M.; Zhou, H.; Yang, J. Activation of c-Jun/JNK signaling predicts poor prognosis in nasopharyngeal carcinoma. Int. J. Clin. Exp. Pathol. 2018, 11, 2699. [Google Scholar]
- Tomás-Loba, A.; Manieri, E.; González-Terán, B.; Mora, A.; Leiva-Vega, L.; Santamans, A.M.; Romero-Becerra, R.; Rodríguez, E.; Pintor-Chocano, A.; Feixas, F. p38γ is essential for cell cycle progression and liver tumorigenesis. Nature 2019, 568, 557–560. [Google Scholar] [CrossRef] [Green Version]
- Tan, F.L.; Ooi, A.; Huang, D.; Wong, J.C.; Qian, C.; Chao, C.; Ooi, L.; Tan, Y.; Chung, A.; Cheow, P. p38 delta/MAPK13 as a diagnostic marker for cholangiocarcinoma and its involvement in cell motility and invasion. Int. J. Cancer 2010, 126, 2353–2361. [Google Scholar]
- Browne, A.; Göbel, A.; Thiele, S.; Hofbauer, L.; Rauner, M.; Rachner, T. p38 MAPK regulates the Wnt inhibitor Dickkopf-1 in osteotropic prostate cancer cells. Cell Death Dis. 2016, 7, 2119. [Google Scholar] [CrossRef] [Green Version]
- Suarez-Cuervo, C.; Merrell, M.A.; Watson, L.; Harris, K.W.; Rosenthal, E.L.; Väänänen, H.K.; Selander, K.S. Breast cancer cells with inhibition of p38α have decreased MMP-9 activity and exhibit decreased bone metastasis in mice. Clin. Exp. Metastasis 2004, 21, 525. [Google Scholar] [CrossRef] [PubMed]
- Wada, M.; Canals, D.; Adada, M.; Coant, N.; Salama, M.F.; Helke, K.L.; Arthur, J.; Shroyer, K.R.; Kitatani, K.; Obeid, L.M. p38 delta MAPK promotes breast cancer progression and lung metastasis by enhancing cell proliferation and cell detachment. Oncogene 2017, 36, 6649–6657. [Google Scholar] [CrossRef] [Green Version]
- Wagner, E.F.; Nebreda, Á.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Harb, O.A.; Haggag, R.; Ali, M.M.; El Shorbagy, S.; Abdelbary, A.M.; Abdelaziz, L.A.; Salim, R.A.; Abdel Wahab, K.M. The prognostic role of NEDD9 and p38 protein expression levels in urinary bladder transitional cell carcinoma. J. Oncol. 2017, 2017, 6095205. [Google Scholar] [CrossRef]
- Kumar, B.; Koul, S.; Petersen, J.; Khandrika, L.; Hwa, J.S.; Meacham, R.B.; Wilson, S.; Koul, H.K. p38 mitogen-activated protein kinase-driven MAPKAPK2 regulates invasion of bladder cancer by modulation of MMP-2 and MMP-9 activity. Cancer Res. 2010, 70, 832–841. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, A.K.; Basu, S.; Hu, J.; Yie, T.; Tchou-Wong, K.M.; Rom, W.N.; Lee, T.C. Selective p38 activation in human non-small cell lung cancer. Am. J. Respir. Cell Mol. Biol. 2002, 26, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Vitos-Faleato, J.; Real, S.M.; Gutierrez-Prat, N.; Villanueva, A.; Llonch, E.; Drosten, M.; Barbacid, M.; Nebreda, A.R. Requirement for epithelial p38 alpha in KRAS-driven lung tumor progression. Proc. Natl. Acad. Sci. USA 2020, 117, 2588–2596. [Google Scholar] [CrossRef]
- Pomerance, M.; Quillard, J.; Chantoux, F.; Young, J.; Blondeau, J. High-level expression, activation, and subcellular localization of p38-MAP kinase in thyroid neoplasms. J. Pathol. 2006, 209, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Stramucci, L.; Pranteda, A.; Stravato, A.; Amoreo, C.A.; Pennetti, A.; Diodoro, M.G.; Bartolazzi, A.; Milella, M.; Bossi, G. MKK3 sustains cell proliferation and survival through p38 delta MAPK activation in colorectal cancer. Cell Death Dis. 2019, 10, 842. [Google Scholar] [CrossRef]
- Paillas, S.; Boissiere, F.; Bibeau, F.; Denouel, A.; Mollevi, C.; Causse, A.; Denis, V.; Vezzio-Vie, N.; Marzi, L.; Cortijo, C.; et al. Targeting the p38 MAPK pathway inhibits irinotecan resistance in colon adenocarcinoma. Cancer Res. 2011, 71, 1041–1049. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Wan, X.; Fu, X.; Wu, P.; Chen, D.; Wang, P.; Jiang, L.; Wang, D.; Chen, Z.; Huang, Y. Phosphorylated p38, a negative prognostic biomarker, complements TNM staging prognostication in colorectal cancer. Tumor. Biol. 2014, 35, 10487–10495. [Google Scholar] [CrossRef]
