
Incorporation of a FRET Pair into a Riboswitch RNA to Measure Mg2+ Concentration and RNA Conformational Change in Cell

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
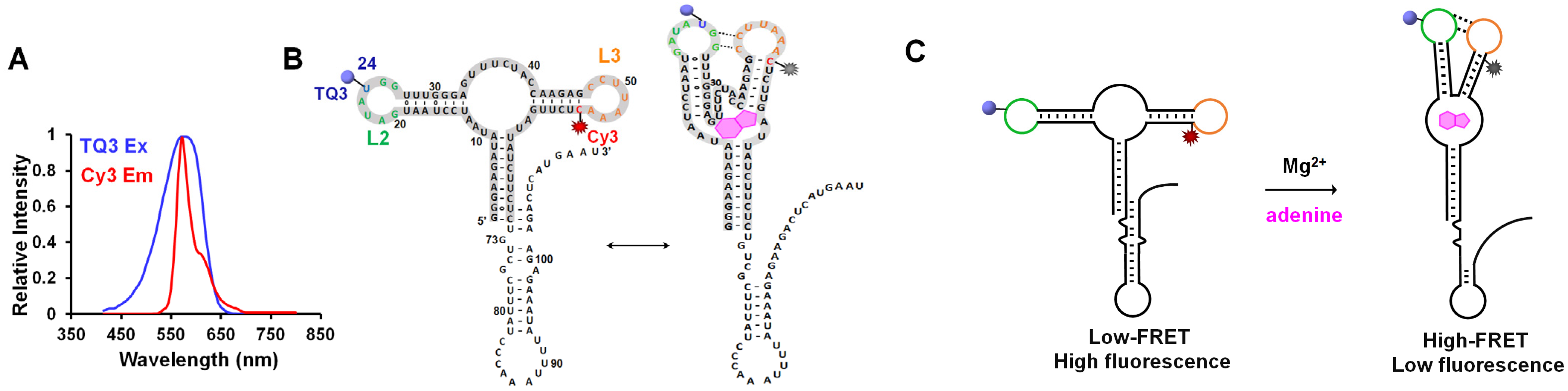
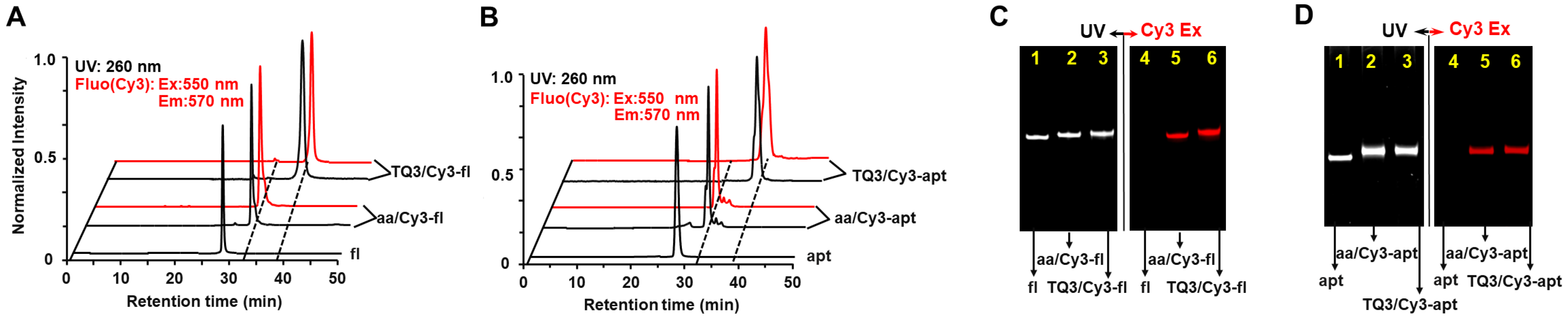
2.1. Site-Specific Labeling of TQ3/Cy3-apt and TQ3/Cy3-fl
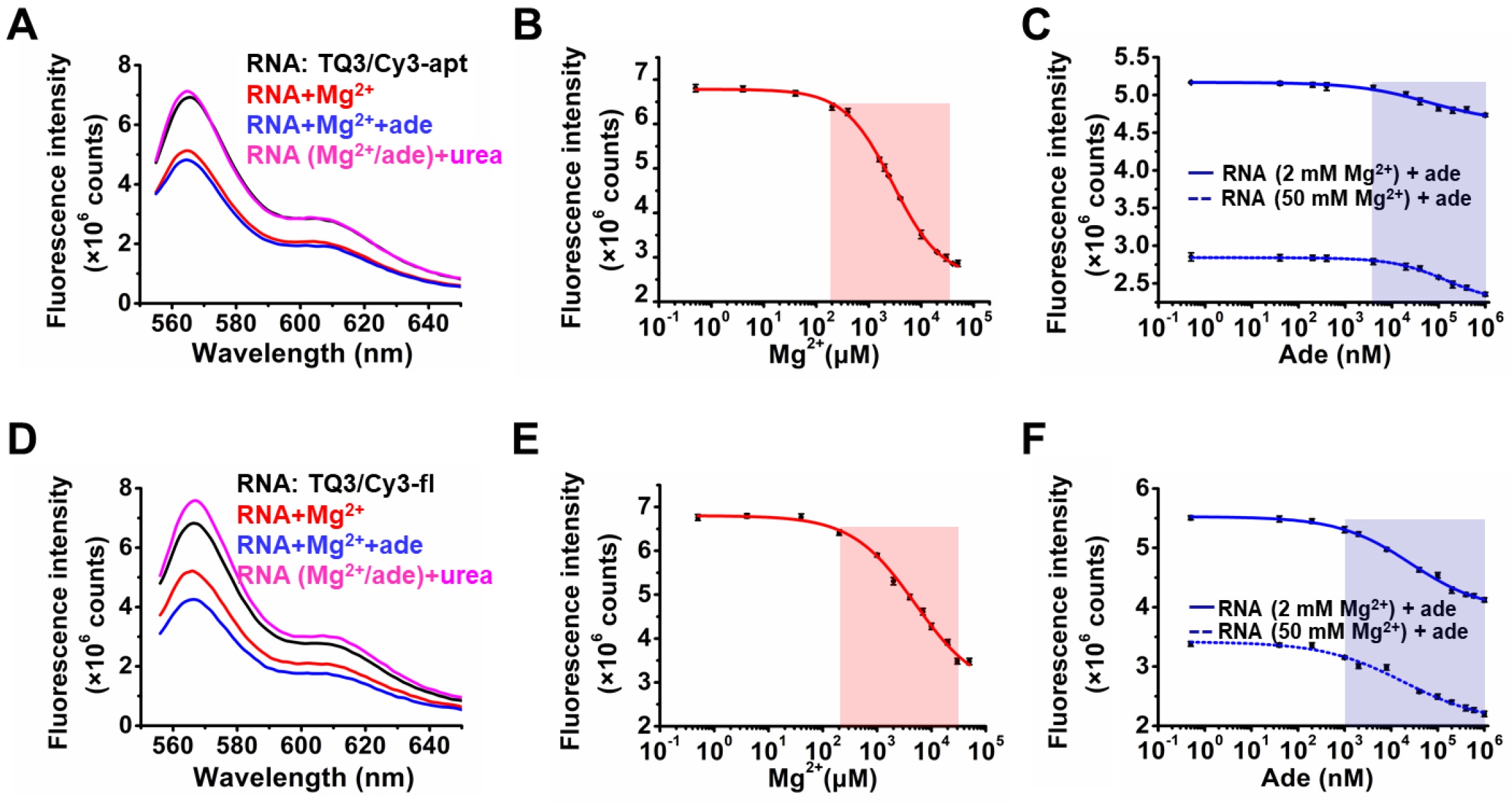
2.2. Influence of Mg2+ and Adenine on the Structural Dynamics of TQ3/Cy3-apt and TQ3/Cy3-fl In Vitro
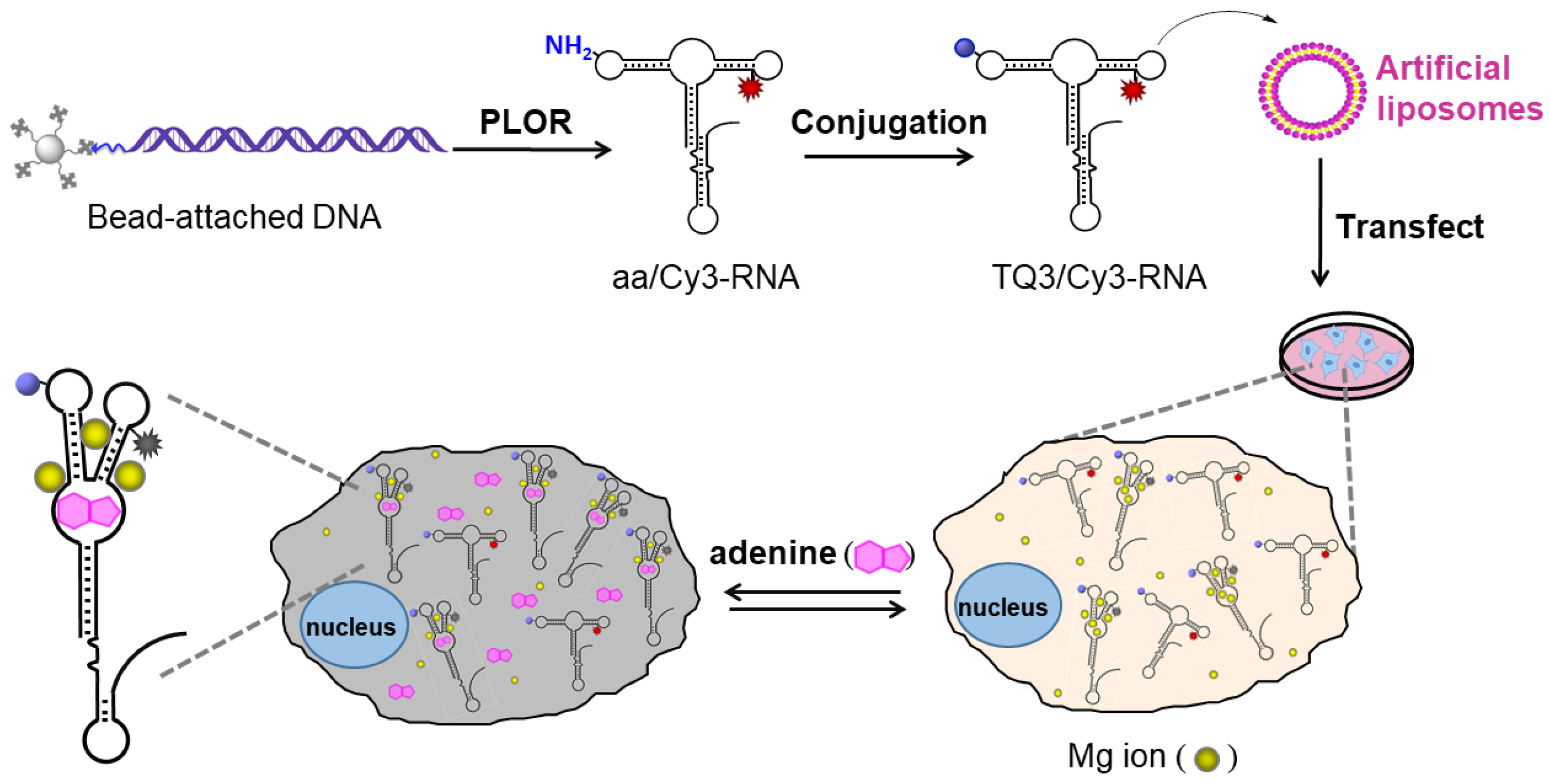
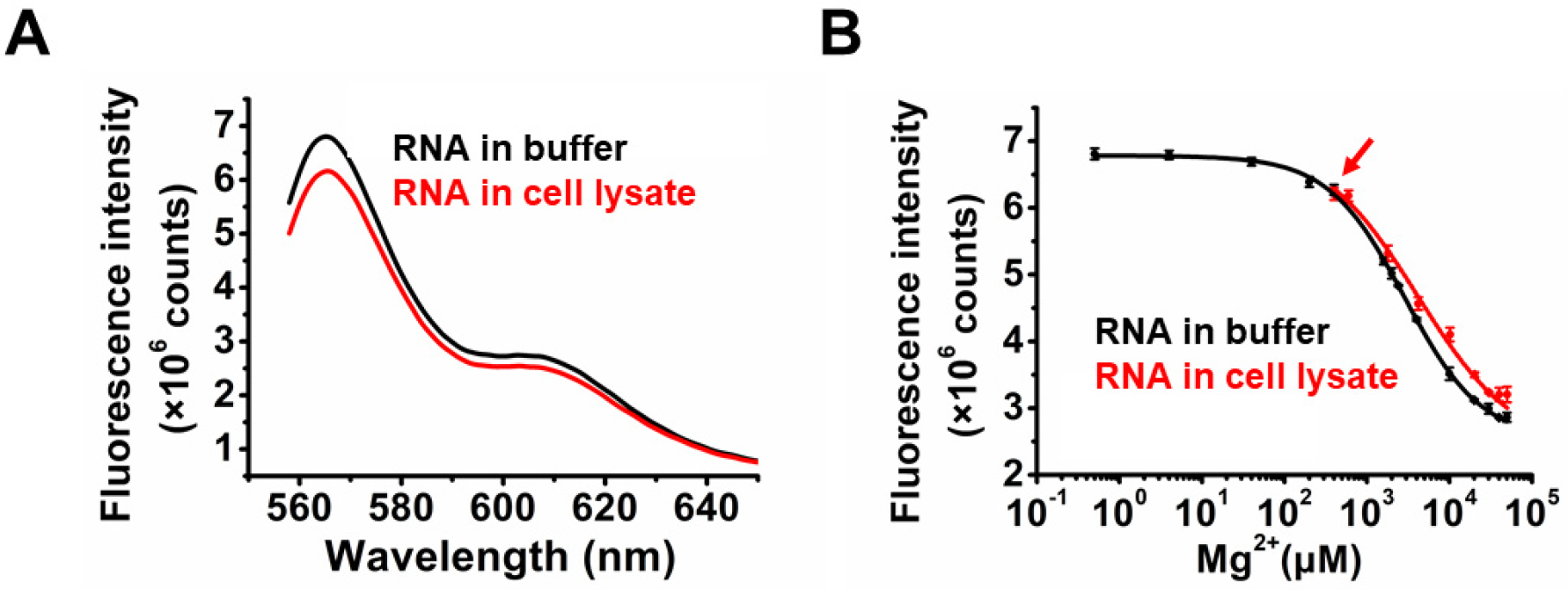
2.3. Application of TQ3/Cy3-RNA as a Biosensor of Cellular Mg2+ Concentration
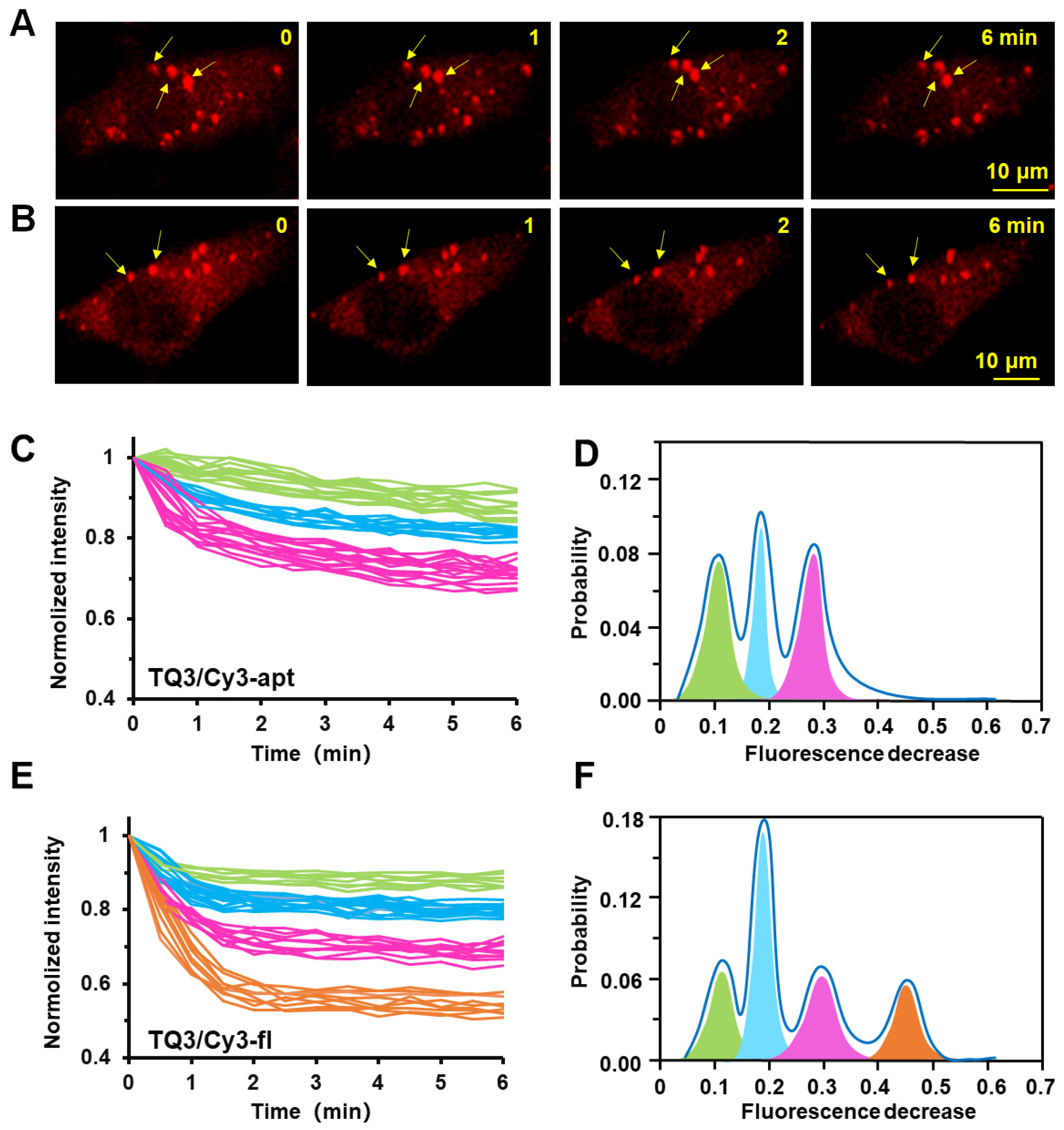
2.4. Conformational Change of TQ3/Cy3-apt and TQ3/Cy3-fl in Mammalian Cells
3. Discussion
4. Materials and Methods
4.1. Preparation of DNA Templates for PLOR Reactions
4.2. PLOR Generation of aa/Cy3-apt and aa/Cy3-fl
4.3. Post-Synthesis Incorporation of TQ3 into aa/Cy3-apt and aa/Cy3-fl
4.4. Steady-State Fluorescence Detection
4.5. Cell Culture and RNA Transfection
4.6. Cell Imaging Using Confocal Microscopy
4.7. Measurement of Cellular Mg2+ Concentration
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Serganov, A.; Nudler, E. A decade of riboswitches. Cell 2013, 152, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montange, R.K.; Batey, R.T. Riboswitches: Emerging themes in RNA structure and function. Annu. Rev. Biophys. 2008, 37, 117–133. