
The T850D Phosphomimetic Mutation in the Androgen Receptor Ligand Binding Domain Enhances Recruitment at Activation Function 2

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
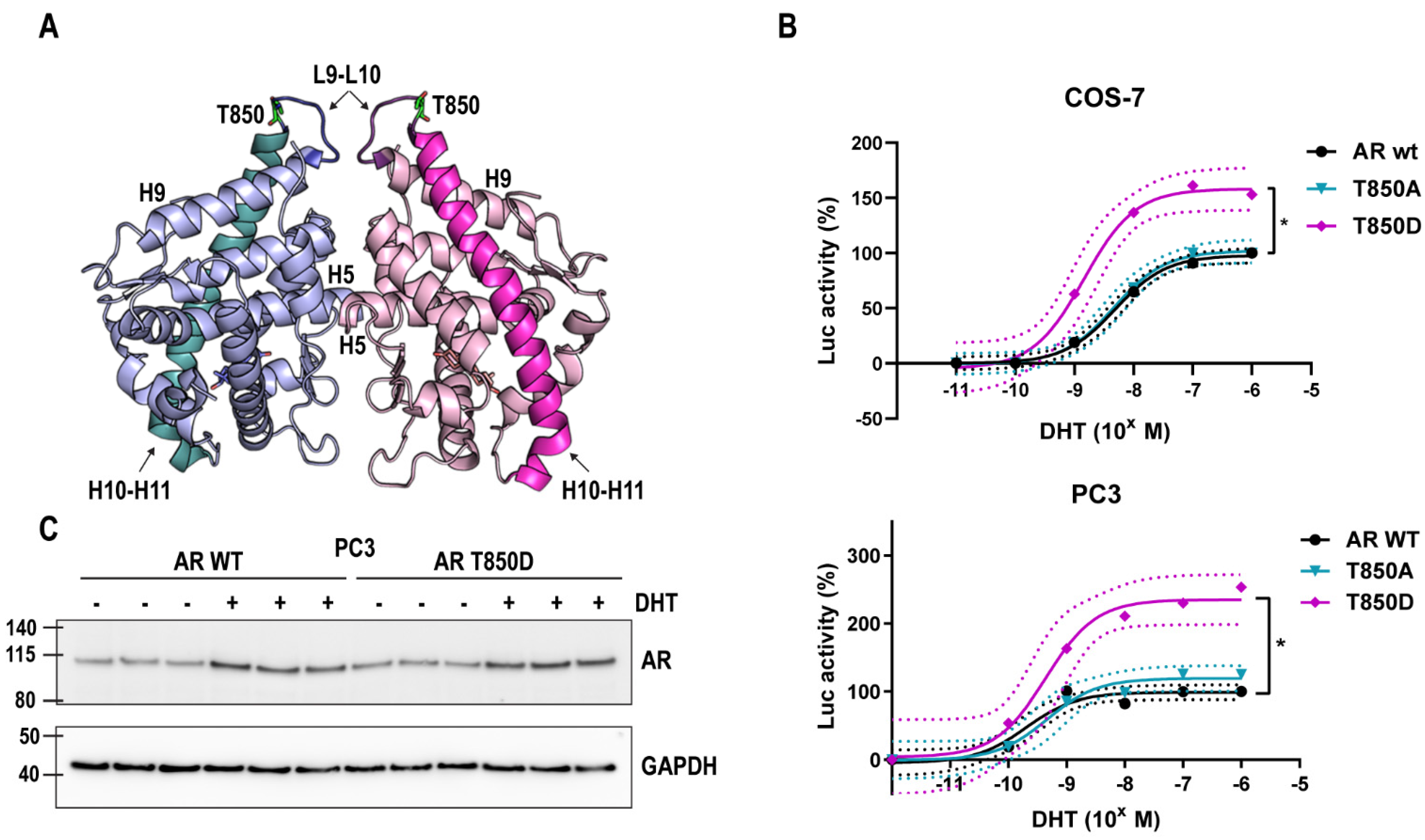
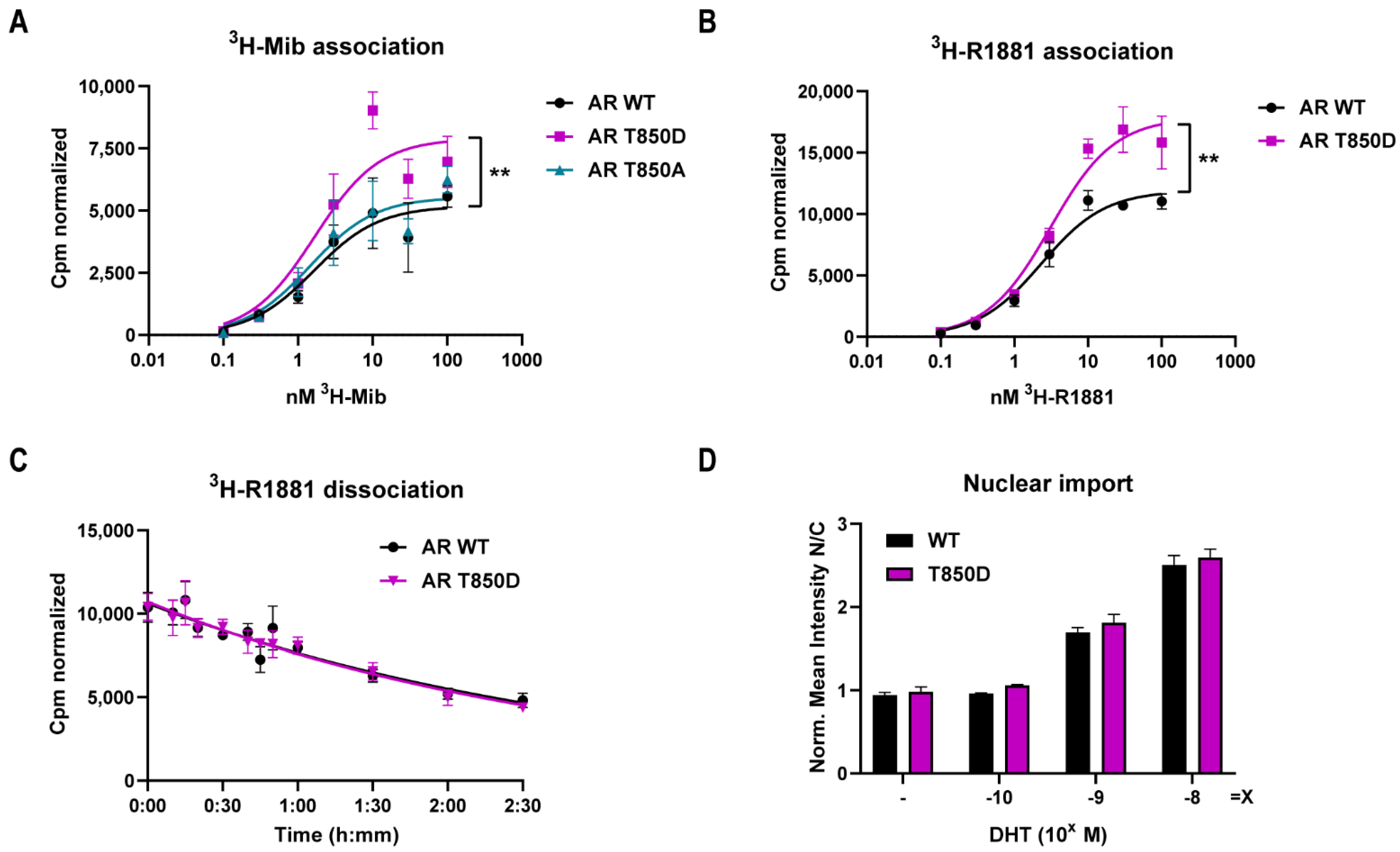
2.1. T850D Increases AR Activity
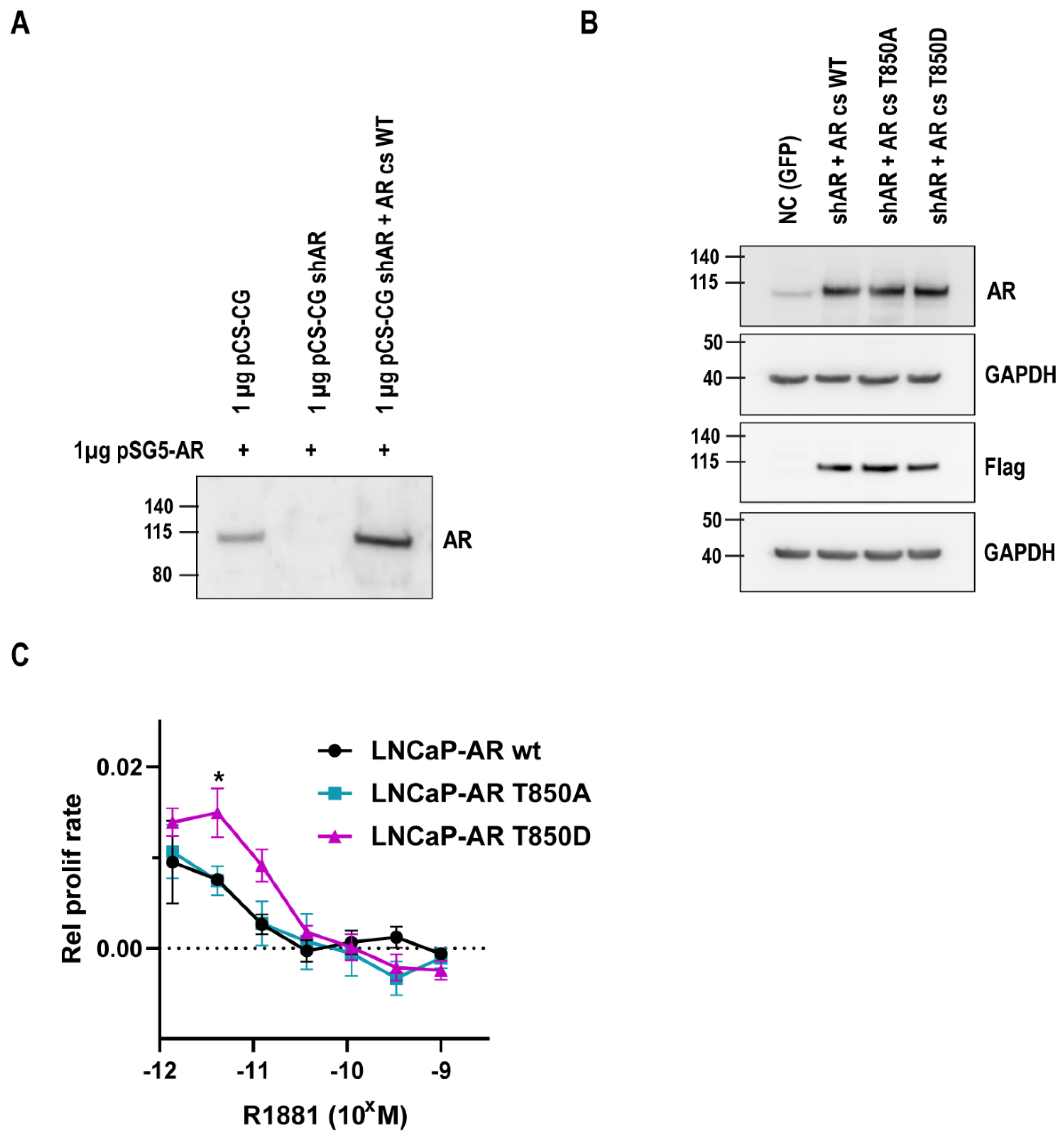
2.2. Relevance of AR T850D in PCa Cells
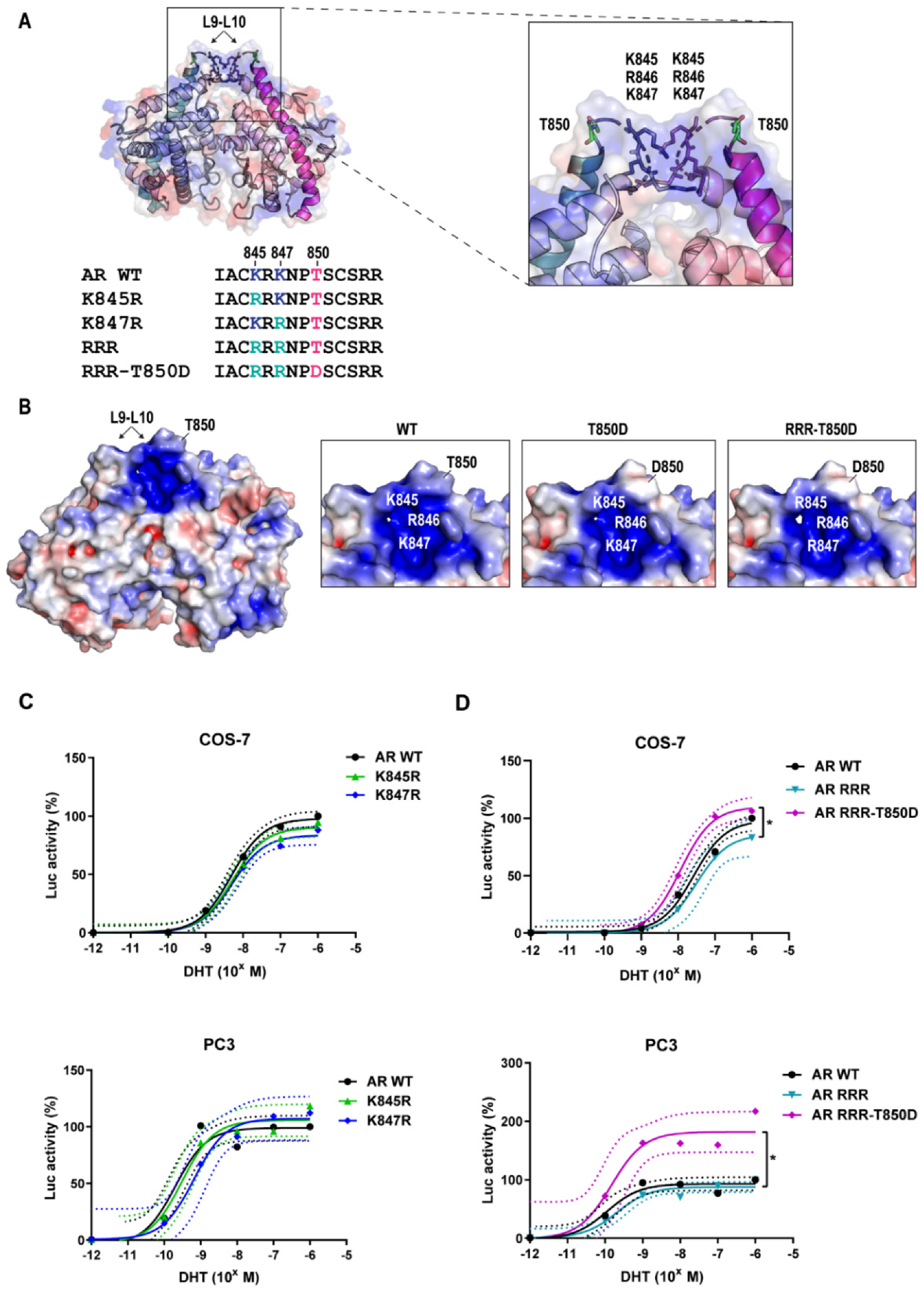
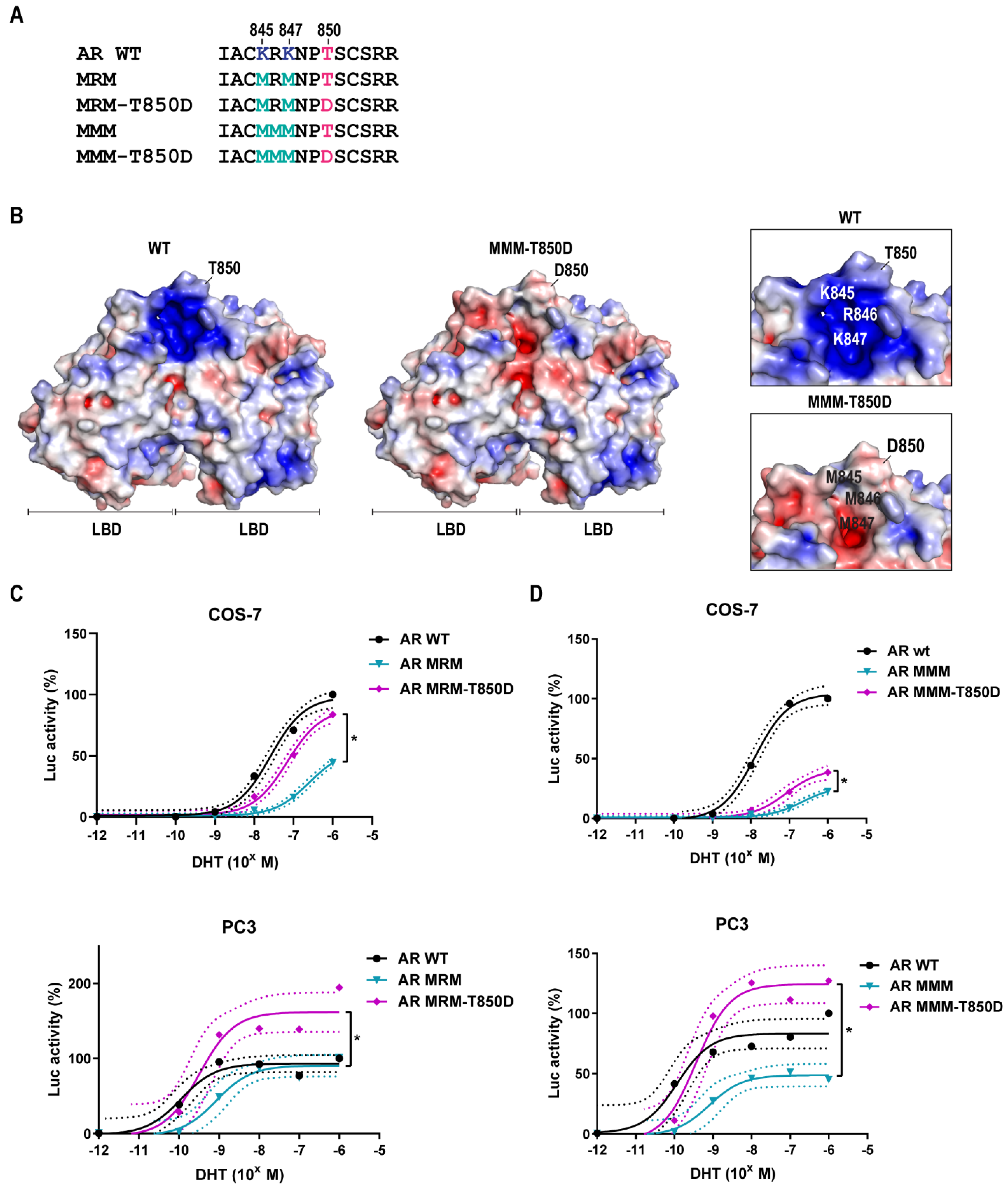
2.3. AR Gain-of-Function by T850D Is Independent from the Ubiquitination Sites K845 and K847
2.4. Is T850D Part of the LBD Dimerization Interface?
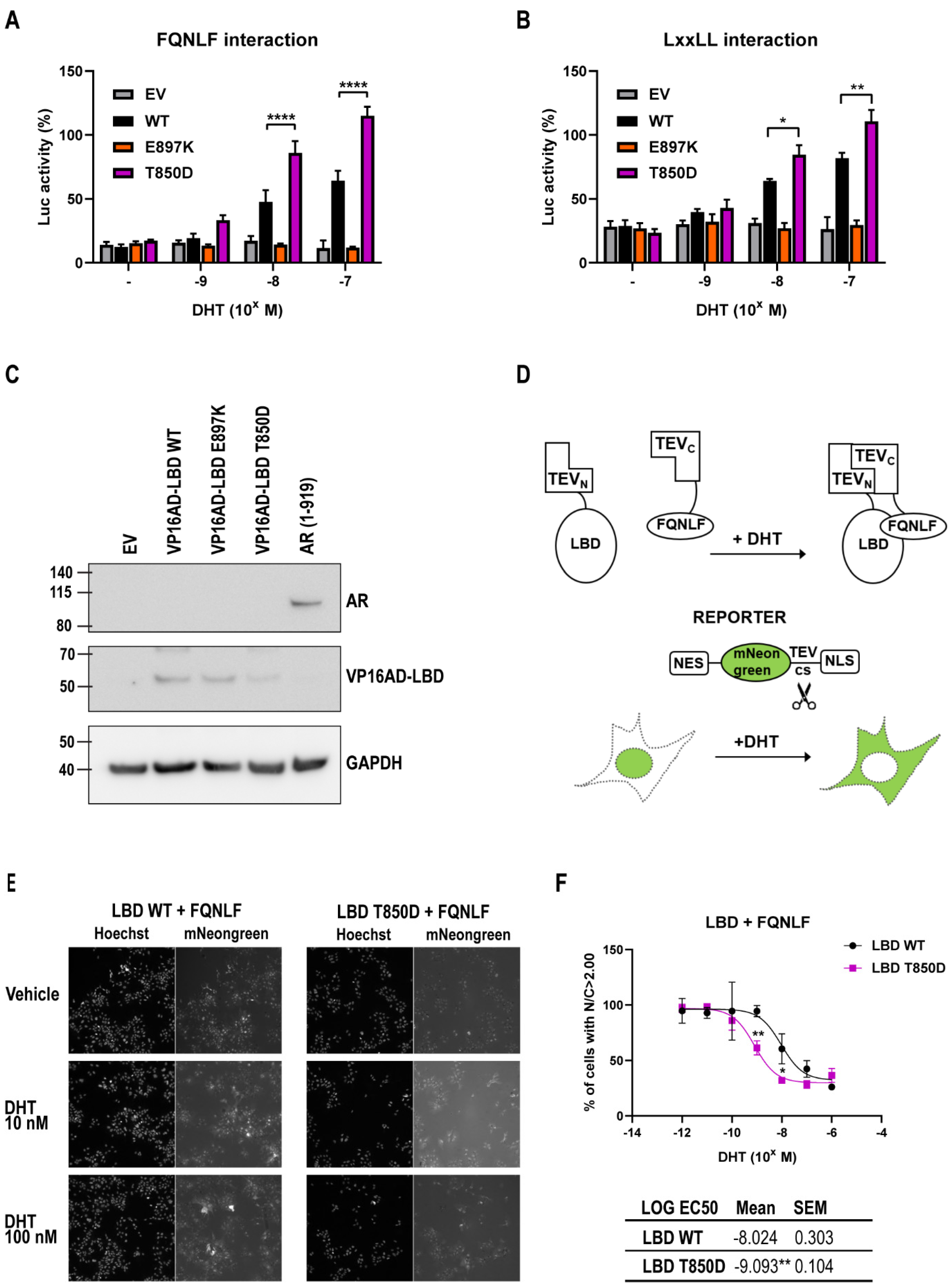
2.5. AR T850D Gain of Function Due to Increased AF2 Interactions
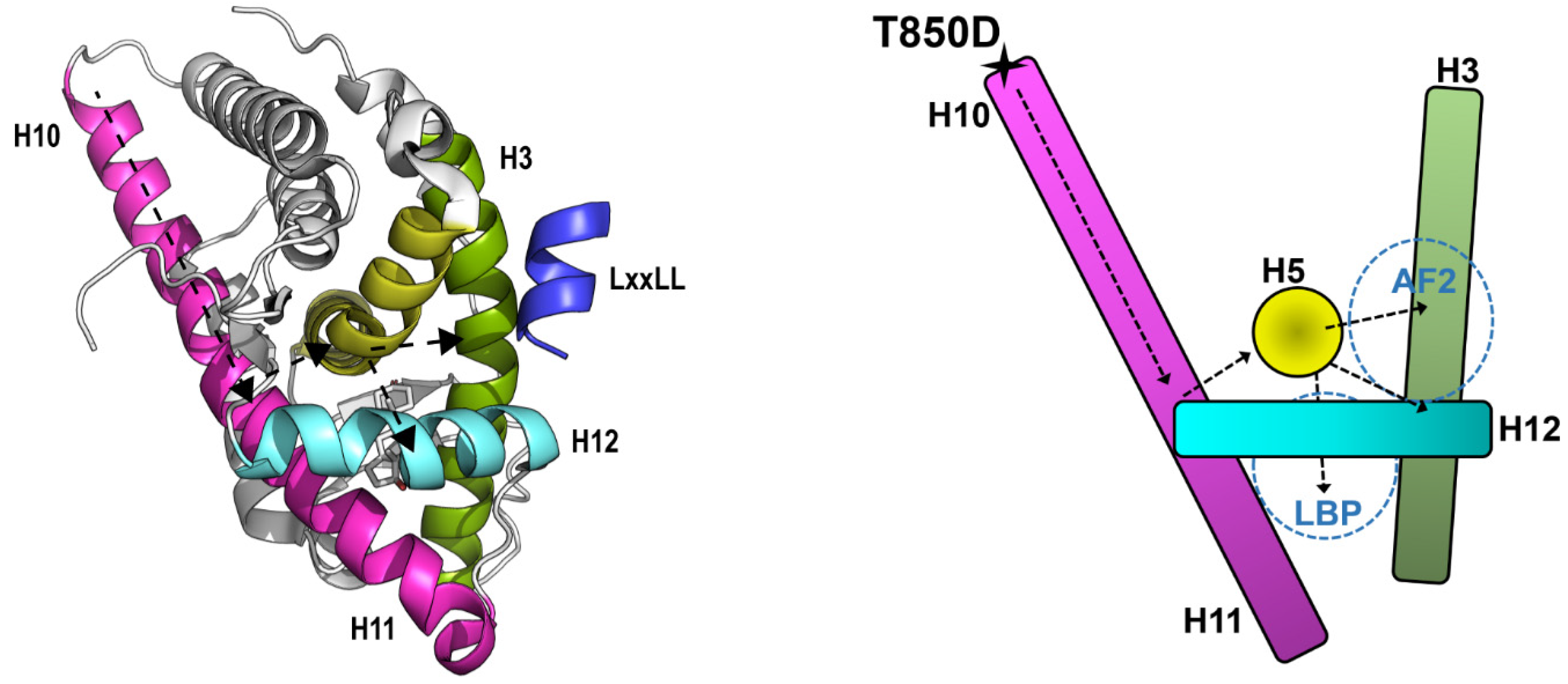
2.6. Effects of T850D and T850 Phosphorylation on H10 and LBD Structure
3. Discussion
3.1. T850D Potentiates AR Independent from Reported Mechanisms
3.2. T850D Facilitates AF2 Binding by Allosteric Signaling
3.3. T850 Phosphorylation in PCa
4. Materials and Methods
4.1. Cells and Plasmids
4.2. Transactivation Study
4.3. Western Blot
4.4. Generation of Lentiviral LNCaP Cell Lines
4.5. Proliferation Measurements
4.6. Ligand Binding and Dissociation
4.7. Nuclear Translocation
4.8. Mammalian Double Hybrid for LBD Interaction with FQNLF or LXXLL
4.9. Split-TEV Assay for LBD-FQNLF Interaction
4.10. Circular Dichroism Spectroscopy
4.11. MD Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baker, M.E. Steroid receptors and vertebrate evolution. Mol. Cell. Endocrinol. 2019, 496, 110526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.H.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.-L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centenera, M.M.; Harris, J.M.; Tilley, W.D.; Butler, L.M. The contribution of different androgen receptor domains to receptor dimerization and signaling. Mol. Endocrinol. 