
Development of a CAPS Marker and a LAMP Assay for Rapid Detection of Xylella fastidiosa Subsp. multiplex and Differentiation from X. fastidiosa Subsp. fastidiosa on Blueberry



Abstract
:1. Introduction
2. Results
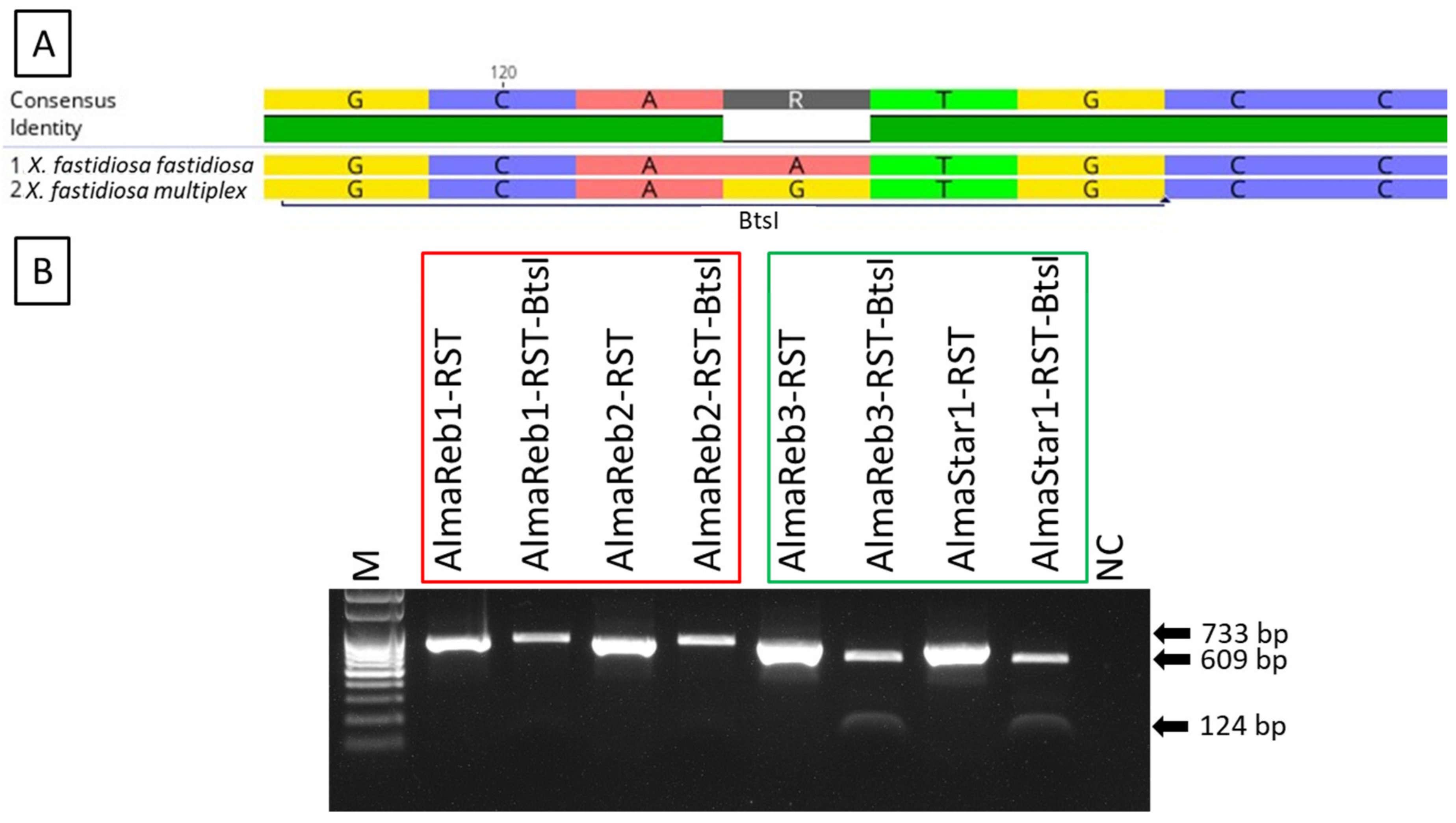
2.1. Detection and Differentiation of Xfm from Xff Using CAPS Marker
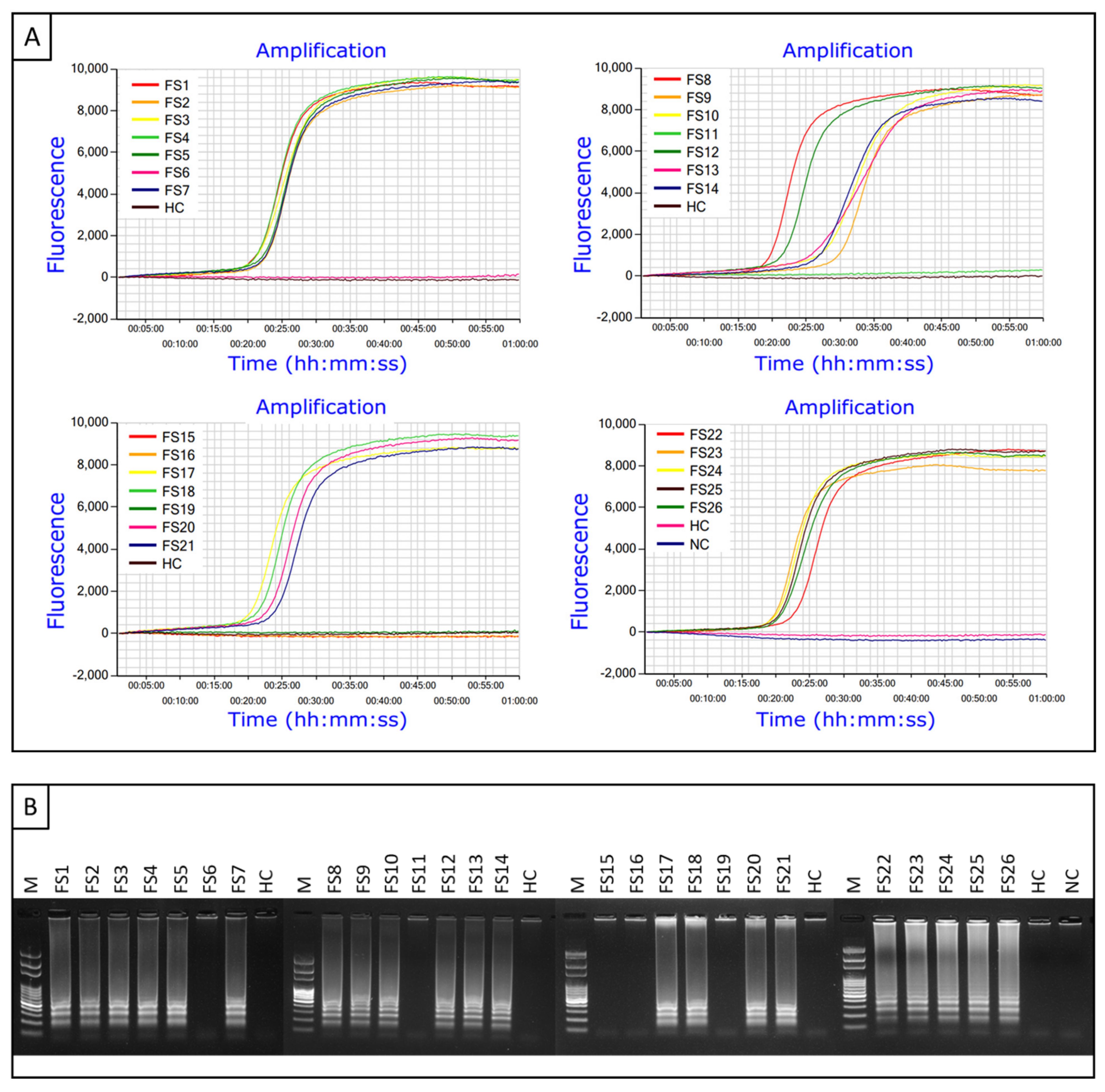
2.2. Detection and Differentiation of Infected Field Samples Using CAPS Marker
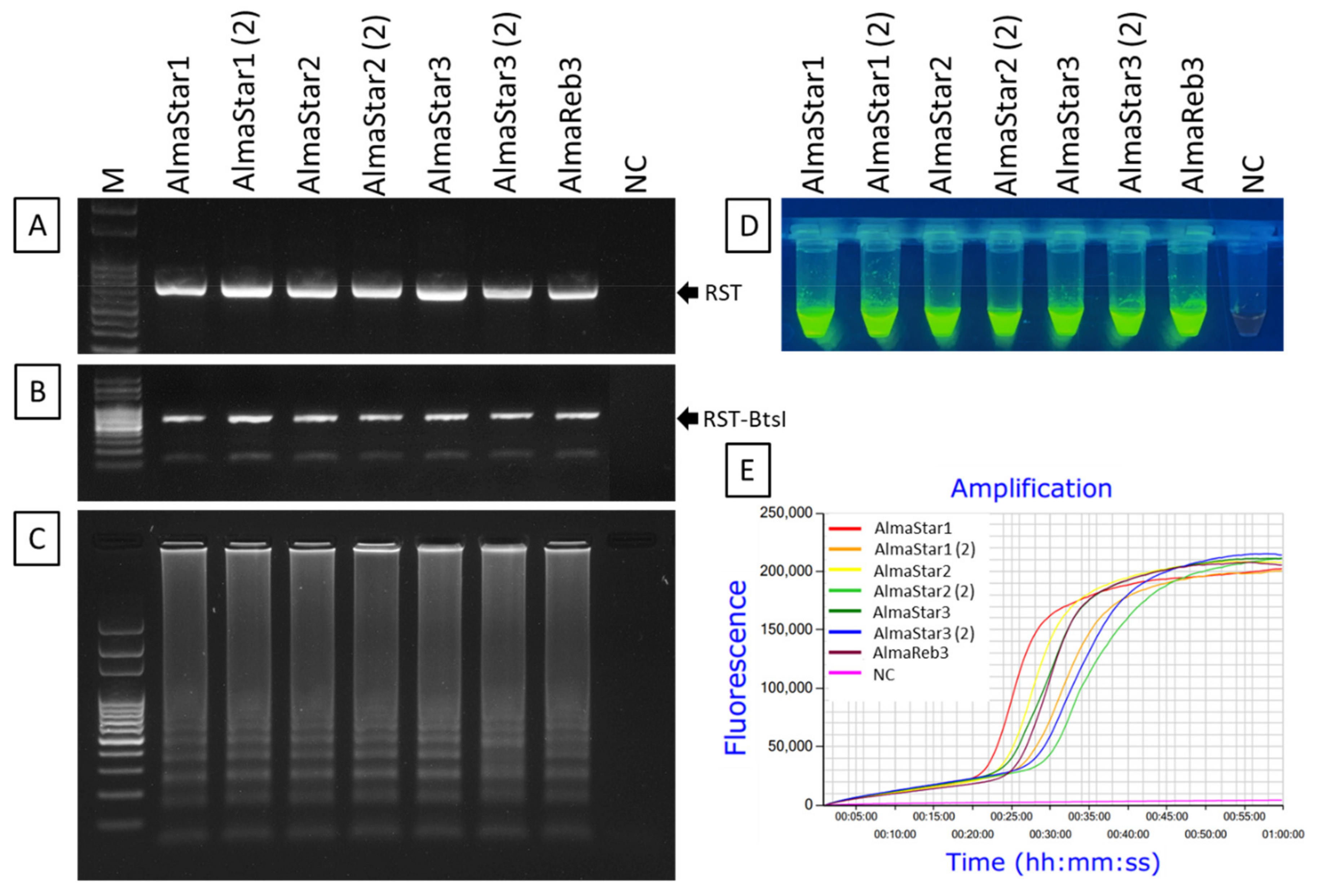
2.3. LAMP Condition Optimization for Xfm Detection
2.4. Amplification of Xfm Using LAMP Assay
2.5. Sensitivity of LAMP Detection of Xfm
2.6. Specificity of LAMP Detection of Xfm
2.7. Detection of Xfm from Greenhouse-Grown and Infected Field Samples Using Probe-Based LAMP
3. Discussion
4. Materials and Methods
4.1. Bacterial Isolates and Growth Conditions
4.2. Plant Materials and Tissue Preparation
4.3. DNA Isolation
4.4. PCR Amplification of the Xf-Specific rpoD Gene
4.5. CAPS Marker Development and Analysis of the PCR Product
4.6. CAPS Analysis of PCR Product from Pure Bacterial Cultures and Infected Plant Samples
4.7. LAMP Primer Design
4.8. Optimization of LAMP Conditions
4.9. Reaction Conditions of LAMP
4.10. Specificity Analysis of LAMP
4.11. Sensitivity Analysis of LAMP
4.12. Evaluation of Infected Field Samples Using CAPS Marker and LAMP Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- EFSA (European Food Safety Authority). Update of the Xylella spp. host plant database–systematic literature searches up to 30 June 2019. EFSA J. 2020, 18, 6114. [Google Scholar]
- Su, C.C.; Deng, W.L.; Jan, F.J.; Chang, C.J.; Huang, H.; Shih, H.T.; Chen, J. Xylella taiwanensis sp. nov., causing pear leaf scorch disease. Int. J. Syst. Evol. Microbiol. 2016, 66, 4766–4771. [Google Scholar] [CrossRef] [PubMed]
- Saponari, M.; Boscia, D.; Altamura, G.; Loconsole, G.; Zicca, S.; D’Attoma, G.; Morelli, M.; Palmisano, F.; Saponari, A.; Tavano, D. Isolation and pathogenicity of Xylella fastidiosa associated to the olive quick decline syndrome in southern Italy. Sci. Rep. 2017, 7, 17723. [Google Scholar] [CrossRef] [PubMed]