- Junttila, M.; Ala-Aho, R.; Jokilehto, T.; Peltonen, J.; Kallajoki, M.; Grenman, R.; Jaakkola, P.; Westermarck, J.; Kähäri, V. p38α and p38δ mitogen-activated protein kinase isoforms regulate invasion and growth of head and neck squamous carcinoma cells. Oncogene 2007, 26, 5267–5279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leelahavanichkul, K.; Amornphimoltham, P.; Molinolo, A.A.; Basile, J.R.; Koontongkaew, S.; Gutkind, J.S. A role for p38 MAPK in head and neck cancer cell growth and tumor-induced angiogenesis and lymphangiogenesis. Mol. Oncol. 2014, 8, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Haider, A.S.; Peters, S.B.; Kaporis, H.; Cardinale, I.; Fei, J.; Ott, J.; Blumenberg, M.; Bowcock, A.M.; Krueger, J.G.; Carucci, J.A. Genomic analysis defines a cancer-specific gene expression signature for human squamous cell carcinoma and distinguishes malignant hyperproliferation from benign hyperplasia. J. Investig. Derm. 2006, 126, 869–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asnir, R.; Yudhistira, A.; Daulay, E.; Muzakkir, M.; Yulius, S. p38 mitogen-activated protein kinase (p38 MAPK) overexpression in clinical staging of nasopharyngeal carcinoma. In IOP Conference Series: Earth and Environmental Sciencep38 Mitogen-Activated Protein Kinase (p38 MAPK) Overexpression in Clinical Staging of Nasopharyngeal Carcinoma; IOP Publishing: Medan, Indonesia, 2018. [Google Scholar]
- Farhat, F.; Daulay, E.R.; Chrestella, J.; Asnir, R.A.; Yudhistira, A.; Susilo, R.R. Correlation of p38 mitogen-activated protein kinase expression to clinical stage in nasopharyngeal carcinoma. Open Access Maced. J. Med. Sci. 2018, 6, 1982–1985. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Chen, K.; Lin, G.; Wan, F.; Chen, L.; Zhu, X. Silencing c-Jun inhibits autophagy and abrogates radioresistance in nasopharyngeal carcinoma by activating the PI3K/AKT/mTOR pathway. Ann. Transl. Med. 2021, 9, 1085. [Google Scholar] [CrossRef]
- Huang, C.; Wang, J.; Kikkawa, U.; Mukai, H.; Shen, M.; Morita, I.; Chen, B.; Chang, W. Calcineurin-mediated dephosphorylation of c-Jun Ser-243 is required for c-Jun protein stability and cell transformation. Oncogene 2008, 27, 2422–2429. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Q.; Shi, M.; Chen, J.; Liao, W. The Ras signaling pathway mediates cetuximab resistance in nasopharyngeal carcinoma. Biomed. Pharmacother. 2011, 65, 168–174. [Google Scholar] [CrossRef]
- Lu, J.; Luo, H.; Liu, X.; Peng, Y.; Zhang, B.; Wang, L.; Xu, X.; Peng, X.; Li, G.; Tian, W. miR-9 targets CXCR4 and functions as a potential tumor suppressor in nasopharyngeal carcinoma. Carcinogenesis 2014, 35, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Vatte, C.; Al Amri, A.M.; Cyrus, C.; Chathoth, S.; Ahmad, A.; Alsayyah, A.; Ali, A.A. Epstein-Barr virus infection mediated TP53 and BCL2 expression in nasopharyngeal carcinoma pathogenesis. Mol. Clin. Oncol. 2021, 15, 1–7. [Google Scholar]
- Fendri, A.; Kontos, C.K.; Khabir, A.; Mokdad-Gargouri, R.; Scorilas, A. BCL2L12 is a novel biomarker for the prediction of short-term relapse in nasopharyngeal carcinoma. Mol. Med. 2011, 17, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Yang, S.; Lai, J.; Yeh, K.; Yang, J.; Chen, L.; Chen, H. Expression of BCL2 correlates with poor prognosis and modulates migration of nasopharyngeal carcinoma cells. Clin. Chim. Acta 2010, 411, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Liu, Z.; Yang, H.; Yu, X.; Wu, Q.; Hua, S.; Long, X.; Jiang, Q.; Song, Y.; Cheng, C. Tumor suppressor PDCD4 modulates miR-184-mediated direct suppression of c-Myc and BCL2 blocking cell growth and survival in nasopharyngeal carcinoma. Cell Death Dis. 2013, 4, 872. [Google Scholar] [CrossRef]
- Song, Q.; Wang, G.; Chu, Y.; Zhou, L.; Jiang, M.; He, Q.; Liu, M.; Qin, J.; Hu, J. TNF-alpha up-regulates cellular inhibitor of apoptosis protein 2 (c-IAP2) via c-Jun N-terminal kinase (JNK) pathway in nasopharyngeal carcinoma. Int. Immunopharmacol. 2013, 16, 148–153. [Google Scholar] [CrossRef]
- Xie, M.; Yi, X.; Wang, R.; Wang, L.; He, G.; Zhu, M.; Qi, C.; Liu, Y.; Ye, Y.; Tan, S.; et al. 14-Thienyl methylene matrine (YYJ18), the derivative from matrine, induces apoptosis of human nasopharyngeal carcinoma cells by targeting MAPK and PI3K/Akt pathways in vitro. Cell Physiol. Biochem. 2014, 33, 1475–1483. [Google Scholar]
- Ngan, H.; Wang, L.; Lo, K.; Lui, V.W.Y. Genomic landscapes of EBV-associated nasopharyngeal carcinoma vs. HPV-associated head and neck cancer. Cancers 2018, 10, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein-Barr virus infection and nasopharyngeal carcinoma. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160270. [Google Scholar] [CrossRef]
- Cao, Y.; Xie, L.; Shi, F.; Tang, M.; Li, Y.; Hu, J.; Zhao, L.; Zhao, L.; Yu, X.; Luo, X. Targeting the signaling in Epstein-Barr virus-associated diseases: Mechanism, regulation, and clinical study. Signal Transduct. Target. Ther. 2021, 6, 1–33. [Google Scholar] [CrossRef]