2008, 22, 2373–2382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helsen, C.; Kerkhofs, S.; Clinckemalie, L.; Spans, L.; Laurent, M.; Boonen, S.; Vanderschueren, D.; Claessens, F. Structural basis for nuclear hormone receptor DNA binding. Mol. Cell. Endocrinol. 2012, 348, 411–417. [Google Scholar] [CrossRef] [PubMed]
- El Kharraz, S.; Dubois, V.; Royen, M.E.; Houtsmuller, A.B.; Pavlova, E.; Atanassova, N.; Nguyen, T.; Voet, A.; Eerlings, R.; Handle, F.; et al. The androgen receptor depends on ligand-binding domain dimerization for transcriptional activation. EMBO Rep. 2021, 22, e52764. [Google Scholar] [CrossRef] [PubMed]
- Pereira de Jésus-Tran, K.; Côté, P.-L.; Cantin, L.; Blanchet, J.; Labrie, F.; Breton, R. Comparison of crystal structures of human androgen receptor ligand-binding domain complexed with various agonists reveals molecular determinants responsible for binding affinity. Protein Sci. 2006, 15, 987–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohl, C.E.; Wu, Z.; Miller, D.D.; Bell, C.E.; Dalton, J.T. Crystal structure of the T877A human androgen receptor ligand-binding domain complexed to cyproterone acetate provides insight for ligand-induced conformational changes and structure-based drug design. J. Biol. Chem. 2007, 282, 13648–13655. [Google Scholar] [CrossRef] [Green Version]
- Bohl, C.E.; Gao, W.; Miller, D.D.; Bell, C.E.; Dalton, J.T. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 6201–6206. [Google Scholar] [CrossRef] [Green Version]
- Helsen, C.; Broeck, T.V.D.; Voet, A.; Prekovic, S.; Van Poppel, H.; Joniau, S.; Claessens, F. Androgen receptor antagonists for prostate cancer therapy. Endocr. Relat. Cancer 2014, 21, T105–T118. [Google Scholar] [CrossRef] [Green Version]