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Kladwang, W.; Das, R. Allosteric mechanism of the V. vulnificus adenine riboswitch resolved by four-dimensional chemical mapping. eLife 2018, 7, e29602. [Google Scholar] [CrossRef] [PubMed]
- Reining, A.; Nozinovic, S.; Schlepckow, K.; Buhr, F.; Furtig, B.; Schwalbe, H. Three-state mechanism couples ligand and temperature sensing in riboswitches. Nature 2013, 499, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Stagno, J.R.; Liu, Y.; Bhandari, Y.R.; Conrad, C.E.; Panja, S.; Swain, M.; Fan, L.; Li, C.; Wendel, D.R.; White, T.A.; et al. Structures of riboswitch RNA reaction states by mix-and-inject XFEL serial crystallography. Nature 2017, 541, 242–246. [Google Scholar] [CrossRef]
- Lin, J.C.; Thirumalai, D. Kinetics of allosteric transitions in S-adenosylmethioine riboswitch are accurately predicted from the folding landscape. J. Am. Chem. Soc. 2013, 135, 16641–16650. [Google Scholar] [CrossRef] [Green Version]
- Bennett, B.D.; Kimball, E.H.; Gao, M.; Osterhout, R.; Dien, S.J.V.; Rabinowitz, J.D. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 2009, 5, 593–599. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, R.; Grossberger, R.; Pichler, A.; Waldsich, C. RNA folding in vivo. Curr. Opin. Struct. Biol. 2002, 12, 296–300. [Google Scholar] [CrossRef]