- Almeida, R.P.; Purcell, A.H. Biological traits of Xylella fastidiosa strains from grapes and almonds. Appl. Environ. Microbiol. 2003, 69, 7447–7452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldi, P.; La Porta, N. Xylella fastidiosa: Host range and advance in molecular identification techniques. Front. Plant Sci. 2017, 8, 944. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, J.F.; Matthews, M.A.; Rost, T.L. The developmental anatomy of Pierce’s disease symptoms in grapevines: Green islands and matchsticks. Plant Dis. 2005, 89, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-J.; Donaldson, R.; Brannen, P.; Krewer, G.; Boland, R. Bacterial leaf scorch, a new blueberry disease caused by Xylella fastidiosa. HortScience 2009, 44, 413–417. [Google Scholar] [CrossRef]
- Harmon, P.; Hopkins, D. First report of bacterial leaf scorch caused by Xylella fastidiosa on southern highbush blueberry in Florida. Plant Dis. 2009, 93, 1220. [Google Scholar] [CrossRef] [PubMed]
- Brannen, P.M.; Krewer, G.W.; Robert, T., Jr.; Horton, D.L.; Chang, C.-J. Bacterial leaf scorch of blueberry. Univ. Ga. Coop. Ext. 2016. [Google Scholar]
- Blua, M.J.; Redak, R.A.; Morgan, D.J.W.; Costa, H.S. Seasonal flight activity of two Homalodisca species (Homoptera: Cicadellidae) that spread Xylella fastidiosa in southern California. J. Econ. Entomol. 2001, 94, 1506–1510. [Google Scholar] [CrossRef]
- Almeida, R.P.; Nunney, L. How do plant diseases caused by Xylella fastidiosa emerge? Plant Dis. 2015, 99, 1457–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tertuliano, M.; Srinivasan, R.; Scherm, H. Settling behavior of the glassy-winged sharpshooter, Homalodisca vitripennis, vector of Xylella fastidiosa, on southern highbush blueberry cultivars. Entomol. Exp. Appl. 2012, 143, 67–73. [Google Scholar] [CrossRef]
- Denancé, N.; Briand, M.; Gaborieau, R.; Gaillard, S.; Jacques, M.-A. Identification of genetic relationships and subspecies signatures in Xylella fastidiosa. BMC Genom. 2019, 20, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunney, L.; Hopkins, D.L.; Morano, L.D.; Russell, S.E.; Stouthamer, R. Intersubspecific recombination in Xylella fastidiosa strains native to the United States: Infection of novel hosts associated with an unsuccessful invasion. Appl. Environ. Microbiol. 