- Nadal, M.; Prekovic, S.; Gallastegui, N.; Helsen, C.; Abella, M.; Zielinska, K.; Gay, M.; Vilaseca, M.; Taulès, M.; Houtsmuller, A.B.; et al. Structure of the homodimeric androgen receptor ligand-binding domain. Nat Commun. 2017, 8, 14388. [Google Scholar] [CrossRef]
- Estebanez-Perpina, E.; Arnold, L.; Nguyen, P.; Rodrigues, E.; Mar, E.; Bateman, R.; Pallai, P.; Shokat, K.M.; Baxter, J.D.; Guy, R.; et al. A surface on the androgen receptor that allosterically regulates coactivator binding. Proc. Natl. Acad. Sci. USA 2007, 104, 16074–16079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubbink, H.E.J.; Hersmus, R.; Verma, C.; Van Der Korput, H.; Berrevoets, C.; Van Tol, J.; Der Made, A.Z.-V.; Brinkmann, A.; Pike, A.; Trapman, J. Distinct recognition modes of FXXLF and LXXLL motifs by the androgen receptor. Mol. Endocrinol. 2004, 18, 2132–2150. [Google Scholar] [CrossRef] [Green Version]
- Nemoto, T.; Ohara-Nemoto, Y.; Ota, M. Association of the 90-kDa heat shock protein does not affect the ligand-binding ability of androgen receptor. J. Steroid. Biochem. Mol. Biol. 1992, 42, 803–812. [Google Scholar] [CrossRef]
- He, B.; Kemppainen, J.A.; Wilson, E.M. FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J. Biol. Chem. 2000, 275, 22986–22994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doesburg, P.; Kuil, C.W.; Berrevoets, C.A.; Steketee, K.; Faber, P.W.; Mulder, E.; Brinkmann, A.A.O.; Trapman, J. Functional in vivo interaction between the amino-terminal, transactivation domain and the ligand binding domain of the androgen receptor. Biochemistry 1997, 36, 1052–1064. [Google Scholar] [CrossRef]