- Lindell, M.; Romby, P.; Wagner, E.G.H. Lead (II) as a probe for investigating RNA structure in vivo. RNA 2002, 8, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Kertesz, M.; Wan, Y.; Mazor, E.; Rinn, J.L.; Nutter, R.C.; Chang, H.Y.; Segal, E. Genome-wide measurement of RNA secondary structure in yeast. Nature 2010, 467, 103–107. [Google Scholar] [CrossRef]
- Sowa, S.W.; Anderson, J.V.; Clark, C.A.; Pena, R.D.L.; Dunn, K.; Fung, E.K.; Khoury, M.J.; Contreras, L.M. Exploiting post-transcriptional regulation to probe RNA structures in vivo via fluorescence. Nucleic Acids Res. 2015, 43, e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Kwork, C.K.; Tang, Y.; Bevilacqua, P.C.; Assmann, S.M. Genome-wide profiling of in vivo RNA structure at single-nucleotide resolution using structure-seq. Nat. Protoc. 2015, 10, 1050–1066. [Google Scholar] [CrossRef] [PubMed]
- Tomezsko, P.J.; Corbin, V.D.A.; Gupta, P.; Swaminathan, H.; Glasgow, M.; Persad, S.; Edwards, M.D.; Mcintosh, L.; Papenfuss, A.T.; Emery, A.; et al. Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature 2020, 582, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Win, M.N.; Smolke, C.D. A modular and extensible RNA-based gene-regulatory platform for engineering cellular function. Proc. Natl. Acad. Sci. USA 2007, 104, 14283–14288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Jaffrey, S.R. A fluorogenic RNA-based sensor activated by metabolite- induced RNA dimerization. Cell Chem. Biol. 2019, 26, 1725–1731. [Google Scholar] [CrossRef]