- Lo, A.K.; Dawson, C.W.; Lung, H.L.; Wong, K.; Young, L.S. The role of EBV-encoded LMP1 in the NPC tumour microenvironment: From function to therapy. Front. Oncol. 2021, 11, 262. [Google Scholar] [CrossRef]
- Song, X.; Ai, M.; Chen, X.; Deng, X.; Tao, Y.; Gong, J.; Wu, Q.; Cao, Y. Regulation of c-Jun/JunB heterodimers mediated by Epstein-Barr virus encoded latent membrane protein 1 on Pchinese. Sci. Bull. 2004, 49, 676–683. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Zeng, L.; Tao, Y.G.; Tang, F.Q.; Wang, H.; Luo, F.J.; Yi, W.; Cao, Y. EB virus-encoded latent membrane protein 1 activates the JNK signalling pathway via a mechanism involving TRADD and TRAF in nasopharyngeal carcinoma cell. Prog. Biochem. Biophys. 2002, 29, 562–566. [Google Scholar]
- Johansson, P.; Jansson, A.; Ruetschi, U.; Rymo, L. The p38 signaling pathway upregulates expression of the Epstein-Barr virus LMP1 oncogene. J. Virol. 2010, 84, 2787–2797. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chen, L.; Liang, Y.; Tsang, N.; Chang, Y. Epstein-Barr virus latent membrane protein 1 induces the chemotherapeutic target, thymidine phosphorylase, via NF-kB and p38 MAPK pathways. Cell. Signal. 2010, 22, 1132–1142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yu, X.; Zhou, Z.; Li, B.; Peng, J.; Wu, X.; Luo, X.; Yang, L. LMP1-positive extracellular vesicles promote radioresistance in nasopharyngeal carcinoma cells through p38 MAPK signaling. Cancer Med. 2019, 8, 6082–6094. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Liu, L.; Xu, Z.; Liao, W.; Feng, D.; Dong, X.; Xu, S.; Xiao, L.; Lu, J.; Luo, X.; et al. EBV-LMP1 targeted DNAzyme enhances radiosensitivity by inhibiting tumor angiogenesis via the JNKs/HIF-1 pathway in nasopharyngeal carcinoma. Oncotarget 2015, 6, 5804–5817. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.; Chang, H.; Hung, W. Transcriptional repression of tissue inhibitor of metalloproteinase-3 by Epstein-Barr virus latent membrane protein 1 enhances invasiveness of nasopharyngeal carcinoma cells. Oral Oncol. 2008, 44, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and cancer: Triggers, mechanisms, and consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Al-Taei, S.; Salimu, J.; Spary, L.K.; Clayton, A.; Lester, J.F.; Tabi, Z. Prostaglandin E2-mediated adenosinergic effects on CD14 cells: Self-amplifying immunosuppression in cancer. Oncoimmunology 2017, 6, 1268308. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, Y.W.; Li, C.F.; Chi, J.Y.; Tseng, J.T.; Chang, Y.; Hsu, L.J.; Lee, C.H.; Chang, T.H.; Wang, S.M.; Wang, D.D.; et al. CCAAT/enhancer binding protein delta in macrophages contributes to immunosuppression and inhibits phagocytosis in nasopharyngeal carcinoma. Sci. Signal 2013, 6, 59. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, D.; Srivastava, S.K.; Chaudhuri, T.K.; Upadhyay, G. Multifaceted role of matrix metalloproteinases (MMPs). Front. Mol. Biosci. 2015, 2, 19. [Google Scholar] [CrossRef]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-mesenchymal transition in cancer: A historical overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef]
- Roomi, M.W.; Bhanap, B.; Niedzwiecki, A.; Rath, M. In vitro modulation of MMP-2 and MMP-9 secretion by cytokines, inducers, and inhibitors in head and neck squamous carcinoma cells (FaDu) and tongue carcinoma cells (SCC-25). J. Otolaryngol. Rhinol. 2017, 3, 29. [Google Scholar] [CrossRef]
- Wong, T.; Kwong, D.; Sham, J.; Wei, W.; Kwong, Y.; Yuen, A. Clinicopathologic significance of plasma matrix metalloproteinase-2 and-9 levels in patients with undifferentiated nasopharyngeal carcinoma. Eur. J. Surg. Oncol. 2004, 30, 560–564. [Google Scholar] [CrossRef]
- Lin, M.; Lu, Y.; Chung, J.; Wang, S.; Lin, H.; Kang, S.; Tang, C.; Ko, J.; Chen, S. Down-regulation of MMP-2 through the p38 MAPK-NF-κB-dependent pathway by aloe-emodin leads to inhibition of nasopharyngeal carcinoma cell invasion. Mol. Carcinog. 2010, 49, 783–797. [Google Scholar] [CrossRef]
- Xu, J.; Ying, Y.; Xiong, G.; Lai, L.; Wang, Q.; Yang, Y. Amyloid β precursor protein silencing attenuates epithelial-mesenchymal transition of nasopharyngeal carcinoma cells via inhibition of the MAPK pathway. Mol. Med. Rep. 2019, 20, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Ghosh Dastidar, D.; Ghosh, D.; Chakrabarti, G. Tumour vasculature targeted anti-cancer therapy. Vessel Plus 2020, 4, 14. [Google Scholar] [CrossRef]