- Matsushita, S.; Suzuki, K.; Murashima, A.; Kajioka, D.; Acebedo, A.R.; Miyagawa, S.; Haraguchi, R.; Ogino, Y.; Yamada, G. Regulation of masculinization: Androgen signalling for external genitalia development. Nat. Rev. Urol. 2018, 15, 358–368. [Google Scholar] [CrossRef]
- O’Hara, L.; Smith, L.B. Androgen receptor roles in spermatogenesis and infertility. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 595–605. [Google Scholar] [CrossRef] [Green Version]
- Monks, D.A.; Holmes, M.M. Androgen receptors and muscle: A key mechanism underlying life history trade-offs. J. Comp. Physiol. A Neuroethol. Sens. Neural. Behav. Physiol. 2018, 204, 51–60. [Google Scholar] [CrossRef]
- Chen, J.-F.; Lin, P.-W.; Tsai, Y.-R.; Yang, Y.-C.; Kang, H.-Y. Androgens and Androgen Receptor Actions on Bone Health and Disease: From Androgen Deficiency to Androgen Therapy. Cells 2019, 8, 1318. [Google Scholar] [CrossRef] [Green Version]
- Mhaouty-Kodja, S. Role of the androgen receptor in the central nervous system. Mol. Cell. Endocrinol. 2018, 465, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Steinkamp, M.; O’Mahony, O.A.; Brogley, M.; Rehman, H.; LaPensee, E.W.; Dhanasekaran, S.M.; Hofer, M.D.; Kuefer, R.; Chinnaiyan, A.; Rubin, M.; et al. Treatment-dependent androgen receptor mutations in prostate cancer exploit multiple mechanisms to evade therapy. Cancer Res. 2009, 69, 4434–4442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyytinen, E.-R.; Haapala, K.; Thompson, J.; Lappalainen, I.; Roiha, M.; Rantala, I.; Helin, H.J.; Jänne, O.A.; Vihinen, M.; Palvimo, J.; et al. Pattern of somatic androgen receptor gene mutations in patients with hormone-refractory prostate cancer. Lab. Investig. 2002, 82, 1591–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, B.; Beitel, L.K.; Nadarajah, A.; Paliouras, M.; Trifiro, M. The androgen receptor gene mutations database: 2012 update. Hum. Mutat. 