2014, 80, 1159–1169. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.K.; Havird, J.C.; De La Fuente, L. Differentiation of Xylella fastidiosa strains via multilocus sequence analysis of environmentally mediated genes (MLSA-E). Appl. Environ. Microbiol. 2012, 78, 1385–1396. [Google Scholar] [CrossRef] [Green Version]
- Oliver, J.; Cobine, P.; De La Fuente, L. Xylella fastidiosa isolates from both subsp. multiplex and fastidiosa cause disease on southern highbush blueberry (Vaccinium sp.) under greenhouse conditions. Phytopathology 2015, 105, 855–862. [Google Scholar] [CrossRef] [Green Version]
- Di Genova, D.; Lewis, K.J.; Oliver, J.E. Natural Infection of Southern Highbush Blueberry (Vaccinium corymbosum Interspecific Hybrids) by Xylella fastidiosa subsp. fastidiosa. Plant Dis. 2020, 104, 2598–2605. [Google Scholar] [CrossRef] [PubMed]
- Nunney, L.; Ortiz, B.; Russell, S.A.; Sánchez, R.R.; Stouthamer, R. The complex biogeography of the plant pathogen Xylella fastidiosa: Genetic evidence of introductions and subspecific introgression in Central America. PLoS ONE 2014, 9, e112463. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, D.; Purcell, A. Xylella fastidiosa: Cause of Pierce’s disease of grapevine and other emergent diseases. Plant Dis. 2002, 86, 1056–1066. [Google Scholar] [CrossRef] [Green Version]
- Killiny, N.; Almeida, R.P. Gene regulation mediates host specificity of a bacterial pathogen. Environ. Microbiol. Rep. 2011, 3, 791–797. [Google Scholar] [CrossRef]
- Hopkins, D.; Harmon, P.; Brannen, P. Host range of Xylella fastidiosa strains that cause blueberry leaf scorch. Proc. Phytopathol. 2012, 102, 55. [Google Scholar]
- Oliver, J.; Sefick, S.; Parker, J.; Arnold, T.; Cobine, P.; De La Fuente, L. Ionome changes in Xylella fastidiosa—Infected Nicotiana tabacum correlate with virulence and discriminate between subspecies of bacterial isolates. Mol. Plant Microbe Interact. 2014, 27, 1048–1058. [Google Scholar] [CrossRef] [Green Version]
- Pooler, M.R.; Hartung, J.S. Specific PCR detection and identification of Xylella fastidiosa strains causing citrus variegated chlorosis. Curr. Microbiol. 1995, 31, 377–381. [Google Scholar] [CrossRef]