- Rivera-Soto, R.; Damania, B. Modulation of angiogenic processes by the human gammaherpesviruses, Epstein-Barr virus and Kaposi’s sarcoma-associated herpesvirus. Front. Microbiol. 2019, 10, 1544. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Chen, J.; Xue, K.; Wang, Z.; Yu, D. Clinicopathologic and prognostic significance of VEGF, JAK2 and STAT3 in patients with nasopharyngeal carcinoma. Cancer Cell Int. 2018, 18, 110. [Google Scholar] [CrossRef] [Green Version]
- Yoshizuka, N.; Chen, R.M.; Xu, Z.; Liao, R.; Hong, L.; Hu, W.; Yu, G.; Han, J.; Chen, L.; Sun, P. A novel function of p38-regulated/activated kinase in endothelial cell migration and tumor angiogenesis. Mol. Cell Biol. 2012, 32, 606–618. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Bian, D.; Mahanivong, C.; Cheng, R.K.; Zhou, W.; Huang, S. p38 mitogen-activated protein kinase regulation of endothelial cell migration depends on urokinase plasminogen activator expression. J. Biol. Chem. 2004, 279, 50446–50454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamalice, L.; Le Boeuf, F.; Huot, J. Endothelial cell migration during angiogenesis. Circ. Res. 2007, 100, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Jittreetat, T.; Shin, Y.S.; Hwang, H.S.; Lee, B.; Kim, Y.S.; Sannikorn, P.; Kim, C. Tolfenamic acid inhibits the proliferation, migration, and invasion of nasopharyngeal carcinoma: Involvement of p38-mediated down-regulation of Slug. Yonsei Med. J. 2016, 57, 588–598. [Google Scholar] [CrossRef]
- Chiang, K.; Yang, S.; Chang, K.; Feng, T.; Chang, K.; Tsui, K.; Shin, Y.; Chen, C.; Chao, M.; Juang, H. Caffeic acid phenethyl ester induces N-myc downstream regulated gene 1 to inhibit cell proliferation and invasion of human nasopharyngeal cancer cells. Int. J. Mol. Sci. 2018, 19, 1397. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chuang, Y.; Lo, Y.; Lin, C.; Hsi, Y.; Hsieh, M.; Chen, M. Asiatic acid, extracted from Centella asiatica and induces apoptosis pathway through the phosphorylation p38 mitogen-activated protein kinase in cisplatin-resistant nasopharyngeal carcinoma cells. Biomolecules 2020, 10, 184. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, M.; Wang, C.; Lin, J.; Chuang, Y.; Hsi, Y.; Lo, Y.; Lin, C.; Chen, M. Celastrol, a plant-derived triterpene, induces cisplatin-resistance nasopharyngeal carcinoma cancer cell apoptosis though ERK1/2 and p38 MAPK signaling pathway. Phytomedicine 2019, 58, 152805. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, P.; Lin, C.; Yang, W.; Ho, Y.; Yang, S.; Chuang, C. Cantharidic acid induces apoptosis in human nasopharyngeal carcinoma cells through p38-mediated upregulation of caspase activation. Environ. Toxicol. 2020, 35, 619–627. [Google Scholar] [CrossRef]
- Chuang, C.; Tang, C.; Ho, H.; Hsin, C.; Weng, C.; Yang, S.; Chen, P.; Lin, C. Licochalcone A induces apoptotic cell death via JNK/p38 activation in human nasopharyngeal carcinoma cells. Environ. Toxicol. 2019, 34, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.J.; Chien, S.Y.; Chou, Y.E.; Chen, C.J.; Chen, J.; Chen, M.K. Hispolon from Phellinus linteus possesses mediate caspases activation and induces human nasopharyngeal carcinomas cells apoptosis through ERK1/2, JNK1/2 and p38 MAPK pathway. Phytomedicine 2014, 21, 1746–1752, Erratum in Phytomedicine 2018, 45, 121. [Google Scholar] [CrossRef]
- Vergote, I.; Heitz, F.; Buderath, P.; Powell, M.A.; Sehouli, J.; Lee, C.M.; Hamilton, A.L.; Fiorica, J.; Moore, K.N.; Teneriello, M. A randomized, double-blind, placebo-controlled phase Ib/II study of ralimetinib, a p38 MAPK inhibitor, plus gemcitabine (G) and carboplatin (C) versus GC for women with recurrent platinum-sensitive ovarian cancer. Gynecol. Oncol. 2019, 1, 23–31. [Google Scholar] [CrossRef]
- Tawil, A.; Mellion, M.; Ronco, L.; Raines, S.; Tracewell, W.; Rahilly, A.; Rojas, A.; Hage, M.; Wagner, K.; Statland, J. Design of a Phase 2, Randomized, Double-Blind, Placebo-Controlled, 24-Week, Parallel-Group Study of the Efficacy and Safety of Losmapimod in Treating Subjects with Facioscapulohumeral Muscular Dystrophy (FSHD): ReDUX4 (1592). 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04003974 (accessed on 30 March 2021).