2012, 33, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, G.; Sun, T. Dissecting the roles of the androgen receptor in prostate cancer from molecular perspectives. Tumour Biol. 2017, 39, 1010428317692259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, D.T.; Iczkowski, K.A.; Kilari, D.; See, W.; Nevalainen, M.T. Androgen receptor-dependent and -independent mechanisms driving prostate cancer progression: Opportunities for therapeutic targeting from multiple angles. Oncotarget 2017, 8, 3724–3745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koryakina, Y.; Ta, H.Q.; Gioeli, D. Androgen receptor phosphorylation: Biological context and functional consequences. Endocr. Relat. Cancer 2014, 21, T131–T145. [Google Scholar] [CrossRef]
- Linn, D.E.; Yang, X.; Xie, Y.; Alfano, A.; Deshmukh, D.; Wang, X.; Shimelis, H.; Chen, H.; Li, W.; Xu, K.; et al. Differential regulation of androgen receptor by PIM-1 kinases via phosphorylation-dependent recruitment of distinct ubiquitin E3 ligases. J. Biol. Chem. 2012, 287, 22959–22968. [Google Scholar] [CrossRef] [Green Version]
- Blanco-Aparicio, C.; Carnero, A. Pim kinases in cancer: Diagnostic, prognostic and treatment opportunities. Biochem. Pharmacol. 2013, 85, 629–643. [Google Scholar] [CrossRef]
- Bullock, A.N.; Debreczeni, J.; Amos, A.L.; Knapp, S.; Turk, B.E. Structure and substrate specificity of the Pim-1 kinase. J. Biol. Chem. 2005, 280, 41675–41682. [Google Scholar] [CrossRef] [Green Version]
- Holder, S.L.; Abdulkadir, S.A. PIM1 kinase as a target in prostate cancer: Roles in tumorigenesis, castration resistance, and docetaxel resistance. Curr. Cancer. Drug Targets 2014, 14, 105–114. [Google Scholar] [CrossRef]