- Chen, J.; Chang, C.; Jarret, R.; Gawel, N. Genetic variation among Xylella fastidiosa strains. Phytopathology 1992, 82, 973–977. [Google Scholar] [CrossRef]
- Chen, J.; Lamikanra, O.; Chang, C.; Hopkins, D. Randomly amplified polymorphic DNA analysis of Xylella fastidiosa Pierce’s disease and oak leaf scorch pathotypes. Appl. Environ. Microbiol. 1995, 61, 1688–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Hartung, J.S.; Chang, C.-J.; Vidaver, A.K. An evolutionary perspective of Pierce’s disease of grapevine, citrus variegated chlorosis, and mulberry leaf scorch diseases. Curr. Microbiol. 2002, 45, 0423–0428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, D.; Albibi, R.; Chen, J.; Lamikanra, O.; Jarret, R.L.; Smith, B.J. Specific detection of Xylella fastidiosa Pierce’s disease strains. Curr. Microbiol. 1999, 39, 85–88. [Google Scholar] [CrossRef]
- Huang, Q. Specific detection and identification of Xylella fastidiosa strains causing oleander leaf scorch using polymerase chain reaction. Curr. Microbiol. 2009, 58, 393–398. [Google Scholar] [CrossRef]
- Guan, W.; Shao, J.; Elbeaino, T.; Davis, R.E.; Zhao, T.; Huang, Q. Specific detection and identification of American mulberry-infecting and Italian olive-associated strains of Xylella fastidiosa by polymerase chain reaction. PLoS ONE 2015, 10, e0129330. [Google Scholar] [CrossRef] [Green Version]
- Bleve, G.; Marchi, G.; Ranaldi, F.; Gallo, A.; Cimaglia, F.; Francesco Logrieco, A.; Mita, G.; Ristori, J.; Surico, G. Molecular characteristics of a strain (Salento-1) of Xylella fastidiosa isolated in Apulia (Italy) from an olive plant with the quick decline syndrome. In Phytopathologia Mediterranea; Firenze University Press: Florence, Italy, 2016; pp. 139–146. [Google Scholar]
- Pooler, M.; Myung, I.; Bentz, J.; Sherald, J.; Hartung, J. Detection of Xylella fastidiosa in potential insect vectors by immunomagnetic separation and nested polymerase chain reaction. Lett. Appl. Microbiol. 1997, 25, 123–126. [Google Scholar] [CrossRef] [Green Version]
- Buzkan, N.; Krivanek, A.F.; Eskalen, A.; Walker, M.A. Improvements in sample preparation and polymerase chain reaction techniques for detection of Xylella fastidiosa in grapevine tissue. Am. J. Enol. Vitic. 2003, 54, 307–312. [Google Scholar]