- Ding, C. Drug evaluation: VX-702, a MAP kinase inhibitor for rheumatoid arthritis and acute coronary syndrome. Curr. Opin. Investig. Drugs 2006, 7, 1020–1025. [Google Scholar] [PubMed]
- MacNee, W.; Allan, R.J.; Jones, I.; De Salvo, M.C.; Tan, L.F. Efficacy and safety of the oral p38 inhibitor PH-797804 in chronic obstructive pulmonary disease: A randomised clinical trial. Thorax 2013, 68, 738–745. [Google Scholar] [CrossRef] [Green Version]
- Dotan, I.; Rachmilewitz, D.; Schreiber, S.; Eliakim, R.; Van der Woude, C.J.; Kornbluth, A.; Buchman, A.L.; Bar-Meir, S.; Bokemeyer, B.; Goldin, E.; et al. A randomised placebo-controlled multicentre trial of intravenous semapimod HCl for moderate to severe Crohn’s disease. Gut 2010, 59, 760–766. [Google Scholar] [CrossRef]
- Messoussi, A.; Feneyrolles, C.; Bros, A.; Deroide, A.; Daydé-Cazals, B.; Chevé, G.; Van Hijfte, N.; Fauvel, B.; Bougrin, K.; Yasri, A. Recent progress in the design, study, and development of c-Jun N-terminal kinase inhibitors as anticancer agents. Chem. Biol. 2014, 21, 1433–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Wu, W.; Jacevic, V.; Franca, T.C.; Wang, X.; Kuca, K. Selective inhibitors for JNK signalling: A potential targeted therapy in cancer. J. Enzym. Inhib. Med. Chem. 2020, 35, 574–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patnaik, A.; Haluska, P.; Tolcher, A.W.; Erlichman, C.; Papadopoulos, K.P.; Lensing, J.L.; Beeram, M.; Molina, J.R.; Rasco, D.W.; Arcos, R.R.; et al. A first-in-human phase I study of the oral p38 MAPK inhibitor, ralimetinib (LY2228820 dimesylate), in patients with advanced cancer. Clin. Cancer Res. 2016, 22, 1095–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicenas, J.; Zalyte, E.; Rimkus, A.; Dapkus, D.; Noreika, R.; Urbonavicius, S. JNK, p38, ERK, and SGK1 inhibitors in cancer. Cancers 2018, 10, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kim, T.H.; Kang, H.S.; Ro, J.; Kim, H.S.; Yoon, S. SP600125, an inhibitor of Jnk pathway, reduces viability of relatively resistant cancer cells to doxorubicin. Biochem. Biophys. Res. Commun. 2009, 387, 450–455. [Google Scholar] [CrossRef]
- Kim, J.; Chae, M.; Choi, A.; Kim, H.S.; Yoon, S. SP600125 overcomes antimitotic drug-resistance in cancer cells by increasing apoptosis with independence of P-gp inhibition. Eur. J. Pharm. 2014, 723, 141–147. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, T.; Wang, X.; Qu, J.; Chen, M. The JNK inhibitor SP600125 enhances dihydroartemisinin-induced apoptosis by accelerating BAX translocation into mitochondria in human lung adenocarcinoma cells. FEBS Lett. 2010, 584, 4019–4026. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Zhang, B.; Liang, H.; Lu, Y.; Ai, X.; Zhang, B.; Chen, X. JNK inhibitor SP600125 enhances TGF-β-induced apoptosis of RBE human cholangiocarcinoma cells in a Smad-dependent manner. Mol. Med. Rep. 2013, 8, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Jemaa, M.; Vitale, I.; Kepp, O.; Berardinelli, F.; Galluzzi, L.; Senovilla, L.; Marino, G.; Malik, S.A.; Rello-Varona, S.; Lissa, D. Selective killing of p53-deficient cancer cells by SP. EMBO Mol. Med. 2012, 4, 500–514. [Google Scholar] [CrossRef]
- Konno, T.; Ninomiya, T.; Kohno, T.; Kikuchi, S.; Sawada, N.; Kojima, T. c-Jun N-terminal kinase inhibitor SP600125 enhances barrier function and elongation of human pancreatic cancer cell line HPAC in a Ca-switch model. Histochem. Cell Biol. 2015, 143, 471–479. [Google Scholar] [CrossRef]
- Li, J.; Huang, J.; Xing, B.; Ren, K.; Li, M.; Wei, D.; Gu, P.; Chen, G.; Gu, B.; Zhang, G. SP600125, a JNK inhibitor, suppresses growth of JNK-inactive glioblastoma cells through cell-cycle G2/M phase arrest. Die Pharm.—Int. J. Pharm. Sci. 2012, 67, 942–946. [Google Scholar]
- Gao, Y.; Cheng, J.; Zeng, Q.; Xu, Z.; Decosterd, I.; Xu, X.; Ji, R. Selective inhibition of JNK with a peptide inhibitor attenuates pain hypersensitivity and tumor growth in a mouse skin cancer pain model. Exp. Neurol. 2009, 219, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Inesta-Vaquera, F.; Niepel, M.; Zhang, J.; Ficarro, S.B.; Machleidt, T.; Xie, T.; Marto, J.A.; Kim, N.; Sim, T. Discovery of potent and selective covalent inhibitors of JNK. Chem. Biol. 2012, 19, 140–154. [Google Scholar] [CrossRef] [Green Version]