- Zemskova, M.; Sahakian, E.; Bashkirova, S.; Lilly, M. The PIM1 kinase is a critical component of a survival pathway activated by docetaxel and promotes survival of docetaxel-treated prostate cancer cells. J. Biol. Chem. 2008, 283, 20635–20644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Xu, K.; Dai, B.; Guo, Z.; Jiang, T.; Chen, H.; Qiu, Y. The 44 kDa Pim-1 kinase directly interacts with tyrosine kinase Etk/BMX and protects human prostate cancer cells from apoptosis induced by chemotherapeutic drugs. Oncogene 2006, 25, 70–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.-L.; Leong, P.-K.; Ho, Y.-F.; Hsu, L.-C.; Lu, P.-H.; Chen, C.-S.; Guh, J.-H. Pim-1 knockdown potentiates paclitaxel-induced apoptosis in human hormone-refractory prostate cancers through inhibition of NHEJ DNA repair. Cancer Lett. 2012, 319, 214–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The PyMOL Molecular Graphics System, Version 2.4.0 Schrödinger, LLC. Available online: www.Pymol.org (accessed on 27 October 2021).
- Xu, K.; Shimelis, H.; Linn, D.E.; Jiang, R.; Yang, X.; Sun, F.; Guo, Z.; Chen, H.; Li, W.; Chen, H.; et al. Regulation of androgen receptor transcriptional activity and specificity by RNF6-induced ubiquitination. Cancer Cell 2009, 15, 270–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Wijngaart, D.J.; Dubbink, H.J.; van Royen, M.E.; Trapman, J.; Jenster, G. Androgen receptor coregulators: Recruitment via the coactivator binding groove. Mol. Cell. Endocrinol. 2012, 352, 57–69. [Google Scholar] [CrossRef]
- He, B.; Wilson, E.M. The NH(2)-terminal and carboxyl-terminal interaction in the human androgen receptor. Mol. Genet. Metab. 2002, 75, 293–298. [Google Scholar] [CrossRef]