- Autour, A.; Jeng, S.C.Y.; Cawte, A.D.; Abdolahzadeh, A.; Galli, A.; Panchapakesan, S.S.S.; Rueda, D.; Ryckelynck, M.; Unrau, P.J. Fluorogenic RNA Mango aptamers for imaging small non-coding RNAs in mammalian cells. Nat. Commun. 2018, 9, 656. [Google Scholar] [CrossRef]
- Paige, J.S.; Wu, K.R.; Jaffrey, S.R. RNA mimics of green fluorescent protein. Science 2011, 333, 642–646. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, D.; Su, N.; Bao, B.; Xie, X.; Zuo, F.; Yang, L.; Wang, H.; Jiang, L.; Lin, Q.; et al. Visualizing RNA dynamics in live cells with bright and stable fluorescent RNAs. Nat. Biotechnol. 2019, 37, 1287–1293. [Google Scholar] [CrossRef]
- Jepsen, M.D.E.; Sparvath, S.M.; Nielsen, T.B.; Langvad, A.H.; Grossi, G.; Gothelf, K.V.; Andersen, E.S. Development of a genetically encodable FRET system using fluorescent RNA aptamers. Nat. Commun. 2018, 9, 18. [Google Scholar] [CrossRef]
- Neubacher, S.; Hennig, S. RNA structure and cellular applications of fluorescent light-up aptamers. Angew. Chem. Int. Ed. Engl. 2019, 58, 1266–1279. [Google Scholar] [CrossRef]
- Cawte, A.D.; Unrau, P.J.; Rueda, D.S. Live cell imaging of single molecules with fluorogenic Mango II arrays. Nat. Commun. 