
Cellular and Molecular Signatures of Oxidative Stress in Bronchial Epithelial Cell Models Injured by Cigarette Smoke Extract

 ,
,  ,
,  ,
,  , and
, and
Abstract
:1. Introduction
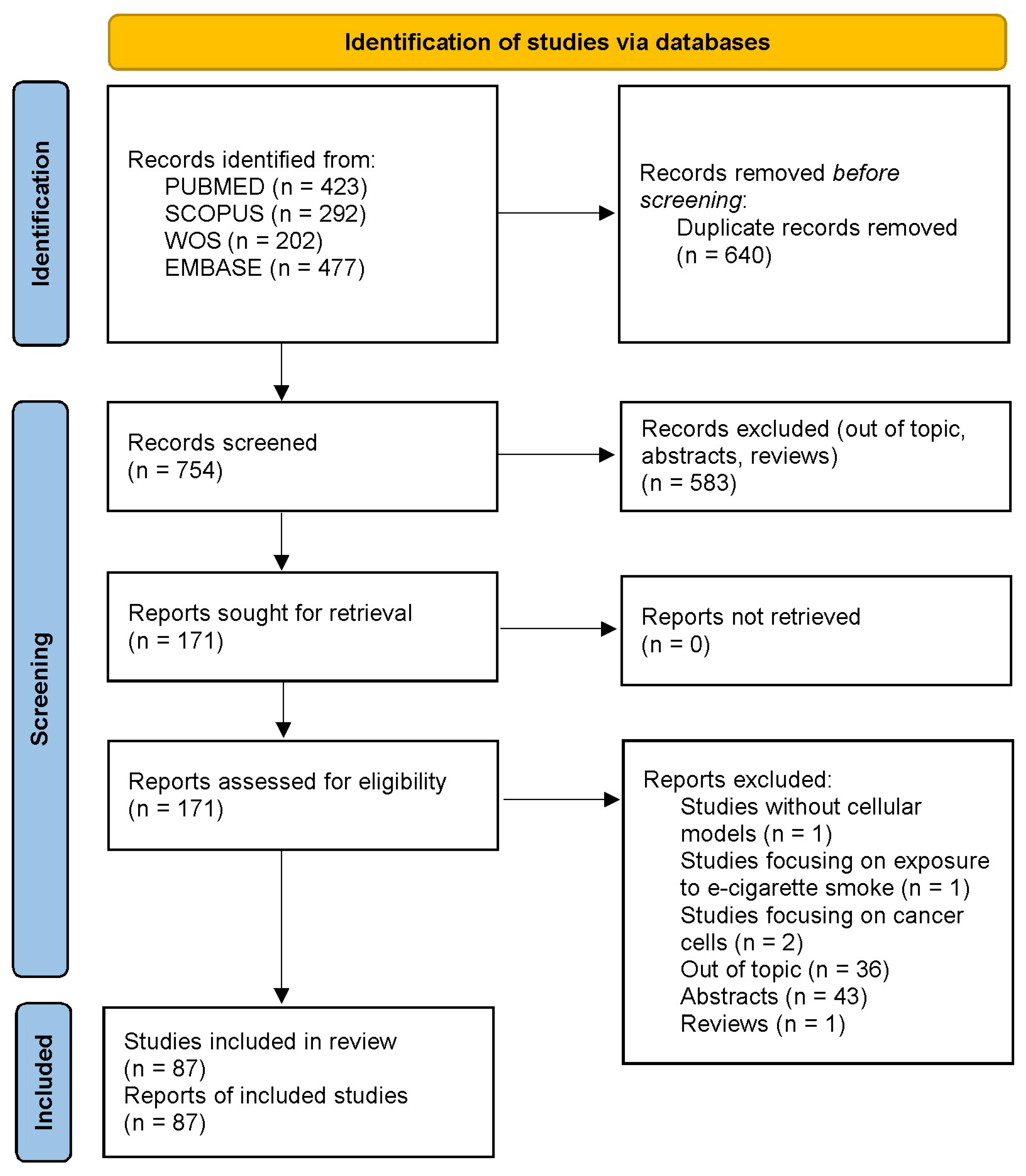
2. Materials and Methods
3. Results
3.1. Cellular Models of Bronchial Epithelium Exposed to CSE: Primary Cell Cultures and Cell Lines, 2D and 3D Models
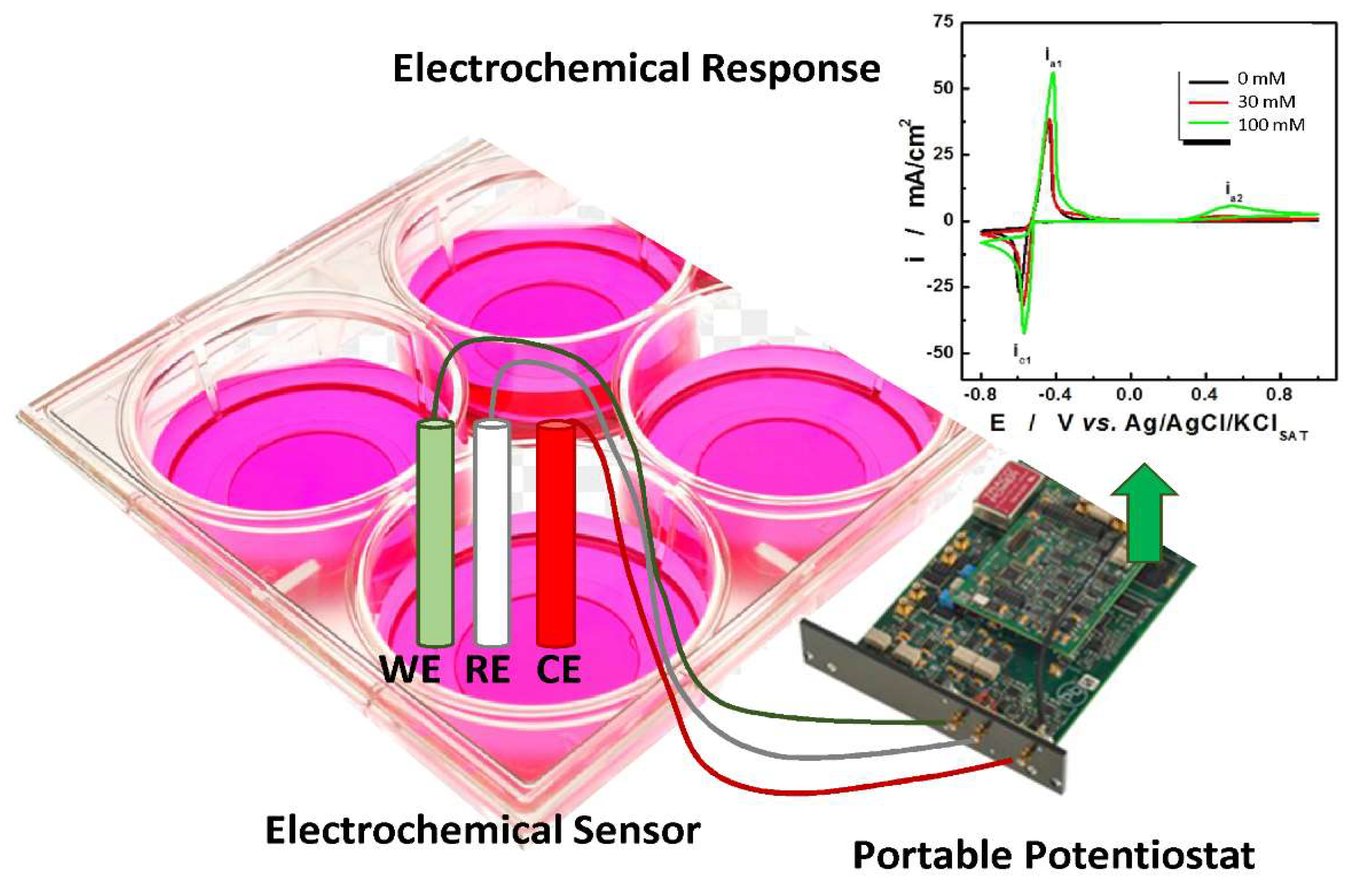
3.2. Methods for Detecting Oxidative Stress
Innovative Methods for the Detection of Oxidative Stress
3.3. Molecular Alterations Induced by CS Exposure
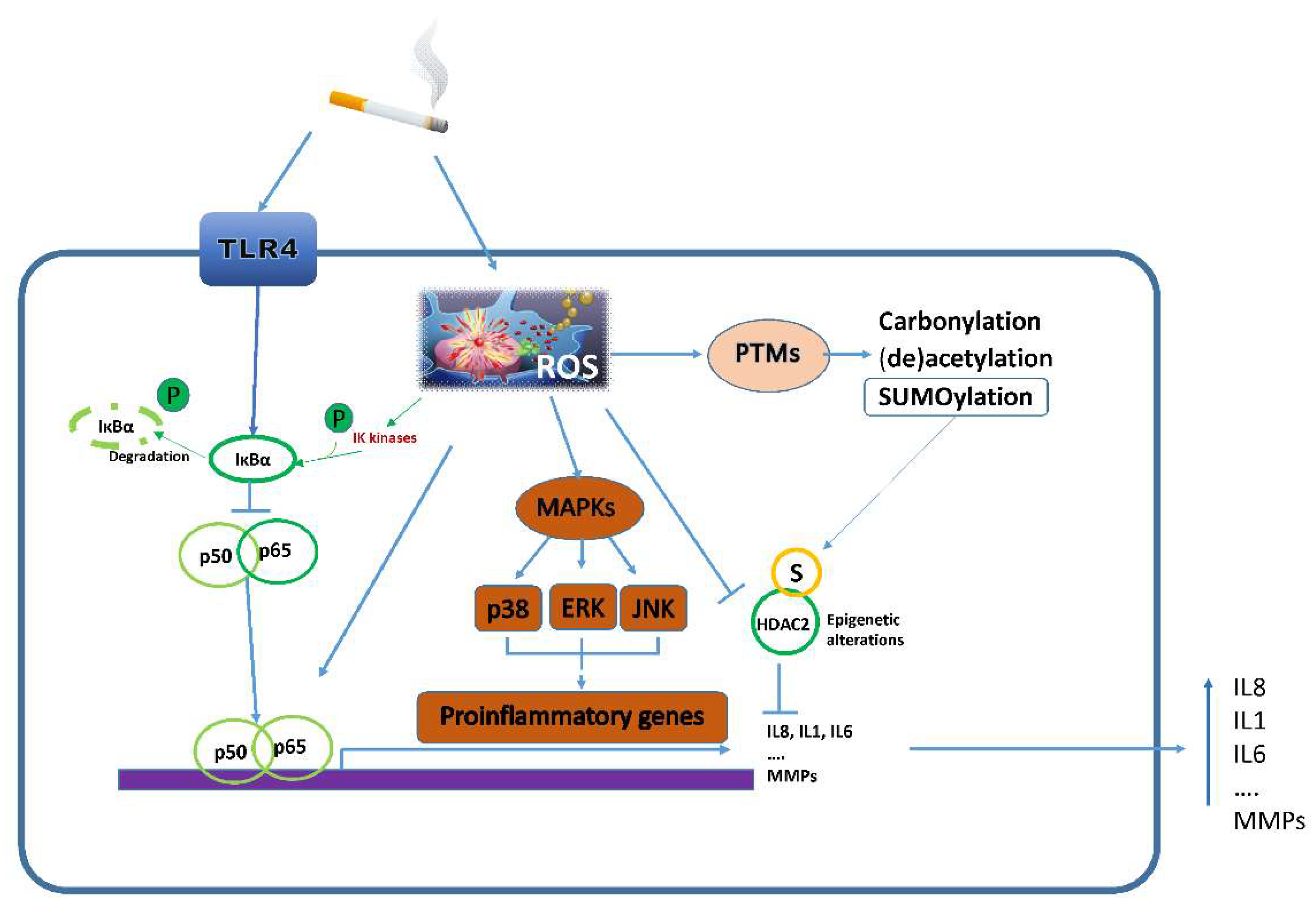
3.3.1. Inflammatory Pathways
3.3.2. Non-Coding RNAs
3.3.3. Post-Translational Modifications
3.3.4. Epigenetic Alterations
3.4. Impact of CS on Cellular Homeostasis
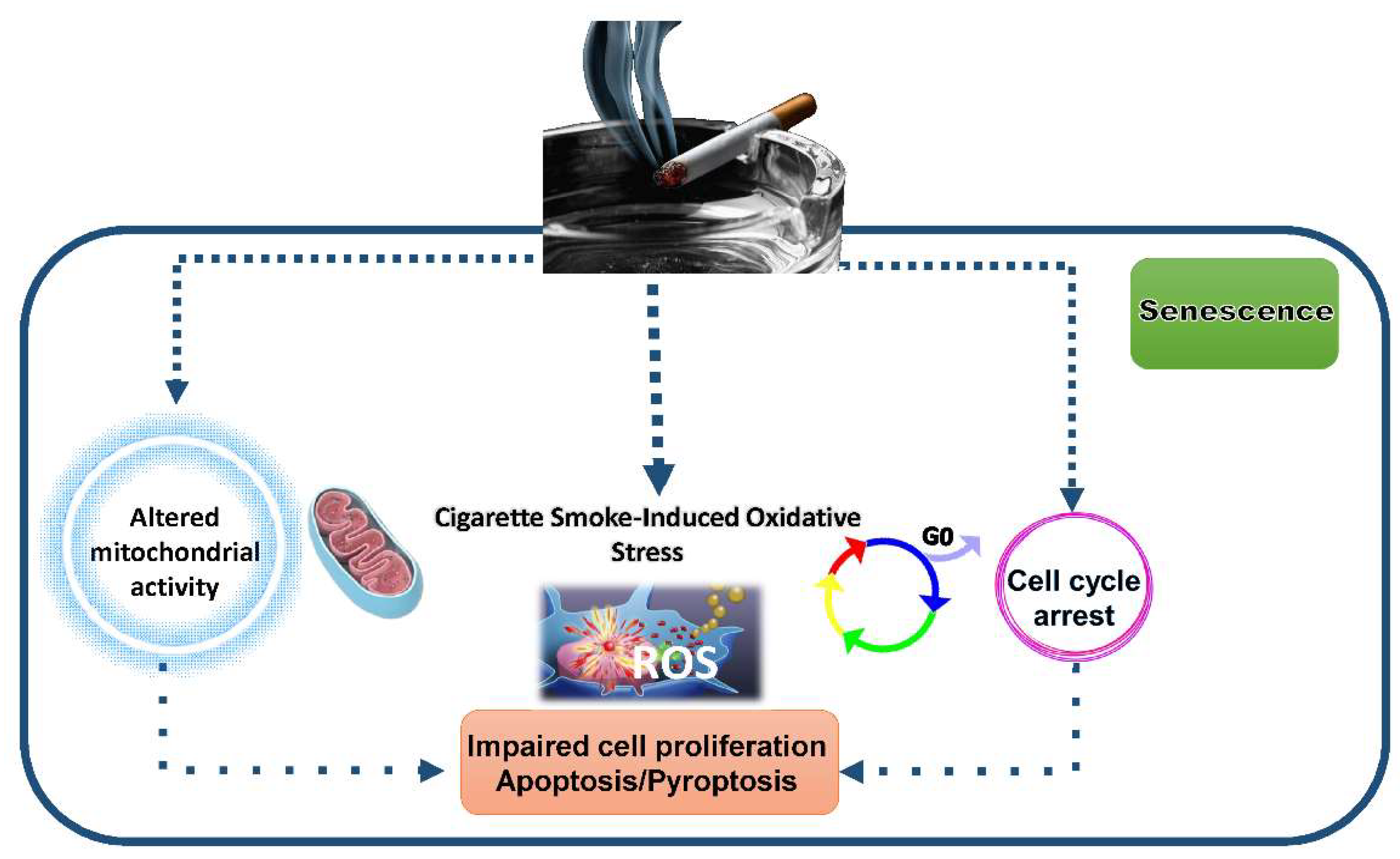
3.4.1. Impact of CSE on Autophagy, Mitophagy and Mitochondrial Activity
3.4.2. Impact of CSE on Senescence and Cell Death
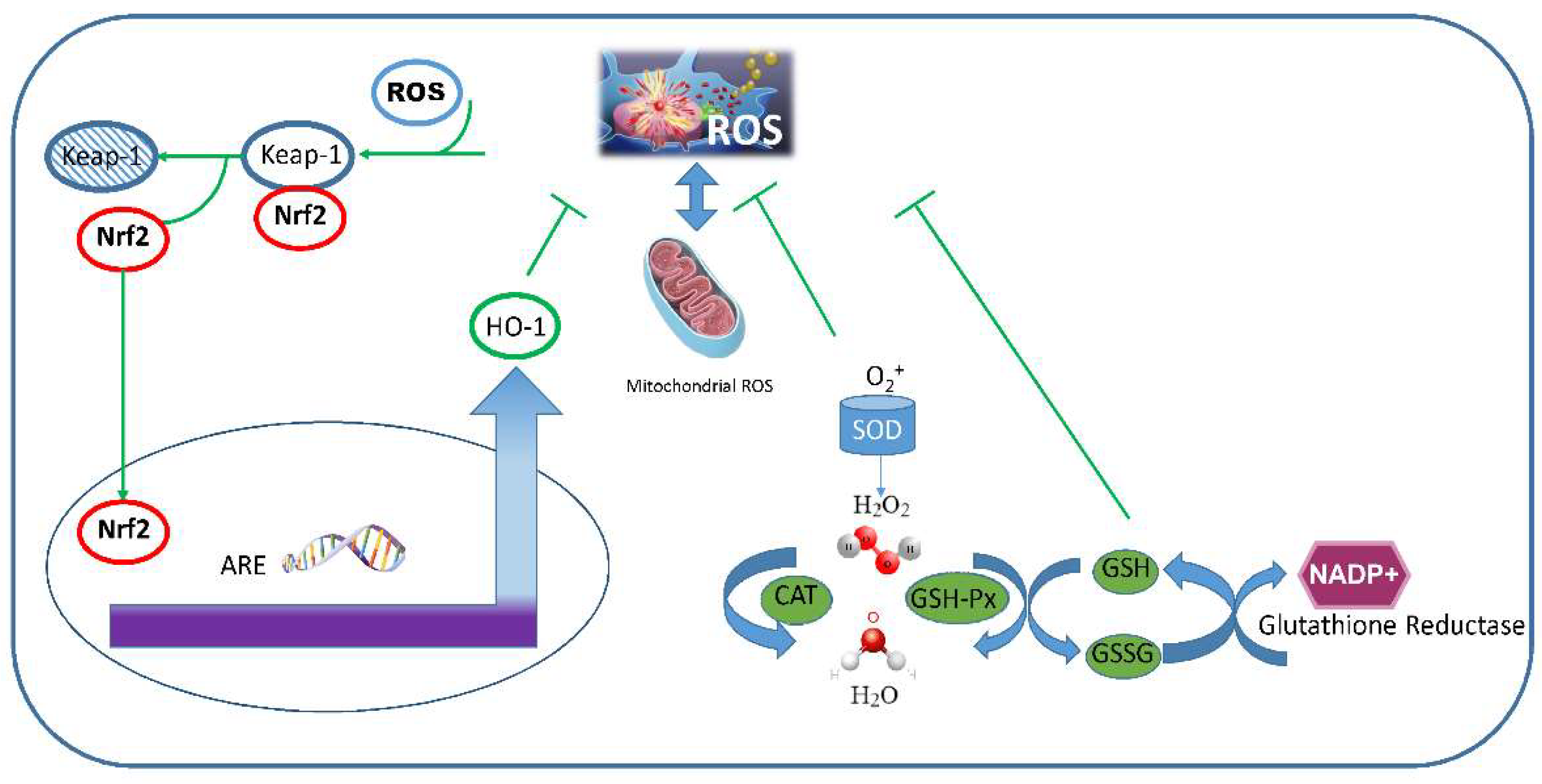
3.5. Cellular Antioxidant Responses
3.5.1. Enzymatic Antioxidant Systems
3.5.2. Non-Enzymatic Antioxidant Activities
3.5.3. Natural and Synthetic Compounds Able to Counteract CS-Induced Oxidative Stress
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Soriano, J.B.; Kendrick, P.J.; Paulson, K.R.; Gupta, V.; Abrams, E.M.; Adedoyin, R.A.; Adhikari, T.B.; Advani, S.M.; Agrawal, A.; Ahmadian, E.; et al. Prevalence and Attributable Health Burden of Chronic Respiratory Diseases, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet Respir. Med. 2020, 8, 585–596. [Google Scholar] [CrossRef]
- Liu, A.; Zhang, X.; Li, R.; Zheng, M.; Yang, S.; Dai, L.; Wu, A.; Hu, C.; Huang, Y.; Xie, M.; et al. Overexpression of the SARS-CoV-2 Receptor ACE2 Is Induced by Cigarette Smoke in Bronchial and Alveolar Epithelia. J. Pathol. 2021, 253, 17–30. [Google Scholar] [CrossRef]
- Remigante, A.; Spinelli, S.; Trichilo, V.; Loddo, S.; Sarikas, A.; Pusch, M.; Dossena, S.; Marino, A.; Morabito, R. D-Galactose Induced Early Aging in Human Erythrocytes: Role of Band 3 Protein. J. Cell. Physiol. 2021, 236, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Morabito, R.; Romano, O.; La Spada, G.; Marino, A. H2O2-Induced Oxidative Stress Affects SO4= Transport in Human Erythrocytes. PLoS ONE 2016, 11, e0146485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marotta, F.; Arunachalam, J.; Banerjee, A.; Catanzaro, R.; Adalti, S.; Das, A.; Kolyada, A.; Pathak, S. Oxidative Stress and Smoke-Related Lung Diseases: A Tentative Approach Through the Blood, Lungs, and Gut. In Oxidative Stress in Lung Diseases; Springer: Berlin, Germany, 2019; pp. 27–50. [Google Scholar]
- Madamanchi, N.R.; Vendrov, A.; Runge, M.S. Oxidative Stress and Vascular Disease. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Liang, Y.; Ip, M.S.M.; Zhang, K.Y.; Mak, J.C.W. Amelioration of Cigarette Smoke-Induced Mucus Hypersecretion and Viscosity by Dendrobium Officinale Polysaccharides in Vitro and in Vivo. Oxidative Med. Cell. Longev. 2020, 2020, 8217642. [Google Scholar] [CrossRef]
- Chang, W.-H.; Thai, P.; Xu, J.; Yang, D.C.; Wu, R.; Chen, C.-H. Cigarette Smoke Regulates the Competitive Interactions between NRF2 and BACH1 for Heme Oxygenase-1 Induction. Int. J. Mol. Sci. 2017, 18, 2386. [Google Scholar] [CrossRef] [Green Version]
- Geraghty, P.; Baumlin, N.; Salathe, M.A.; Foronjy, R.F.; D’Armiento, J.M. Glutathione Peroxidase-1 Suppresses the Unfolded Protein Response upon Cigarette Smoke Exposure. Mediat. Inflamm. 2016, 2016, 9461289. [Google Scholar] [CrossRef]
- Hara, H.; Araya, J.; Takasaka, N.; Fujii, S.; Kojima, J.; Yumino, Y.; Shimizu, K.; Ishikawa, T.; Numata, T.; Kawaishi, M.; et al. Involvement of Creatine Kinase B in Cigarette Smoke-Induced Bronchial Epithelial Cell Senescence. Am. J. Respir. Cell Mol. Biol. 2012, 46, 306–312. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, W.; Xu, X.; Li, B.; He, W.; Padilla, M.T.; Jang, J.-H.; Nyunoya, T.; Amin, S.; Wang, X.; et al. RIP1 Potentiates BPDE-Induced Transformation in Human Bronchial Epithelial Cells through Catalase-Mediated Suppression of Excessive Reactive Oxygen Species. Carcinogenesis 2013, 34, 2119–2128. [Google Scholar] [CrossRef]
- Sundar, I.K.; Maremanda, K.P.; Rahman, I. Mitochondrial Dysfunction Is Associated with Miro1 Reduction in Lung Epithelial Cells by Cigarette Smoke. Toxicol. Lett. 2019, 317, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, Y.; Wang, Z.; Yang, Y.; Li, M.; Yuan, D.; Zhang, X.; Li, Y. CD147 Promoted Epithelial Mesenchymal Transition in Airway Epithelial Cells Induced by Cigarette Smoke via Oxidative Stress Signaling Pathway. COPD J. Chronic Obstr. Pulm. Dis. 2020, 17, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhang, L.; Li, Y.; Wu, G.; Zhu, H.; Zhang, H.; Su, J.-K.; Guo, L.; Zhou, Q.; Xiong, F.; et al. Cigarette Smoke Extract Stimulates Bronchial Epithelial Cells to Undergo a SUMOylation Turnover. BMC Pulm. Med. 2020, 20, 276. [Google Scholar] [CrossRef] [PubMed]
- Mercado, N.; Colley, T.; Baker, J.R.; Vuppussetty, C.; Kono, Y.; Clarke, C.; Tooze, S.; Johansen, T.; Barnes, P.J. Bicaudal D1 Impairs Autophagosome Maturation in Chronic Obstructive Pulmonary Disease. FASEB Bioadv. 2019, 1, 688–705. [Google Scholar] [CrossRef]