- Ciapina, L.; Carareto Alves, L.; Lemos, E. A nested-PCR assay for detection of Xylella fastidiosa in citrus plants and sharpshooter leafhoppers. J. Appl. Microbiol. 2004, 96, 546–551. [Google Scholar] [CrossRef] [Green Version]
- Mang, S.M.; Frisullo, S.; Elshafie, H.S.; Camele, I. Diversity evaluation of Xylella fastidiosa from infected olive trees in Apulia (Southern Italy). Plant Pathol. J. 2016, 32, 102–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, M.; Lin, H.; Cabrera-La Rosa, J.; Doddapaneni, H.; Civerolo, E.L. Genome-based PCR primers for specific and sensitive detection and quantification of Xylella fastidiosa. Eur. J. Plant Pathol. 2006, 115, 203–213. [Google Scholar] [CrossRef]
- Bextine, B.; Child, B. Xylella fastidiosa genotype differentiation by SYBR® Green-based QRT-PCR. FEMS Microbiol. Lett. 2007, 276, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.; Shao, J.; Singh, R.; Davis, R.E.; Zhao, T.; Huang, Q. A TaqMan-based real time PCR assay for specific detection and quantification of Xylella fastidiosa strains causing bacterial leaf scorch in oleander. J. Microbiol. Methods 2013, 92, 108–112. [Google Scholar] [CrossRef]
- Li, W.; Teixeira, D.C.; Hartung, J.S.; Huang, Q.; Duan, Y.; Zhou, L.; Chen, J.; Lin, H.; Lopes, S.; Ayres, A.J. Development and systematic validation of qPCR assays for rapid and reliable differentiation of Xylella fastidiosa strains causing citrus variegated chlorosis. J. Microbiol. Methods 2013, 92, 79–89. [Google Scholar] [CrossRef]
- Harper, S.; Ward, L.; Clover, G. Development of LAMP and real-time PCR methods for the rapid detection of Xylella fastidiosa for quarantine and field applications. Phytopathology 2010, 100, 1282–1288. [Google Scholar] [CrossRef]
- Dupas, E.; Legendre, B.; Olivier, V.; Poliakoff, F.; Manceau, C.; Cunty, A. Comparison of real-time PCR and droplet digital PCR for the detection of Xylella fastidiosa in plants. J. Microbiol. Methods 2019, 162, 86–95. [Google Scholar] [CrossRef]