2020, 11, 1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Holmstrom, E.; Zhang, J.; Yu, P.; Wang, J.; Dyba, M.A.; Chen, D.; Ying, J.; Lockett, S.; Nesbitt, D.J.; et al. Synthesis and applications of RNAs with position-selective labeling and mosaic composition. Nature 2015, 522, 368–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Holmstrom, E.; Yu, P.; Tan, K.; Zuo, X.; Nesbitt, D.J.; Sousa, R.; Stagno, J.R.; Wang, Y. Incorporation of isotopic, fluorescent, and heavy-atom-modified nucleotides into RNAs by position-selective labeling of RNA. Nat. Protoc. 2018, 13, 987–1005. [Google Scholar] [CrossRef] [PubMed]
- Algar, W.R.; Ancona, M.G.; Malanoski, A.P.; Susumu, K.; Medintz, I.L. Assembly of a concentric förster resonance energy transfer relay on a quantum dot scaffold: Characterization and application to multiplexed protease sensing. ACS Nano 2012, 6, 11044–11058. [Google Scholar] [CrossRef]
- Zhang, S.; Metelev, V.; Tabatadze, D.; Zamecnik, P.C.; Bogdanov, A., Jr. Fluorescence resonance energy transfer in near-infrared fluorescent oligonucleotide probes for detecting protein-DNA interactions. Proc. Natl. Acad. Sci. USA 2008, 105, 4156–4161. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chetnani, B.; Cormack, E.D.; Alonso, D.; Liu, W.; Mondragon, A.; Fei, J. Specific structural elements of the T-box riboswitch drive the two-step binding of the tRNA ligand. eLife 2018, 7, e39518. [Google Scholar] [CrossRef]