- Baskoro, H.; Sato, T.; Karasutani, K.; Suzuki, Y.; Mitsui, A.; Arano, N.; Nurwidya, F.; Kato, M.; Takahashi, F.; Kodama, Y.; et al. Regional Heterogeneity in Response of Airway Epithelial Cells to Cigarette Smoke. BMC Pulm. Med. 2018, 18, 148. [Google Scholar] [CrossRef]
- Reddy, A.T.; Lakshmi, S.P.; Banno, A.; Reddy, R.C. Role of GPx3 in PPARγ-Induced Protection against COPD-Associated Oxidative Stress. Free Radic. Biol. Med. 2018, 126, 350–357. [Google Scholar] [CrossRef]
- Wartenberg, M.; Andrault, P.-M.; Schamberger, A.C.; Chazeirat, T.; Sizaret, D.; Renault, J.; Petit, A.; Saidi, A.; Guyetant, S.; Courty, Y.; et al. Cigarette Smoke Induces Overexpression of Cathepsin S in Active Smokers with and without COPD. Eur. Respir. J. 2018, 52, L625–L638. [Google Scholar] [CrossRef]
- Zulueta, A.; Caretti, A.; Campisi, G.M.; Brizzolari, A.; Abad, J.L.; Paroni, R.; Signorelli, P.; Ghidoni, R. Inhibitors of Ceramide de Novo Biosynthesis Rescue Damages Induced by Cigarette Smoke in Airways Epithelia. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2017, 390, 753–759. [Google Scholar] [CrossRef]
- Schumacher, F.-R.; Schubert, S.; Hannus, M.; Sönnichsen, B.; Ittrich, C.; Kreideweiss, S.; Kurz, T.; Rippmann, J.F. RNAi Screen for NRF2 Inducers Identifies Targets That Rescue Primary Lung Epithelial Cells from Cigarette Smoke Induced Radical Stress. PLoS ONE 2016, 11, e0166352. [Google Scholar] [CrossRef] [Green Version]
- Boylston, J.A.; Brenner, C. A Knockdown with Smoke Model Reveals FHIT as a Repressor of Heme Oxygenase 1. Cell Cycle 2014, 13, 2913–2930. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.-H.; Wu, Y.-L.; Liu, M.-H.; Lee, T.-S.; Kou, Y.R. Glucosamine Attenuates Cigarette Smoke-Induced Lung Inflammation via Its Antioxidant Function and by Inhibiting ROS-Sensitive Inflammatory Signaling. FASEB J. 2014, 28, 208–218. [Google Scholar] [CrossRef]
- Zhang, H.; Shih, A.; Rinna, A.; Forman, H.J. Exacerbation of Tobacco Smoke Mediated Apoptosis by Resveratrol: An Unexpected Consequence of Its Antioxidant Action. Int. J. Biochem. Cell Biol. 2011, 43, 1059–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakshmi, S.P.; Reddy, A.T.; Kodidhela, L.D.; Varadacharyulu, N.C. Epigallocatechin Gallate Diminishes Cigarette Smoke-Induced Oxidative Stress, Lipid Peroxidation, and Inflammation in Human Bronchial Epithelial Cells. Life Sci. 2020, 259, 118260. [Google Scholar] [CrossRef]
- Ding, S.; Hou, X.; Yuan, J.; Tan, X.; Chen, J.; Yang, N.; Luo, Y.; Jiang, Z.; Jin, P.; Dong, Z.; et al. Wedelolactone Protects Human Bronchial Epithelial Cell Injury against Cigarette Smoke Extract-Induced Oxidant Stress and Inflammation Responses through Nrf2 Pathway. Int. Immunopharmacol. 2015, 29, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Jin, P.; Feng, L.; Song, J.; Sun, E.; Liu, W.; Shu, L.; Jia, X. Protective Effect of Luteolin on Cigarette Smoke Extract-Induced Cellular Toxicity and Apoptosis in Normal Human Bronchial Epithelial Cells via the Nrf2 Pathway. Oncol. Rep. 2014, 31, 1855–1862. [Google Scholar] [CrossRef] [Green Version]
- Brito, A.; Santos, T.; Herculano, K.; Miranda, M.; Sá, A.; Carvalho, J.L.; Albertini, R.; Castro-Faria-Neto, H.; Ligeiro-de-Oliveira, A.P.; Aimbire, F. The MAPKinase Signaling and the Stimulatory Protein-1 (Sp1) Transcription Factor Are Involved in the Phototherapy Effect on Cytokines Secretion from Human Bronchial Epithelial Cells Stimulated with Cigarette Smoke Extract. Inflammation 2021, 44, 1643–1661. [Google Scholar] [CrossRef] [PubMed]
- Reis, R.; Orak, D.; Yilmaz, D.; Cimen, H.; Sipahi, H. Modulation of Cigarette Smoke Extract-Induced Human Bronchial Epithelial Damage by Eucalyptol and Curcumin. Hum. Exp. Toxicol. 2021, 40, 1445–1462. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.-Q.; Zuo, Q.-N.; Wang, T.; Xu, D.; Lian, L.; Gao, L.-J.; Wan, C.; Chen, L.; Wen, F.-Q.; Shen, Y.-C. Mitochondrial-Targeting Antioxidant SS-31 Suppresses Airway Inflammation and Oxidative Stress Induced by Cigarette Smoke. Oxidative Med. Cell. Longev. 2021, 2021, 6644238. [Google Scholar] [CrossRef]
- Dang, X.; He, B.; Ning, Q.; Liu, Y.; Guo, J.; Niu, G.; Chen, M. Alantolactone Suppresses Inflammation, Apoptosis and Oxidative Stress in Cigarette Smoke-Induced Human Bronchial Epithelial Cells through Activation of Nrf2/HO-1 and Inhibition of the NF-ΚB Pathways. Respir. Res. 2020, 21, 95. [Google Scholar] [CrossRef]
- Dera, A.A.; Al Fayi, M.; Otifi, H.; Alshyarba, M.; Alfhili, M.; Rajagopalan, P. Thymoquinone (Tq) Protects Necroptosis Induced by Autophagy/Mitophagy-Dependent Oxidative Stress in Human Bronchial Epithelial Cells Exposed to Cigarette Smoke Extract (CSE). J. Food Biochem. 2020, 44, e13366. [Google Scholar] [CrossRef]