- He, B.; Gampe, R.T.; Hnat, A.T.; Faggart, J.L.; Minges, J.T.; French, F.S.; Wilson, E.M. Probing the functional link between androgen receptor coactivator and ligand-binding sites in prostate cancer and androgen insensitivity. J. Biol. Chem. 2006, 281, 6648–6663. [Google Scholar] [CrossRef] [Green Version]
- Munoz, V.; Serrano, L. Elucidating the folding problem of helical peptides using empirical parameters. II. Helix macrodipole effects and rational modification of the helical content of natural peptides. J. Mol. Biol. 1995, 245, 275–296. [Google Scholar] [CrossRef] [Green Version]
- Sommese, R.F.; Sivaramakrishnan, S.; Baldwin, R.L.; Spudich, J.A. Helicity of short E-R/K peptides. Protein Sci. 2010, 19, 2001–2005. [Google Scholar] [CrossRef] [Green Version]
- Bianchetti, L.; Sinar, D.; Depenveiller, C.; Dejaegere, A. Insights into mineralocorticoid receptor homodimerization from a combined molecular modeling and bioinformatics study. Proteins 2021. [CrossRef]
- Kojetin, D.J.; Matta-Camacho, E.; Hughes, T.; Srinivasan, S.; Nwachukwu, J.; Cavett, V.; Nowak, J.; Chalmers, M.J.; Marciano, D.; Kamenecka, T.M.; et al. Structural mechanism for signal transduction in RXR nuclear receptor heterodimers. Nat. Commun. 2015, 6, 8013. [Google Scholar] [CrossRef] [Green Version]
- Nettles, K.W.; Greene, G.L. Ligand control of coregulator recruitment to nuclear receptors. Annu. Rev. Physiol. 2005, 67, 309–333. [Google Scholar] [CrossRef] [PubMed]
- Shulman, A.I.; Larson, C.; Mangelsdorf, D.J.; Ranganathan, R. Structural determinants of allosteric ligand activation in RXR heterodimers. Cell 2004, 116, 417–429. [Google Scholar] [CrossRef] [Green Version]
- Gnome, A.D. Available online: https://gnomad.broadinstitute.org/ (accessed on 27 October 2021).
- McGill Androgen Receptor Gene Mutation Database. Available online: http://androgendb.mcgill.ca/ (accessed on 27 October 2021).