- Chen, J.; Tsia, A.; Petrov, A.; Puglisi, J.D. Non-fluorescent quenchers to correlate single-molecule conformational and compositional dynamics. J. Am. Chem. Soc. 2012, 134, 5734–5737. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, J.J.; Quake, S.R. Single molecule measurement of the “speed limit” of DNA polymerase. Proc. Natl. Acad. Sci. USA 2009, 106, 20294–20299. [Google Scholar] [CrossRef] [Green Version]
- Warhaut, S.; Mertinkus, K.R.; Höllthaler, P.; Fürtig, B.; Heilemann, M.; Hengesbach, M.; Schwalbe, H. Ligand-modulated folding of the full-length adenine riboswitch probed by NMR and single-molecule FRET spectroscopy. Nucleic Acids Res. 2017, 45, 5512–5522. [Google Scholar] [CrossRef] [Green Version]
- Lemay, J.F.; Penedo, J.C.; Tremblay, R.; Lillty, D.M.J.; Lafontaine, D.A. Folding of the adenine riboswitch. Chem. Biol. 2006, 13, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Noeske, J.; Schwalbe, H.; Wöhnert, J. Metal-ion binding and metal-ion induced folding of the adenine-sensing riboswitch aptamer domain. Nucleic Acids Res. 2007, 35, 5262–5273. [Google Scholar] [CrossRef] [PubMed]
- Onuma, S.; Manabe, A.; Yoshino, Y.; Matsunaga, T.; Asai, T.; Ikari, A. Upregulation of chemoresistance by Mg2+ deficiency through elevation of ATP binding casssette subfamily B member 1 expression in human lung adenocarcinoma A549 cells. Cells 2021, 10, 1179. [Google Scholar] [CrossRef] [PubMed]
- Bruton, J.; Cheng, A.J.; Westerblad, H. Measuring Ca2+ in living cells. Adv. Exp. Med. Biol. 2020, 1131, 7–26. [Google Scholar] [PubMed]