- Hong, J.; Chen, Q.; Wang, Y.; Lin, S.; Su, G. Dexmedetomidine Alleviates Smoke-Induced Bronchial and Alveolar Epithelial Cell Injury. Gen. Physiol. Biophys. 2020, 39, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Son, E.S.; Park, J.-W.; Kim, Y.J.; Jeong, S.H.; Hong, J.H.; Kim, S.-H.; Kyung, S.Y. Effects of Antioxidants on Oxidative Stress and Inflammatory Responses of Human Bronchial Epithelial Cells Exposed to Particulate Matter and Cigarette Smoke Extract. Toxicol. In Vitro 2020, 67, 104883. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, Y.; Roth, M.; Zhang, L.; Shi, R.; Yang, X.; Li, Y.; Zhang, J. Sirtuin 3 Inhibits Airway Epithelial Mitochondrial Oxidative Stress in Cigarette Smoke-Induced COPD. Oxidative Med. Cell. Longev. 2020, 2020, 7582980. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, Y.; Xu, M.; Zhang, H.; Chen, Y.; Chung, K.F.; Adcock, I.M.; Li, F. Roles of TRPA1 and TRPV1 in Cigarette Smoke -Induced Airway Epithelial Cell Injury Model. Free Radic. Biol. Med. 2019, 134, 229–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.-H.; Lee, J.; Jeong, J.; Woo, J.; Lee, C.-H.; Yoo, C.-G. Cigarette Smoke Extract Enhances Neutrophil Elastase-Induced IL-8 Production via Proteinase-Activated Receptor-2 Upregulation in Human Bronchial Epithelial Cells. Exp. Mol. Med. 2018, 50, 79. [Google Scholar] [CrossRef] [PubMed]
- Munakata, S.; Ishimori, K.; Kitamura, N.; Ishikawa, S.; Takanami, Y.; Ito, S. Oxidative Stress Responses in Human Bronchial Epithelial Cells Exposed to Cigarette Smoke and Vapor from Tobacco- and Nicotine-Containing Products. Regul. Toxicol. Pharmacol. 2018, 99, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.S.D.; Liao, W.; Peh, H.Y.; Vila, M.; Dong, J.; Shen, H.-M.; Wong, W.S.F. Andrographolide Simultaneously Augments Nrf2 Antioxidant Defense and Facilitates Autophagic Flux Blockade in Cigarette Smoke-Exposed Human Bronchial Epithelial Cells. Toxicol. Appl. Pharmacol. 2018, 360, 120–130. [Google Scholar] [CrossRef]
- Yanagisawa, S.; Baker, J.R.; Vuppusetty, C.; Koga, T.; Colley, T.; Fenwick, P.; Donnelly, L.E.; Barnes, P.J.; Ito, K. The Dynamic Shuttling of SIRT1 between Cytoplasm and Nuclei in Bronchial Epithelial Cells by Single and Repeated Cigarette Smoke Exposure. PLoS ONE 2018, 13, e0193921. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Tang, J.; Shan, H.; Zhang, Q.; Yang, X.; Zhang, J.; Li, Y. P66Shc Mediates Mitochondrial Dysfunction Dependent on PKC Activation in Airway Epithelial Cells Induced by Cigarette Smoke. Oxidative Med. Cell. Longev. 2018, 2018, 5837123. [Google Scholar] [CrossRef] [Green Version]
- Benedikter, B.J.; Volgers, C.; van Eijck, P.H.; Wouters, E.F.M.; Savelkoul, P.H.M.; Reynaert, N.L.; Haenen, G.R.M.M.; Rohde, G.G.U.; Weseler, A.R.; Stassen, F.R.M. Cigarette Smoke Extract Induced Exosome Release Is Mediated by Depletion of Exofacial Thiols and Can Be Inhibited by Thiol-Antioxidants. Free Radic. Biol. Med. 2017, 108, 334–344. [Google Scholar] [CrossRef]
- Cui, Y.; Liu, K.W.K.; Liang, Y.; Ip, M.S.M.; Mak, J.C.W. Inhibition of Monoamine Oxidase-B by Selegiline Reduces Cigarette Smoke-Induced Oxidative Stress and Inflammation in Airway Epithelial Cells. Toxicol. Lett. 2017, 268, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-H.; Jeong, J.; Koo, Y.-J.; Jang, A.-H.; Lee, C.-H.; Yoo, C.-G. Exogenous Neutrophil Elastase Enters Bronchial Epithelial Cells and Suppresses Cigarette Smoke Extract–Induced Heme Oxygenase-1 by Cleaving Sirtuin 1. J. Biol. Chem. 2017, 292, 11970–11979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Tong, D.; Liu, J.; Chen, F.; Shen, Y. Oroxylin A Attenuates Cigarette Smoke-Induced Lung Inflammation by Activating Nrf2. Int. Immunopharmacol. 2016, 40, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Fu, X.; Wang, X.; Wang, W.; Cao, W.; Zhang, W. Protective Effects of Sesaminol on BEAS-2B Cells Impaired by Cigarette Smoke Extract. Cell Biochem. Biophys. 2015, 71, 1207–1213. [Google Scholar] [CrossRef]
- Lee, K.-H.; Lee, C.-H.; Jeong, J.; Jang, A.-H.; Yoo, C.-G. Neutrophil Elastase Differentially Regulates Interleukin 8 (IL-8) and Vascular Endothelial Growth Factor (VEGF) Production by Cigarette Smoke Extract. J. Biol. Chem. 2015, 290, 28438–28445. [Google Scholar] [CrossRef] [Green Version]
- Peña, N.; Carrillo, D.; Muñoz, J.P.; Chnaiderman, J.; Urzúa, U.; León, O.; Tornesello, M.L.; Corvalán, A.H.; Soto-Rifo, R.; Aguayo, F. Tobacco Smoke Activates Human Papillomavirus 16 P97 Promoter and Cooperates with High-Risk E6/E7 for Oxidative DNA Damage in Lung Cells. PLoS ONE 2015, 10, e0123029. [Google Scholar] [CrossRef] [Green Version]
- Aug, A.; Altraja, A.; Altraja, S.; Laaniste, L.; Mahlapuu, R.; Soomets, U.; Kilk, K. Alterations of Bronchial Epithelial Metabolome by Cigarette Smoke Are Reversible by an Antioxidant, O-Methyl-L-Tyrosinyl-γ-L-Glutamyl-L-Cysteinylglycine. Am. J. Respir. Cell Mol. Biol. 2014, 51, 586–594. [Google Scholar] [CrossRef]
- Lin, X.-X.; Yang, X.-F.; Jiang, J.-X.; Zhang, S.-J.; Guan, Y.; Liu, Y.-N.; Sun, Y.-H.; Xie, Q.-M. Cigarette Smoke Extract-Induced BEAS-2B Cell Apoptosis and Anti-Oxidative Nrf-2 up-Regulation Are Mediated by ROS-Stimulated P38 Activation. Toxicol. Mechan. Methods 2014, 24, 575–583. [Google Scholar] [CrossRef]
- Mercado, N.; Kizawa, Y.; Ueda, K.; Xiong, Y.; Kimura, G.; Moses, A.; Curtis, J.M.; Ito, K.; Barnes, P.J. Activation of Transcription Factor Nrf2 Signalling by the Sphingosine Kinase Inhibitor SKI-II Is Mediated by the Formation of Keap1 Dimers. PLoS ONE 2014, 9, e88168. [Google Scholar] [CrossRef]
- Altraja, S.; Mahlapuu, R.; Soomets, U.; Altraja, A. Cigarette Smoke-Induced Differential Regulation of Glutathione Metabolism in Bronchial Epithelial Cells Is Balanced by an Antioxidant Tetrapeptide UPF1. Exp. Toxicol. Pathol. 2013, 65, 711–717. [Google Scholar] [CrossRef]