- van der Poel, H.G.; Zevenhoven, J.; Bergman, A.M. Pim1 regulates androgen-dependent survival signaling in prostate cancer cells. Urol. Int. 2010, 84, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Markou, A.; Tzanikou, E.; Strati, A.; Zavridou, M.; Mastoraki, S.; Bournakis, E.; Lianidou, E. PIM-1 Is Overexpressed at a High Frequency in Circulating Tumor Cells from Metastatic Castration-Resistant Prostate Cancer Patients. Cancers 2020, 12, 1188. [Google Scholar] [CrossRef] [PubMed]
- De Velasco, M.A.; Kura, Y.; Ando, N.; Sako, N.; Banno, E.; Fujita, K.; Nozawa, M.; Yoshimura, K.; Sakai, K.; Yoshikawa, K.; et al. Context-Specific Efficacy of Apalutamide Therapy in Preclinical Models of Pten-Deficient Prostate Cancer. Cancers 2021, 13, 3975. [Google Scholar] [CrossRef] [PubMed]
- McCall, P.; Adams, C.E.; Willder, J.M.; Bennett, L.; Qayyum, T.; Orange, C.; Underwood, M.A.; Edwards, J. Androgen receptor phosphorylation at serine 308 and serine 791 predicts enhanced survival in castrate resistant prostate cancer patients. Int. J. Mol. Sci. 2013, 14, 16656–16671. [Google Scholar] [CrossRef] [Green Version]
- Tanner, T.M.; Denayer, S.; Geverts, B.; Van Tilborgh, N.; Kerkhofs, S.; Helsen, C.; Spans, L.; Dubois, V.; Houtsmuller, A.B.; Claessens, F.; et al. A 629RKLKK633 motif in the hinge region controls the androgen receptor at multiple levels. Cell. Mol. Life Sci. 2010, 67, 1919–1927. [Google Scholar] [CrossRef]
- Denayer, S.; Helsen, C.; Thorrez, L.; Haelens, A.; Claessens, F. The rules of DNA recognition by the androgen receptor. Mol. Endocrinol. 2010, 24, 898–913. [Google Scholar] [CrossRef] [Green Version]
- Baniahmad, A.; Kohne, A.C.; Renkawitz, R. A transferable silencing domain is present in the thyroid hormone receptor, in the v-erbA oncogene product and in the retinoic acid receptor. EMBO J. 1992, 11, 1015–1023. [Google Scholar] [CrossRef]
- Kalkhoven, E.; Valentine, J.E.; Heery, D.M.; Parker, M.G. Isoforms of steroid receptor co-activator 1 differ in their ability to potentiate transcription by the oestrogen receptor. EMBO J. 1998, 17, 232–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alen, P.; Claessens, F.; Verhoeven, G.; Rombauts, W.; Peeters, B. The androgen receptor amino-terminal domain plays a key role in p160 coactivator-stimulated gene transcription. Mol. Cell. Biol. 1999, 19, 6085–6097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, H.; Blömer, U.; Takahashi, M.; Gage, F.H.; Verma, I.M. Development of a self-inactivating lentivirus vector. J. Virol. 1998, 72, 8150–8157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Dai, B.; Jiang, T.; Xu, K.; Xie, Y.; Kim, O.; Nesheiwat, I.; Kong, X.; Melamed, J.; Handratta, V.D.; et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell. 2006, 10, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reya, T.; Duncan, A.; Ailles, L.; Domen, J.; Scherer, D.C.; Willert, K.; Hintz, L.; Nusse, R.; Weissman, I.L. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 2003, 423, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE), C.C.G.U. 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7. 2020. Available online: https://www.chemcomp.com/Products.htm (accessed on 27 October 2021).
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single-Crystals—A New Molecular-Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Hess, B. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald—An N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Grosdidier, S.; Carbó, L.R.; Buzón, V.; Brooke, G.; Nguyen, P.; Baxter, J.D.; Bevan, C.; Webb, P.; Estébanez-Perpiñá, E.; Fernández-Recio, J. Allosteric conversation in the androgen receptor ligand-binding domain surfaces. Mol. Endocrinol. 2012, 26, 1078–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3H-Mib Binding | 3H-R1881 Binding | 3H-R1881 Dissociation | |||
|---|---|---|---|---|---|
| KD (nM) | Bmax (cpm) | KD (nM) | Bmax (cpm) | t1/2 (min) | |
| AR WT | 1.65 (±0.92) | 5182 (±602) | 2.32 (±0.43) | 11,905 (±488) | 126.4 (±21.4) |
| AR T850A | 1.45 (±0.72) | 5514 (±561) | |||
| AR T850D | 1.67 (±0.74) | 7902 (±773) ** | 3.11 (±0.72) | 17,849 (±959) ** | 120.8 (±21.7) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Helsen, C.; Nguyen, T.; Vercruysse, T.; Wouters, S.; Daelemans, D.; Voet, A.; Claessens, F. The T850D Phosphomimetic Mutation in the Androgen Receptor Ligand Binding Domain Enhances Recruitment at Activation Function 2. Int. J. Mol. Sci. 2022, 23, 1557. https://doi.org/10.3390/ijms23031557
Helsen C, Nguyen T, Vercruysse T, Wouters S, Daelemans D, Voet A, Claessens F. The T850D Phosphomimetic Mutation in the Androgen Receptor Ligand Binding Domain Enhances Recruitment at Activation Function 2. International Journal of Molecular Sciences. 2022; 23(3):1557. https://doi.org/10.3390/ijms23031557
Chicago/Turabian StyleHelsen, Christine, Tien Nguyen, Thomas Vercruysse, Staf Wouters, Dirk Daelemans, Arnout Voet, and Frank Claessens. 2022. "The T850D Phosphomimetic Mutation in the Androgen Receptor Ligand Binding Domain Enhances Recruitment at Activation Function 2" International Journal of Molecular Sciences 23, no. 3: 1557. https://doi.org/10.3390/ijms23031557
APA StyleHelsen, C., Nguyen, T., Vercruysse, T., Wouters, S., Daelemans, D., Voet, A., & Claessens, F. (2022). The T850D Phosphomimetic Mutation in the Androgen Receptor Ligand Binding Domain Enhances Recruitment at Activation Function 2. International Journal of Molecular Sciences, 23(3), 1557. https://doi.org/10.3390/ijms23031557