- Yaseen, T.; Drago, S.; Valentini, F.; Elbeaino, T.; Stampone, G.; Digiaro, M.; D’ONGHIA, A.M. On-site detection of Xylella fastidiosa in host plants and in” spy insects” using the real-time loop-mediated isothermal amplification method. Phytopathol. Mediterr. 2015, 54, 488–496. [Google Scholar]
- Waliullah, S.; Hudson, O.; Oliver, J.E.; Brannen, P.M.; Ji, P.; Ali, M.E. Comparative analysis of different molecular and serological methods for detection of Xylella fastidiosa in blueberry. PLoS ONE 2019, 14, e0221903. [Google Scholar] [CrossRef] [Green Version]
- Silvester, R.; Alexander, D.; Antony, A.C.; Hatha, M. GroEL PCR-RFLP–An efficient tool to discriminate closely related pathogenic Vibrio species. Microb. Pathog. 2017, 105, 196–200. [Google Scholar] [CrossRef]
- Kularatne, H.G.C.; Lawrie, A.C.; Barber, P.A.; Keane, P.J. A specific primer PCR and RFLP assay for the rapid detection and differentiation in planta of some Mycosphaerella species associated with foliar diseases of Eucalyptus globulus. Mycol. Res. 2004, 108, 1476–1493. [Google Scholar] [CrossRef] [Green Version]
- Waliullah, S.; Ling, K.-S.; Cieniewicz, E.J.; Oliver, J.E.; Ji, P.; Ali, M.E. Development of loop-mediated isothermal amplification assay for rapid detection of Cucurbit leaf crumple virus. Int. J. Mol. Sci. 2020, 21, 1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waliullah, S.; Bell, J.; Jagdale, G.; Stackhouse, T.; Hajihassani, A.; Brenneman, T.; Ali, M.E. Rapid detection of pecan root-knot nematode, Meloidogyne partityla, in laboratory and field conditions using loop-mediated isothermal amplification. PLoS ONE 2020, 15, e0228123. [Google Scholar]
- Kubota, R.; Alvarez, A.; Su, W.; Jenkins, D. FRET-based assimilating probe for sequence-specific real-time monitoring of loop-mediated isothermal amplification (LAMP). Biol. Eng. Trans. 2011, 4, 81–100. [Google Scholar] [CrossRef]
- Hernandez-Martinez, R.; Costa, H.; Dumenyo, C.; Cooksey, D. Differentiation of strains of Xylella fastidiosa infecting grape, almonds, and oleander using a multiprimer PCR assay. Plant Dis. 2006, 90, 1382–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melanson, R.; Sanderlin, R.; McTaggart, A.; Ham, J. A systematic study reveals that Xylella fastidiosa strains from pecan are part of X. fastidiosa subsp. multiplex. Plant Dis. 2012, 96, 1123–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupas, E.; Briand, M.; Jacques, M.