- Hoffmann, R.F.; Zarrintan, S.; Brandenburg, S.M.; Kol, A.; de Bruin, H.G.; Jafari, S.; Dijk, F.; Kalicharan, D.; Kelders, M.; Gosker, H.R.; et al. Prolonged Cigarette Smoke Exposure Alters Mitochondrial Structure and Function in Airway Epithelial Cells. Respir. Res. 2013, 14, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, W.K.W.; Chan, S.C.H.; Law, A.C.K.; Ip, M.S.M.; Mak, J.C.W. The Role of MAPK and Nrf2 Pathways in Ketanserin-Elicited Attenuation of Cigarette Smoke-Induced IL-8 Production in Human Bronchial Epithelial Cells. Toxicol. Sci. 2012, 125, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Mercado, N.; Thimmulappa, R.; Thomas, C.M.R.; Fenwick, P.S.; Chana, K.K.; Donnelly, L.E.; Biswal, S.; Ito, K.; Barnes, P.J. Decreased Histone Deacetylase 2 Impairs Nrf2 Activation by Oxidative Stress. Biochem. Biophys. Res. Commun. 2011, 406, 292–298. [Google Scholar] [CrossRef] [Green Version]
- Tagawa, Y.; Hiramatsu, N.; Kato, H.; Sakoh, T.; Nakajima, S.; Hayakawa, K.; Saito, Y.; Johno, H.; Takahashi, S.; Gu, L.; et al. Induction of CCAAT/Enhancer-Binding Protein-Homologous Protein by Cigarette Smoke through the Superoxide Anion-Triggered PERK-EIF2α Pathway. Toxicology 2011, 287, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Klimecki, W.T. Culture Conditions Profoundly Impact Phenotype in BEAS-2B, a Human Pulmonary Epithelial Model. J. Appl. Toxicol. 2015, 35, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; He, G.; Gong, W.; Wen, W.; Sun, W.; Ning, B.; Huang, S.; Wu, K.; Huang, C.; Wu, M.; et al. Effects of Nickel on Cyclin Expression, Cell Cycle Progression and Cell Proliferation in Human Pulmonary Cells. Cancer Epidemiol. Prev. Biomark. 2009, 18, 1720–1729. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Clancy, H.A.; Kluz, T.; Zavadil, J.; Costa, M. Comparison of Gene Expression Profiles in Chromate Transformed BEAS-2B Cells. PLoS ONE 2011, 6, e17982. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Son, Y.-O.; Chang, Q.; Sun, L.; Hitron, J.A.; Budhraja, A.; Zhang, Z.; Ke, Z.; Chen, F.; Luo, J.; et al. NADPH Oxidase Activation Is Required in Reactive Oxygen Species Generation and Cell Transformation Induced by Hexavalent Chromium. Toxicol. Sci. 2011, 123, 399–410. [Google Scholar] [CrossRef]
- Martin, N.; Ruddick, A.; Arthur, G.K.; Wan, H.; Woodman, L.; Brightling, C.E.; Jones, D.J.; Pavord, I.D.; Bradding, P. Primary Human Airway Epithelial Cell-Dependent Inhibition of Human Lung Mast Cell Degranulation. PLoS ONE 2012, 7, e43545. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Mychajlowycz, M.; Lau, C.; Gutierrez, C.; Scott, J.A.; Chow, C.-W. Spleen Tyrosine Kinase Mediates BEAS-2B Cell Migration and Proliferation and Human Rhinovirus-Induced Expression of Vascular Endothelial Growth Factor and Interleukin-8. J. Pharmacol. Exp. Ther. 2012, 340, 277–285. [Google Scholar] [CrossRef]
- Zhang, T.; Qi, Y.; Liao, M.; Xu, M.; Bower, K.A.; Frank, J.A.; Shen, H.-M.; Luo, J.; Shi, X.; Chen, G. Autophagy Is a Cell Self-Protective Mechanism against Arsenic-Induced Cell Transformation. Toxicol. Sci. 2012, 130, 298–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nymark, P.; Catalán, J.; Suhonen, S.; Järventaus, H.; Birkedal, R.; Clausen, P.A.; Jensen, K.A.; Vippola, M.; Savolainen, K.; Norppa, H. Genotoxicity of Polyvinylpyrrolidone-Coated Silver Nanoparticles in BEAS 2B Cells. Toxicology 2013, 313, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Lechner, J.F.; Haugen, A.; McClendon, I.A.; Shamsuddin, A.M. Induction of Squamous Differentiation of Normal Human Bronchial Epithelial Cells by Small Amounts of Serum. Differentiation 1984, 25, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, M.; Smith, M.W.; Willey, J.C.; Lechner, J.F.; Trump, B.F.; Harris, C.C. Effects of Serum, Transforming Growth Factor Type β, or 12-O-Tetradecanoylphorbol-13-Acetate on Ionized Cytosolic Calcium Concentration in Normal and Transformed Human Bronchial Epithelial Cells. Cancer Res. 1989, 49, 63–67. [Google Scholar]
- Veranth, J.M.; Cutler, N.S.; Kaser, E.G.; Reilly, C.A.; Yost, G.S. Effects of Cell Type and Culture Media on Interleukin-6 Secretion in Response to Environmental Particles. Toxicol. In Vitro 2008, 22, 498–509. [Google Scholar] [CrossRef] [Green Version]
- Cozens, A.L.; Yezzi, M.J.; Kunzelmann, K.; Ohrui, T.; Chin, L.; Eng, K.; Finkbeiner, W.E.; Widdicombe, J.H.; Gruenert, D.C. CFTR Expression and Chloride Secretion in Polarized Immortal Human Bronchial Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 1994, 10, 38–47. [Google Scholar] [CrossRef]
- Manca, M.L.; Ferraro, M.; Pace, E.; Di Vincenzo, S.; Valenti, D.; Fernàndez-Busquets, X.; Peptu, C.A.; Manconi, M. Loading of Beclomethasone in Liposomes and Hyalurosomes Improved with Mucin as Effective Approach to Counteract the Oxidative Stress Generated by Cigarette Smoke Extract. Nanomaterials 2021, 11, 850. [Google Scholar] [CrossRef]
- Zhang, M.-Y.; Jiang, Y.-X.; Yang, Y.-C.; Liu, J.-Y.; Huo, C.; Ji, X.-L.; Qu, Y.-Q. Cigarette Smoke Extract Induces Pyroptosis in Human Bronchial Epithelial Cells through the ROS/NLRP3/Caspase-1 Pathway. Life Sci. 2021, 269, 119090. [Google Scholar] [CrossRef]
- Zheng, C.; Zhang, Y.; Zhao, Y.; Duan, Y.; Mu, Q.; Wang, X. Circ-Osbpl2 Contributes to Smoke-Related Chronic Obstructive Pulmonary Disease by Targeting Mir-193a-5p/Brd4 Axis. Int. J. COPD 2021, 16, 919–931. [Google Scholar] [CrossRef]