- Bischof, H.; Burgstaller, S.; Weiermair, M.W.; Rauter, T.; Schinagl, M.; Muja, J.R.; Graier, W.F.; Malli, R. Live-cell imaging of physiologically relevant metal ions using genetically encoded FRET-based probes. Cells 2019, 8, 492. [Google Scholar] [CrossRef] [Green Version]
- ’t Hoen, C.P.A.; Kort, F.; van Ommen, G.J.B.; Dunnen, J.T. Fluorescent labelling of cRNA for microarray applications. Nucleic Acids Res. 2003, 31, e20. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, Y.; Liu, Y. Incorporation of a FRET Pair into a Riboswitch RNA to Measure Mg2+ Concentration and RNA Conformational Change in Cell. Int. J. Mol. Sci. 2022, 23, 1493. https://doi.org/10.3390/ijms23031493
Xue Y, Liu Y. Incorporation of a FRET Pair into a Riboswitch RNA to Measure Mg2+ Concentration and RNA Conformational Change in Cell. International Journal of Molecular Sciences. 2022; 23(3):1493. https://doi.org/10.3390/ijms23031493
Chicago/Turabian StyleXue, Yanyan, and Yu Liu. 2022. "Incorporation of a FRET Pair into a Riboswitch RNA to Measure Mg2+ Concentration and RNA Conformational Change in Cell" International Journal of Molecular Sciences 23, no. 3: 1493. https://doi.org/10.3390/ijms23031493
APA StyleXue, Y., & Liu, Y. (2022). Incorporation of a FRET Pair into a Riboswitch RNA to Measure Mg2+ Concentration and RNA Conformational Change in Cell. International Journal of Molecular Sciences, 23(3), 1493. https://doi.org/10.3390/ijms23031493