- Ebelt, N.D.; Kaoud, T.S.; Edupuganti, R.; Van Ravenstein, S.; Dalby, K.N.; Van Den Berg, C.L. A c-Jun N-terminal kinase inhibitor, JNK-IN-8, sensitizes triple negative breast cancer cells to lapatinib. Oncotarget 2017, 8, 104894–104912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, M.; Kuramoto, K.; Takeda, H.; Watarai, H.; Sakaki, H.; Seino, S.; Seino, M.; Suzuki, S.; Kitanaka, C. The novel JNK inhibitor AS602801 inhibits cancer stem cells in vitro and in vivo. Oncotarget 2016, 7, 27021–27032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posthumadeboer, J.; Van Egmond, P.W.; Helder, M.N.; De Menezes, R.X.; Cleton-Jansen, A.M.; Belien, J.A.; Verheul, H.M.; Van Royen, B.J.; Kaspers, G.J.; Van Beusechem, V.W. Targeting JNK-interacting-protein-1 (JIP1) sensitises osteosarcoma to doxorubicin. Oncotarget 2012, 3, 1169–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasilevskaya, I.A.; Selvakumaran, M.; Hierro, L.C.; Goldstein, S.R.; Winkler, J.D.; O’Dwyer, P.J. Inhibition of JNK sensitizes hypoxic colon cancer cells to DNA-damaging agents. Clin. Cancer Res. 2015, 21, 4143–4152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Düzgün, Ş.A.; Yerlikaya, A.; Zeren, S.; Bayhan, Z.; Okur, E.; Boyacı, İ. Differential effects of p38 MAP kinase inhibitors SB203580 and SB202190 on growth and migration of human MDA-MB-231 cancer cell line. Cytotechnology 2017, 69, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Yasui, H.; Hideshima, T.; Ikeda, H.; Jin, J.; Ocio, E.M.; Kiziltepe, T.; Okawa, Y.; Vallet, S.; Podar, K.; Ishitsuka, K. BIRB 796 enhances cytotoxicity triggered by bortezomib, heat shock protein (Hsp) 90 inhibitor, and dexamethasone via inhibition of p38 mitogen-activated protein kinase/Hsp27 pathway in multiple myeloma cell lines and inhibits paracrine tumour growth. Br. J. Haematol. 2007, 136, 414–423. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Zhao, X.; Chen, X.; Fang, Y.; Singh, S.; Talele, T.T.; Qiu, H.; Liang, Y.; Wang, X.; Zhang, G. BIRB796, the inhibitor of p38 mitogen-activated protein kinase, enhances the efficacy of chemotherapeutic agents in ABCB1 overexpression cells. PLoS ONE 2013, 8, e54181. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Mo, Q.; Zhang, Y.; Gao, Y.; Wu, Y.; Li, J.; Hao, X.; Ma, D.; Gao, Q.; Chen, P. The p38 MAPK inhibitor BIRB796 enhances the antitumor effects of VX680 in cervical cancer. Cancer Biol. Ther. 2016, 17, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Podar, K.; Chauhan, D.; Ishitsuka, K.; Mitsiades, C.; Tai, Y.; Hamasaki, M.; Raje, N.; Hideshima, H.; Schreiner, G. p38 MAPK inhibition enhances PS-341 (bortezomib)-induced cytotoxicity against multiple myeloma cells. Oncogene 2004, 23, 8766–8776. [Google Scholar] [CrossRef] [Green Version]
- Giafis, N.; Katsoulidis, E.; Sassano, A.; Tallman, M.S.; Higgins, L.S.; Nebreda, A.R.; Davis, R.J.; Platanias, L.C. Role of the p38 mitogen-activated protein kinase pathway in the generation of arsenic trioxide-dependent cellular responses. Cancer Res. 2006, 66, 6763–6771. [Google Scholar] [CrossRef] [Green Version]
- Campbell, R.M.; Anderson, B.D.; Brooks, N.A.; Brooks, H.B.; Chan, E.M.; De Dios, A.; Gilmour, R.; Graff, J.R.; Jambrina, E.; Mader, M.; et al. Characterization of LY2228820 dimesylate, a potent and selective inhibitor of p38 MAPK with antitumor activity. Mol. Cancer 2014, 13, 364–374. [Google Scholar] [CrossRef] [Green Version]
- Yeung, Y.T.; Yin, S.; Lu, B.; Fan, S.; Yang, R.; Bai, R.; Zhang, C.; Bode, A.M.; Liu, K.; Dong, Z. Losmapimod overcomes gefitinib resistance in non-small cell lung cancer by preventing tetraploidization. EBioMedicine 2018, 28, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Bachegowda, L.; Morrone, K.; Winski, S.L.; Mantzaris, I.; Bartenstein, M.; Ramachandra, N.; Giricz, O.; Sukrithan, V.; Nwankwo, G.; Shahnaz, S.; et al. Pexmetinib: A novel dual inhibitor of TIE2 and p38 MAPK with efficacy in preclinical models of myelodysplastic syndromes and acute myeloid leukemia. Cancer Res. 2016, 76, 4841–4849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollenberg, L.A.; Corson, D.T.; Nugent, C.A.; Peterson, F.L.; Ptaszynski, A.M.; Arrigo, A.; Mannila, C.G.; Litwiler, K.S.; Bell, S.J. An exploratory, randomized, parallel-group, open-label, relative bioavailability study with an additional two-period crossover food-effect study exploring the pharmacokinetics of two novel formulations of pexmetinib (ARRY-614). Clin. Pharm. 2015, 7, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.C.; Hsieh, M.J.; Chen, C.J.; Lin, J.T.; Lo, Y.S.; Chuang, Y.C.; Chien, S.Y.; Chen, M.K. Polyphyllin G induce apoptosis and autophagy in human nasopharyngeal cancer cells by modulation of AKT and mitogen-activated protein kinase pathways in vitro and in vivo. Oncotarget 2016, 7, 70276–70289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roulston, A.; Reinhard, C.; Amiri, P.; Williams, L.T. Early activation of c-Jun N-terminal kinase and p38 kinase regulate cell survival in response to tumor necrosis factor α. J. Biol. Chem. 1998, 273, 10232–10239. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Type of Cancer | JNK Status | Clinical Implications | References |