- Zhou, F.; Cao, C.; Chai, H.; Hong, J.; Zhu, M. Circ-HACE1 Aggravates Cigarette Smoke Extract-Induced Injury in Human Bronchial Epithelial Cells via Regulating Toll-like Receptor 4 by Sponging MiR-485-3p. Int. J. COPD 2021, 16, 1535–1547. [Google Scholar] [CrossRef]
- Lin, J.; Peng, J.; Liu, G.; Deng, L. Overexpression of MECP2 Attenuates Cigarette Smoke Extracts Induced Lung Epithelial Cell Injury by Promoting CYP1B1 Methylation. J. Toxicol. Sci. 2020, 45, 177–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Yuan, T.; Zhu, H.; Zhang, H.; Su, J.; Guo, L.; Zhou, Q.; Xiong, F.; Yu, Q.; Yang, P.; et al. Chrysophanol Protects Human Bronchial Epithelial Cells from Cigarette Smoke Extract (Cse)-Induced Apoptosis. Int. J. Mol. Epidemiol. Genet. 2020, 11, 39–45. [Google Scholar] [PubMed]
- Pace, E.; Di Vincenzo, S.; Di Salvo, E.; Genovese, S.; Dino, P.; Sangiorgi, C.; Ferraro, M.; Gangemi, S. MiR-21 Upregulation Increases IL-8 Expression and Tumorigenesis Program in Airway Epithelial Cells Exposed to Cigarette Smoke. J. Cell. Physiol. 2019, 234, 22183–22194. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Chi, D.-Y.; Yu, P.; Lu, J.-J.; Xu, J.-R.; Tan, P.-P.; Wang, B.; Cui, Y.-Y.; Chen, H.-Z. Carbocisteine Improves Histone Deacetylase 2 Deacetylation Activity via Regulating SuMoylation of Histone Deacetylase 2 in Human Tracheobronchial Epithelial Cells. Front. Pharmacol. 2019, 10, 166. [Google Scholar] [CrossRef] [Green Version]
- Anzalone, G.; Albano, G.D.; Montalbano, A.M.; Riccobono, L.; Bonanno, A.; Gagliardo, R.; Bucchieri, F.; Marchese, R.; Moscato, M.; Profita, M. IL-17A-Associated IKK-α Signaling Induced TSLP Production in Epithelial Cells of COPD Patients. Exp. Mol. Med. 2018, 50, 131. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Chen, X.; Jin, W.; Xu, X.; Li, X.; Sun, L. Ginsenoside Rb3 Exerts Protective Properties against Cigarette Smoke Extract-Induced Cell Injury by Inhibiting the P38 MAPK/NF-ΚB and TGF-Β1/VEGF Pathways in Fibroblasts and Epithelial Cells. Biomed. Pharmacother. 2018, 108, 1751–1758. [Google Scholar] [CrossRef]
- Ferraro, M.; Gjomarkaj, M.; Siena, L.; Di Vincenzo, S.; Pace, E. Formoterol and Fluticasone Propionate Combination Improves Histone Deacetylation and Anti-Inflammatory Activities in Bronchial Epithelial Cells Exposed to Cigarette Smoke. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1718–1727. [Google Scholar] [CrossRef]
- Pace, E.; Di Vincenzo, S.; Ferraro, M.; Siena, L.; Chiappara, G.; Dino, P.; Vitulo, P.; Bertani, A.; Saibene, F.; Lanata, L.; et al. Effects of Carbocysteine and Beclomethasone on Histone Acetylation/Deacetylation Processes in Cigarette Smoke Exposed Bronchial Epithelial Cells. J. Cell. Physiol. 2017, 232, 2851–2859. [Google Scholar] [CrossRef]
- Vanella, L.; Li Volti, G.; Distefano, A.; Raffaele, M.; Zingales, V.; Avola, R.; Tibullo, D.; Barbagallo, I. A New Antioxidant Formulation Reduces the Apoptotic and Damaging Effect of Cigarette Smoke Extract on Human Bronchial Epithelial Cells. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 5478–5484. [Google Scholar] [CrossRef]
- Anzalone, G.; Gagliardo, R.; Bucchieri, F.; Albano, G.D.; Siena, L.; Montalbano, A.M.; Bonanno, A.; Riccobono, L.; Pieper, M.P.; Gjomarkaj, M.; et al. IL-17A Induces Chromatin Remodeling Promoting IL-8 Release in Bronchial Epithelial Cells: Effect of Tiotropium. Life Sci. 2016, 152, 107–116. [Google Scholar] [CrossRef]
- Pace, E.; Di Vincenzo, S.; Ferraro, M.; Bruno, A.; Dino, P.; Bonsignore, M.R.; Battaglia, S.; Saibene, F.; Lanata, L.; Gjomarkaj, M. Carbocysteine Counteracts the Effects of Cigarette Smoke on Cell Growth and on the SIRT1/FoxO3 Axis in Bronchial Epithelial Cells. Exp. Gerontol. 2016, 81, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Yuan, Y.; Lin, Z.; Lai, T.; Chen, M.; Li, W.; Lv, Q.; Yuan, B.; Li, D.; Wu, B. Cigarette Smoke Extract Induces Placental Growth Factor Release from Human Bronchial Epithelial Cells via ROS/MAPK (ERK-1/2)/Egr-1 Axis. Int. J. Chron. Obs. Pulmon. Dis. 2016, 11, 3031–3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondì, M.L.; Ferraro, M.; Di Vincenzo, S.; Gerbino, S.; Cavallaro, G.; Giammona, G.; Botto, C.; Gjomarkaj, M.; Pace, E. Effects in Cigarette Smoke Stimulated Bronchial Epithelial Cells of a Corticosteroid Entrapped into Nanostructured Lipid Carriers. J. Nanobiotechnology 2014, 12, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cipollina, C.; Di Vincenzo, S.; Gerbino, S.; Siena, L.; Gjomarkaj, M.; Pace, E. Dual Anti-Oxidant and Anti-Inflammatory Actions of the Electrophilic Cyclooxygenase-2-Derived 17-Oxo-DHA in Lipopolysaccharide- and Cigarette Smoke-Induced Inflammation. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 2299–2309. [Google Scholar] [CrossRef]
- Heijink, I.; Van Oosterhout, A.; Kliphuis, N.; Jonker, M.; Hoffmann, R.; Telenga, E.; Klooster, K.; Slebos, D.-J.; Ten Hacken, N.; Postma, D.; et al. Oxidant-Induced Corticosteroid Unresponsiveness in Human Bronchial Epithelial Cells. Thorax 2014, 69, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Bazzini, C.; Rossetti, V.; Civello, D.A.; Sassone, F.; Vezzoli, V.; Persani, L.; Tiberio, L.; Lanata, L.; Bagnasco, M.; Paulmichl, M.; et al. Short-and Long-Term Effects of Cigarette Smoke Exposure on Glutathione Homeostasis in Human Bronchial Epithelial Cells. Cell. Physiol. Biochem. 2013, 32, 129–145. [Google Scholar] [CrossRef] [Green Version]
- Montalbano, A.M.; Anzalone, G.; Albano, G.D.; Sano, C.D.; Gagliardo, R.; Bonanno, A.; Riccobono, L.; Nicolini, G.; Ingrassia, E.; Gjomarkaj, M.; et al. Beclomethasone Dipropionate and Formoterol Reduce Oxidative/Nitrosative Stress Generated by Cigarette Smoke Extracts and IL-17A in Human Bronchial Epithelial Cells. Eur. J. Pharmacol. 2013, 718, 418–427. [Google Scholar] [CrossRef]
- Pace, E.; Ferraro, M.; Di Vincenzo, S.; Cipollina, C.; Gerbino, S.; Balsamo, R.; Lanata, L.; Gjomarkaj, M. Comparative Cytoprotective Effects of Carbocysteine and Fluticasone Propionate in Cigarette Smoke Stimulated Bronchial Epithelial Cells. Am. J. Respir. Crit. Care Med. 2013, 18, 733–743. [Google Scholar] [CrossRef] [Green Version]
- Profita, M.; Albano, G.D.; Montalbano, A.M.; Di Sano, C.; Anzalone, G.; Gagliardo, R.; Riccobono, L.; Bonanno, A.; Siena, L.; Pieper, M.P.; et al. Acetylcholine Leads to Signal Transducer and Activator of Transcription 1 (STAT-1) Mediated Oxidative/Nitrosative Stress in Human Bronchial Epithelial Cell Line. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1949–1958. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, R.; Derakhshan, T.; Liang, Y.; Ritchey, J.; Liu, L.; Gappa-Fahlenkamp, H. A Three-Dimensional Human Tissue-Engineered Lung Model to Study Influenza an Infection. Tissue Eng. Part A 2018, 24, 1468–1480. [Google Scholar] [CrossRef]
- Fuchs, S.; Hollins, A.; Laue, M.; Schaefer, U.; Roemer, K.; Gumbleton, M.; Lehr, C.-M. Differentiation of Human Alveolar Epithelial Cells in Primary Culture: Morphological Characterization and Synthesis of Caveolin-1 and Surfactant Protein-C. Cell Tissue Res. 2003, 311, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Fulcher, M.L.; Gabriel, S.; Burns, K.A.; Yankaskas, J.R.; Randell, S.H. Well-Differentiated Human Airway Epithelial Cell Cultures. In Human Cell Culture Protocols; Springer: Southampton, UK, 2005; pp. 183–206. [Google Scholar]
- Müller, L.; Brighton, L.E.; Carson, J.L.; Fischer, W.A. Culturing of Human Nasal Epithelial Cells at the Air Liquid Interface. J. Vis. Exp. JoVE 2013, 80, e50646. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Boda, B.; Vernaz, J.; Ferreira, E.; Wiszniewski, L.; Constant, S. Establishment and Characterization of an in Vitro Human Small Airway Model (SmallAir™). Eur. J. Pharm. Biopharm. 2017, 118, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Rayner, R.E.; Makena, P.; Prasad, G.L.; Cormet-Boyaka, E. Optimization of Normal Human Bronchial Epithelial (NHBE) Cell 3D Cultures for in Vitro Lung Model Studies. Sci. Rep. 2019, 9, 500. [Google Scholar] [CrossRef] [PubMed]
- Xiong, R.; Wu, Y.; Wu, Q.; Muskhelishvili, L.; Davis, K.; Tripathi, P.; Chen, Y.; Chen, T.; Bryant, M.; Rosenfeldt, H.; et al. Integration of Transcriptome Analysis with Pathophysiological Endpoints to Evaluate Cigarette Smoke Toxicity in an in Vitro Human Airway Tissue Model. Arch. Toxicol. 2021, 95, 1739–1761. [Google Scholar] [CrossRef]
- Ishikawa, S.; Matsumura, K.; Kitamura, N.; Takanami, Y.; Ito, S. Multi-Omics Analysis: Repeated Exposure of a 3D Bronchial Tissue Culture to Whole-Cigarette Smoke. Toxicol. In Vitro 2019, 54, 251–262. [Google Scholar] [CrossRef]
- Sekine, T.; Hirata, T.; Ishikawa, S.; Ito, S.; Ishimori, K.; Matsumura, K.; Muraki, K. Regulation of NRF2, AP-1 and NF-ΚB by Cigarette Smoke Exposure in Three-Dimensional Human Bronchial Epithelial Cells. J. Appl. Toxicol. 2019, 39, 717–725. [Google Scholar] [CrossRef]
- Yamaguchi, M.S.; McCartney, M.M.; Falcon, A.K.; Linderholm, A.L.; Ebeler, S.E.; Kenyon, N.J.; Harper, R.H.; Schivo, M.; Davis, C.E. Modeling Cellular Metabolomic Effects of Oxidative Stress Impacts from Hydrogen Peroxide and Cigarette Smoke on Human Lung Epithelial Cells. J. Breath Res. 2019, 13, 36014. [Google Scholar] [CrossRef]
- Wong, F.H.; AbuArish, A.; Matthes, E.; Turner, M.J.; Greene, L.E.; Cloutier, A.; Robert, R.; Thomas, D.Y.; Cosa, G.; Cantin, A.M.; et al. Cigarette Smoke Activates CFTR through ROS-Stimulated CAMP Signaling in Human Bronchial Epithelial Cells. Am. J. Physiol. Cell Physiol. 2018, 314, C118–C134. [Google Scholar] [CrossRef]
- Amatngalim, G.D.; Broekman, W.; Daniel, N.M.; Van Der Vlugt, L.E.P.M.; Van Schadewijk, A.; Taube, C.; Hiemstra, P.S. Cigarette Smoke Modulates Repair and Innate Immunity Following Injury to Airway Epithelial Cells. PLoS ONE 2016, 11, e0166255. [Google Scholar] [CrossRef]