|---|---|---|---|
| Liver cancer | JNK1 activity upregulated | Higher expression of JNK1, rather than JNK2, was detected as a progenitor cell biomarker and lowered the survival rate of patients with hepatocellular carcinoma (HCC). | [46] |
| Prostate cancer | JNK1,2 activities upregulated in silico | Total JNK expression was upregulated in human malignant prostate epithelium compared to normal or benign hyperplasic (BPH) epithelium. | [47,48] |
| Breast cancer | JNK1,2 activities downregulated | Decreased p-JNK1/2 expression was observed in breast infiltrating ductal carcinoma (IDC) cases and was correlated significantly with increased tumor grade and decreased age at diagnosis. | [49] |
| Bladder cancer | JNK2 activity downregulated | Lower JNK2 expression was associated with poorer overall survival among patients who underwent radical cystectomy. | [50] |
| Lung cancer | JNK1,2 activity downregulated | JNK1/2 was inactivated in human lung squamous cell carcinoma (LSCC) and their activities were positively correlated with survival rates of patients. | [51] |
| Thyroid cancer | JNK activity upregulated | p-JNK was overexpressed in papillary thyroid carcinomas and was significantly associated with the presence of lymph node metastases and advanced TNM stages. | [52] |
| Colorectal cancer | JNK1 activity upregulated | JNK activity was elevated in human colorectal tumors compared to normal intestinal mucosa. p-JNK1 was overexpressed in the multidrug-resistant colon cancer cells. | [53] |
| Head and neck squamous cell carcinoma | JNK1,2 activities downregulated | Higher JNK1/2 activities had better survival rate than those with lower JNK1/2 activities in patients with head and neck squamous cell carcinoma tumors. | [51,54] |
| Skin cancer | JNK1 activity downregulated, JNK2 activity upregulated | JNK1 expression was inhibited by squamous cell carcinoma antigen (SCCA) and blocked UV-induced keratinocyte apoptosis. JNK2 was activated in more than 70% of human squamous cell carcinoma (SCC) and is sufficient to couple with oncogenic Ras to transform primary human epidermal cells into malignancy. | [55,56] |
| Ovarian cancer | JNK1 activities upregulated | JNK1 expression levels were found to be higher in advanced stage (III and IV) cases than in early stage (I and II) cases and inversely associated with the survival of ovarian cancer patients. | [57,58] |
| Nasopharyngeal carcinoma | JNK activity upregulated | Activation of JNK signaling was associated with TNM staging of NPC, as NPC patients with stage III–IV had higher positive expression rates of JNK and p-JNK proteins compared to NPC patients with stage I–II. | [59] |
| Type of Cancer | p38 MAPK Status | Clinical Implications | References |
|---|---|---|---|
| Liver cancer | p38γ, δ activities upregulated | High p38γ expression was associated with a poorer outcome in cases of liver cancer. Overexpression of p38δ was observed in cholangiocarcinoma and responsible for cancer cell motility and invasion. | [60,61] |
| Prostate cancer | p38 MAPKs upregulated | Strong expression of p38 MAPKs was observed in all prostate cancer patients with progressive disease from stages II to IV. | [62] |
| Breast cancer | p38α, δ activities upregulated | High levels of active p38α were correlated with poor prognosis, lymph node metastasis, and tamoxifen resistance in breast cancer patients. High p38δ levels were associated with poor prognosis in breast cancer patients of all tumor subtypes, especially estrogen receptor (ER)-positive/human epidermal growth factor receptor 2 (HER2)-negative types. | [63,64,65] |
| Bladder cancer | p38 activity upregulated | The expression of p38 in transitional cell carcinoma (TCC) of the bladder was positively correlated with depth of muscle invasion, grade, stage, lymph node metastasis, distant metastasis, size, and number of tumors. | [66,67] |