- Kanai, K.; Koarai, A.; Shishikura, Y.; Sugiura, H.; Ichikawa, T.; Kikuchi, T.; Akamatsu, K.; Hirano, T.; Nakanishi, M.; Matsunaga, K.; et al. Cigarette Smoke Augments MUC5AC Production via the TLR3-EGFR Pathway in Airway Epithelial Cells. Respir. Investig. 2015, 53, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Albano, G.D.; Bonanno, A.; Moscato, M.; Anzalone, G.; Di Sano, C.; Riccobono, L.; Wenzel, S.E.; Profita, M. Crosstalk between MAChRM3 and Β2AR, via Acetylcholine PI3/PKC/PBEP1/Raf-1 MEK1/2/ERK1/2 Pathway Activation, in Human Bronchial Epithelial Cells after Long-Term Cigarette Smoke Exposure. Life Sci. 2018, 192, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Fang, L.; Zhou, L.; Molino, A.; Valentino, M.R.; Yang, S.; Zhang, J.; Li, Y.; Roth, M. MAPK15-ULK1 Signaling Regulates Mitophagy of Airway Epithelial Cell in Chronic Obstructive Pulmonary Disease. Free Radic. Biol. Med. 2021, 172, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Rusling, J.F.; Forster, R.J. Biosensors Designed for Clinical Applications. Biomedicines 2021, 9, 702. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.K.; Baruah, R.K.; Bhamra, H.; Kim, Y.-J.; Yoo, H. Recent Advances in Electrode Development for Biomedical Applications. Biomed. Eng. Lett. 2021, 11, 107–115. [Google Scholar] [CrossRef]
- Deng, X.; Zou, Z.; Zhang, Y.; Gao, J.; Liang, T.; Lu, Z.; Li, C.M. Synthesis of Merit-Combined Antimony Tetroxide Nanoflowers/Reduced Graphene Oxide to Synergistically Boost Real-Time Detection of Nitric Oxide Released from Living Cells for High Sensitivity. J. Colloid Interface Sci. 2021, 581, 465–474. [Google Scholar] [CrossRef]
- Patella, B.; Buscetta, M.; Di Vincenzo, S.; Ferraro, M.; Aiello, G.; Sunseri, C.; Pace, E.; Inguanta, R.; Cipollina, C. Electrochemical Sensor Based on RGO/Au Nanoparticles for Monitoring H2O2 Released by Human Macrophages. Sens. Actuators B Chem. 2021, 327, 128901. [Google Scholar] [CrossRef]
- Duanghathaipornsuk, S.; Farrell, E.J.; Alba-Rubio, A.C.; Zelenay, P.; Kim, D.-S. Detection Technologies for Reactive Oxygen Species: Fluorescence and Electrochemical Methods and Their Applications. Biosensors 2021, 11, 30. [Google Scholar] [CrossRef]
- Promphet, N.; Ummartyotin, S.; Ngeontae, W.; Puthongkham, P.; Rodthongkum, N. Non-Invasive Wearable Chemical Sensors in Real-Life Applications. Anal. Chim. Acta 2021, 1179, 338643. [Google Scholar] [CrossRef]
- Chen, P.; Xiao, Z.; Wu, H.; Wang, Y.; Fan, W.; Su, W.; Li, P. Beneficial Effects of Naringenin in Cigarette Smoke-Induced Damage to the Lung Based on Bioinformatic Prediction and In Vitro Analysis. Molecules 2020, 25, 4704. [Google Scholar] [CrossRef]
- Chung, S.; Vu, S.; Filosto, S.; Goldkorn, T. Src Regulates Cigarette Smoke-Induced Ceramide Generation via Neutral Sphingomyelinase 2 in the Airway Epithelium. Am. J. Respir. Cell Mol. Biol. 2015, 52, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Sundar, I.K.; Nevid, M.Z.; Friedman, A.E.; Rahman, I. Cigarette Smoke Induces Distinct Histone Modifications in Lung Cells: Implications for the Pathogenesis of COPD and Lung Cancer. J. Proteome Res. 2014, 13, 982–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, G.; Garavaglia, M.L.; Astori, E.; Giustarini, D.; Rossi, R.; Milzani, A.; Dalle-Donne, I. Protein Carbonylation in Human Bronchial Epithelial Cells Exposed to Cigarette Smoke Extract. Cell Biol. Toxicol. 2019, 35, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, Y.; Xing, S.; Ma, P.; Lin, D. SIRT5 Prevents Cigarette Smoke Extract-Induced Apoptosis in Lung Epithelial Cells via Deacetylation of FOXO3. Cell Stress Chaperones 2015, 20, 805–810. [Google Scholar] [CrossRef] [Green Version]
- Ni, H.-M.; Williams, J.A.; Ding, W.-X. Mitochondrial Dynamics and Mitochondrial Quality Control. Redox. Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Decker, S.T.; Kwon, O.-S.; Zhao, J.; Hoidal, J.R.; Heuckstadt, T.; Richardson, R.S.; Sanders, K.A.; Layec, G. Skeletal Muscle Mitochondrial Adaptations Induced by Long-Term Cigarette Smoke Exposure. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E80–E89. [Google Scholar] [CrossRef]
- Cano, M.; Datta, S.; Wang, L.; Liu, T.; Flores-Bellver, M.; Sachdeva, M.; Sinha, D.; Handa, J.T. Nrf2 Deficiency Decreases NADPH from Impaired IDH Shuttle and Pentose Phosphate Pathway in Retinal Pigmented Epithelial Cells to Magnify Oxidative Stress-Induced Mitochondrial Dysfunction. Aging Cell 2021, 20, e13444. [Google Scholar] [CrossRef]
- Nigro, A.; Mauro, L.; Giordano, F.; Panza, S.; Iannacone, R.; Liuzzi, G.M.; Aquila, S.; De Amicis, F.; Cellini, F.; Indiveri, C.; et al. Recombinant Arabidopsis HSP70 Sustains Cell Survival and Metastatic Potential of Breast Cancer Cells. Mol. Cancer 2016, 15, 1063–1073. [Google Scholar] [CrossRef] [Green Version]
- Pace, E.; Ferraro, M.; Siena, L.; Melis, M.; Montalbano, A.M.; Johnson, M.; Bonsignore, M.R.; Bonsignore, G.; Gjomarkaj, M. Cigarette Smoke Increases Toll-like Receptor 4 and Modifies Lipopolysaccharide-Mediated Responses in Airway Epithelial Cells. Immunology 2008, 124, 401–411. [Google Scholar] [CrossRef]
- Voelkel, N.F.; Vandivier, R.W.; Tuder, R.M. Vascular Endothelial Growth Factor in the Lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L209–L221. [Google Scholar] [CrossRef]
- Thaikoottathil, J.V.; Martin, R.J.; Zdunek, J.; Weinberger, A.; Rino, J.G.; Chu, H.W. Cigarette Smoke Extract Reduces VEGF in Primary Human Airway Epithelial Cells. Eur. Respir. J. 2009, 33, 835–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Yang, T.; Meng, X.; Sun, T. Azithromycin Attenuates Cigarette Smoke Extract-Induced Oxidative Stress Injury in Human Alveolar Epithelial Cells. Mol. Med. Rep. 2015, 11, 3414–3422. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.-F.; Li, B.; Shen, J.; Wang, Q.-F.; Chen, S.-S.; Jiang, X.-C.; Qiang, D. Ghrelin Alleviates Traumatic Brain Injury-Induced Acute Lung Injury through Pyroptosis/NF-ΚB Pathway. Int. Immunopharmacol. 2020, 79, 106175. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. The Thioredoxin Antioxidant System. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-J.; Chang, F.-K. Associations of Exercise, Nutritional Status, and Smoking with Cognitive Decline among Older Adults in Taiwan: Results of a Longitudinal Population-Based Study. Arch. Gerontol. Geriatr. 2019, 82, 133–138. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Effects | References |
|---|---|---|
| Eucalyptol curcumin | ↓apoptosis ↓oxidative damage ↓inflammation ↑antioxidant response (GSH, Nrf2) | [28] |
| Naringenin | ↓apoptosis ↓oxidative damage (SOD, NQO1 and HO-1) ↓inflammation ↑antioxidant response | [112] |
| Dendrobiumofficinale polysaccharides | ↓mucus secretion and viscosity | [7] |
| Alantolactone | ↓inflammation (NFkB) ↑antioxidant response (Nrf2-HO-1 pathway) | [30] |
| Thymoquinone | ↑antioxidant response (SOD, CAT, GR, GSH) ↓mitochondrial dysfunction | [31] |
| Epigallocatechin gallate | ↓oxidative stress (ROS) ↓inflammation (NF-kB) | [24] |
| Chrysophanol | ↓cell apoptosis (Bax, caspases) ↓oxidative stress (CYP1A1) ↓ER stress | [73] |
| Ginsenoside Rb3 | ↓inflammation (Il-8, TNF-alpha, p38 and NFkB) ↑antioxidant responses (SOD, catalase, GSH, GPx) | [77] |
| Andrographolide | ↑autophagy (LC3B-II ↓oxidative stress (ROS) ↑antioxidant responses (Nrf2 and p62-Nrf2 positive feedback) | [38] |
| Oroxylin A | ↑antioxidant responses (Nrf2, GSH, GR, GPx, HO-1) | [44] |
| Luteolin | ↑antioxidant responses (GSH, Nrf2, NQO1 and HO-1) ↓oxidative stress (ROS) ↓cell apoptosis (caspases 3, 8 and 9) | [26] |
| Wedelolactone | ↑antioxidant responses (SOD, catalase, GSH, Nrf2, NQO1 and HO-1) | [25] |
| Sesaminol | ↓inflammation (IL-8, IL-6) ↓apoptosis ↓oxidative stress (ROS) ↑antioxidant responses (SOD and catalase) | [45] |
| 17-0xo-DHA | ↓oxidative stress (ROS) ↑antioxidant responses (GSH, Nrf2, HO-1) | [85] |
| Resveratrol | ↑antioxidant responses (Nrf2) | [23] |
| Name | Effects | References |
|---|---|---|
| Beclomethasone dipropionate loaded into nanoparticles into liposomes and hyalurosomes modified with mucin | ↓oxidative stress (ROS) | [68] |
| Beclomethasone+ formoterol | ↓oxidative stress (ROS) ↓inflammation (STAT-1) | [88] |
| Fluticasone propionate±formoterol | ↓inflammation (HDAC2, ERKSTAT-1) | [78] |
| Fluticasone propionate loaded in nanostructured lipid carriers | ↓oxidative stress (ROS) ↑antioxidant response (GSH) | [84] |
| Dexamethasone | ↓oxidative stress (ROS) ↑antioxidant responses (SOD, catalase) ↓inflammation (NF-kB, COX-2) | [32] |
| Sulforaphane and Sulforaphane N-acetylcysteine | ↓oxidative stress (ROS) ↑antioxidant responses (Nrf2) ↓inflammation (ERK/JNK) CSE exposure | [33] |
| N-Acetyl-cysteine and Curcumin, Vitamin B2, Carnitine | ↓inflammation (IL-1β, IL-6, TNFα) ↑antioxidant responses (Nrf2, HO-1and PPAR-γ) | [80] |
| Cardiac glycosides (Strophanthidin, digoxin, and digoxigenin) | ↑authophagy (p62 and bicaudal D1). | [15] |
| Carbocysteine | ↓oxidative stress (ROS) ↑antioxidant responses (GSH, Nrf2, HO-1, GSH-Px2 and 3, GR and glutamate-cysteine-ligase) ↓inflammation (HDAC-2, IL-8) Senescence (Sirt-1/FoxO3 axis) | [75,87,89] |
| Carbocysteine and beclomethasone | ↓inflammation (pCREB, IL-1 mRNA and neutrophil chemotaxis) | [79] |
| Tiotropium and Tiotropium and long acting b2 agonist | ↓inflammation (IL-8) ↓oxidative stress (ROS) | [90,104] |
| Selegiline | ↓inflammation (IL-8) ↓oxidative stress (ROS) ↑antioxidant responses (GSH/GSSG ratio, SOD, catalase, Nrf2, Bach1, HO-1) | [42] |
| Ketanserin | ↓inflammation (p38, ERK1/2, IL-8) ↑antioxidant responses (Nrf2) | [53] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cipollina, C.; Bruno, A.; Fasola, S.; Cristaldi, M.; Patella, B.; Inguanta, R.; Vilasi, A.; Aiello, G.; La Grutta, S.; Torino, C.; et al. Cellular and Molecular Signatures of Oxidative Stress in Bronchial Epithelial Cell Models Injured by Cigarette Smoke Extract. Int. J. Mol. Sci. 2022, 23, 1770. https://doi.org/10.3390/ijms23031770
Cipollina C, Bruno A, Fasola S, Cristaldi M, Patella B, Inguanta R, Vilasi A, Aiello G, La Grutta S, Torino C, et al. Cellular and Molecular Signatures of Oxidative Stress in Bronchial Epithelial Cell Models Injured by Cigarette Smoke Extract. International Journal of Molecular Sciences. 2022; 23(3):1770. https://doi.org/10.3390/ijms23031770
Chicago/Turabian StyleCipollina, Chiara, Andreina Bruno, Salvatore Fasola, Marta Cristaldi, Bernardo Patella, Rosalinda Inguanta, Antonio Vilasi, Giuseppe Aiello, Stefania La Grutta, Claudia Torino, and et al. 2022. "Cellular and Molecular Signatures of Oxidative Stress in Bronchial Epithelial Cell Models Injured by Cigarette Smoke Extract" International Journal of Molecular Sciences 23, no. 3: 1770. https://doi.org/10.3390/ijms23031770
APA StyleCipollina, C., Bruno, A., Fasola, S., Cristaldi, M., Patella, B., Inguanta, R., Vilasi, A., Aiello, G., La Grutta, S., Torino, C., & Pace, E. (2022). Cellular and Molecular Signatures of Oxidative Stress in Bronchial Epithelial Cell Models Injured by Cigarette Smoke Extract. International Journal of Molecular Sciences, 23(3), 1770. https://doi.org/10.3390/ijms23031770