| Lung cancer | p38α activity upregulated | Higher numbers of both phosphorylated-p38 and p38α-positive cells were observed in lung adenocarcinoma compared to the normal lung parenchyma and correlated with a higher mortality rate as well as with a shorter time to relapse. | [68,69] |
| Thyroid cancer | p38α activity upregulated | High expression of p38α was revealed in malignant thyroid carcinoma, such as human papillary and follicular thyroid carcinomas. | [70] |
| Colorectal cancer | p38α, β, δ activities upregulated | High levels of phosphorylated p38, p38α, and p38β were correlated with chemotherapy resistance and poor overall survival in colon cancer patients. The depletion of p38δ impaired tumor growth in vivo. | [71,72,73] |
| Head and neck squamous cell carcinoma | p38α, δ activities upregulated | Expression of p38α and p38δ by tumor cells was detected in HNSCCs in vivo. Phosphorylated p38 expression was clearly increased in moderately differentiated and even further increased in poorly differentiated HNSCC, with increased angiogenesis and lymph angiogenesis. | [74,75] |
| Skin cancer | p38α, δ activities upregulated | Increased expression levels of p38α and p38δ were detected in human primary cutaneous SCCs. | [76] |
| Nasopharyngeal carcinoma | p38 MAPKs upregulated | p38 MAPKs were overexpressed in non-keratinizing squamous cell carcinoma (most common form in high-risk countries) at T3–T4, N2–N3 and clinical stage III–IV. | [77,78] |
| Name | IC50 (nM) | Potential Usages in Cancer | References | |||
|---|---|---|---|---|---|---|
| JNK1 | JNK2 | JNK3 | JNKs | |||
| SP600125 | 40 | 40 | 90 | ND | Anti-cancer effects in stomach cancer, oral squamous carcinoma, lung adenocarcinoma, cholangiocarcinoma, colon carcinoma, pancreatic cancer, and glioblastoma. | [131,132,133,134,135,136,137,138] |
| JNK-IN-1 | ND | ND | ND | 2.31 | Anti-cancer effects in skin cancer. | [139] |
| JNK-IN-8 | 4.67 | 18.7 | 0.98 | ND | Sensitized triple-negative breast cancer cells to lapatinib. | [140,141] |
| Bentamapimod (AS602801/PGL5001) | 80 | 90 | 230 | ND | Induced apoptosis of cancer stem cells. | [142] |
| BI-78D3 | ND | ND | ND | 280 | Anti-cancer effects in osteosarcoma. | [143] |
| CC-401 | 25–50 | 500–1000 | 25–50 | ND | Anti-cancer effects in colon cancer and acute myeloid leukemia. | [128,144] |
| Name | IC50 (nM) | Potential Usages in Cancer | References | ||||
|---|---|---|---|---|---|---|---|
| p38 MAPKs | p38α | p38β | p38γ | p38δ | |||
| SB203580 | 50 | ND | 500 | ND | ND | Anti-cancer effects in breast cancer. | [145] |
| Doramapimod (BIRB 796) | 0.1 | 38 | 65 | 520 | 200 | Anti-cancer effects in multiple myeloma, oral epidermoid carcinoma, cervical cancer. | [146,147,148] |
| Talmapimod (SCIO-469) | ND | 9 | 90 | ND | ND | Potential chemotherapy for multiple myeloma and leukemia. | [149,150] |
| Ralimetinib (LY2228820) | ND | 5.3 | 3.2 | ND | ND | Potential chemotherapy for melanoma, non-small cell lung cancer, ovarian cancer, glioma, myeloma, breast cancer, colorectal cancer, sarcoma, renal cancer, and pancreatic cancer. | [130,151] |
| Losmapimod (GW856553X) | ND | 8.1 | 7.6 | ND | ND | Overcame gefitinib resistance in non-small cell lung cancer (NSCLC). | [152] |
| Pexmetinib (ARRY-614) | 1 | 35 | 26 | ND | ND | Potential chemotherapy for hematological carcinoma, such as myelodysplastic syndromes. | [153,154] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pua, L.J.W.; Mai, C.-W.; Chung, F.F.-L.; Khoo, A.S.-B.; Leong, C.-O.; Lim, W.-M.; Hii, L.-W. Functional Roles of JNK and p38 MAPK Signaling in Nasopharyngeal Carcinoma. Int. J. Mol. Sci. 2022, 23, 1108. https://doi.org/10.3390/ijms23031108
Pua LJW, Mai C-W, Chung FF-L, Khoo AS-B, Leong C-O, Lim W-M, Hii L-W. Functional Roles of JNK and p38 MAPK Signaling in Nasopharyngeal Carcinoma. International Journal of Molecular Sciences. 2022; 23(3):1108. https://doi.org/10.3390/ijms23031108
Chicago/Turabian StylePua, Lesley Jia Wei, Chun-Wai Mai, Felicia Fei-Lei Chung, Alan Soo-Beng Khoo, Chee-Onn Leong, Wei-Meng Lim, and Ling-Wei Hii. 2022. "Functional Roles of JNK and p38 MAPK Signaling in Nasopharyngeal Carcinoma" International Journal of Molecular Sciences 23, no. 3: 1108. https://doi.org/10.3390/ijms23031108
APA StylePua, L. J. W., Mai, C. -W., Chung, F. F. -L., Khoo, A. S. -B., Leong, C. -O., Lim, W. -M., & Hii, L. -W. (2022). Functional Roles of JNK and p38 MAPK Signaling in Nasopharyngeal Carcinoma. International Journal of Molecular Sciences, 23(3), 1108. https://doi.org/10.3390/ijms23031108