-A.; Cesbron, S. Novel tetraplex quantitative PCR assays for simultaneous detection and identification of Xylella fastidiosa subspecies in plant tissues. Front. Plant Sci. 2019, 10, 1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faino, L.; Scala, V.; Albanese, A.; Modesti, V.; Grottoli, A.; Pucci, N.; Doddi, A.; L’Aurora, A.; Tatulli, G.; Reverberi, M. Nanopore sequencing for the detection and identification of Xylella fastidiosa subspecies and sequence types from naturally infected plant material. Plant Pathol. 2019, 70, 1860–1870. [Google Scholar] [CrossRef]
- Tyler, A.D.; Mataseje, L.; Urfano, C.J.; Schmidt, L.; Antonation, K.S.; Mulvey, M.R.; Corbett, C.R. Evaluation of Oxford Nanopore’s MinION Sequencing Device for Microbial Whole Genome Sequencing Applications. Sci. Rep. 2018, 8, 10931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, M.J.; French, W.J.; Schaad, N.W. Axenic culture of the bacteria associated with phony disease of peach and plum leaf scald. Curr. Microbiol. 1981, 6, 309–314. [Google Scholar] [CrossRef]
- Davis, M.J.; Purcell, A.H.; Thomson, S.V. Isolation media for the Pierce’s disease bacterium. Phytopathology 1980, 70, 425–429. [Google Scholar] [CrossRef]
- Minsavage, G.; Thompson, C.; Hopkins, D.; Leite, R.; Stall, R. Development of a polymerase chain reaction protocol for detection of Xylella fastidiosa in plant tissue. Phytopathology 1994, 84, 456–461. [Google Scholar] [CrossRef]
- Van Horn, C.; Chang, C.-J.; Chen, J. De novo whole-genome sequence of Xylella fastidiosa subsp. multiplex strain BB01 isolated from a blueberry in Georgia, USA. Genome Announc. 2017, 5, e01598-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample no. | SHB Cultivar | Year of Collection | Field Site ID | Geographic Location in Georgia | Subsp. Identity by CAPS Marker and LAMP Assay | Subsp. Identity by Direct Sequencing |
|---|---|---|---|---|---|---|
| FS1 | Rebel | 2019 | Site 1 | Bacon County | Xfm | Xfm |
| FS2 | Rebel | 2019 | Site 1 | Bacon County | Xfm | Xfm |
| FS3 | Rebel | 2019 | Site 1 | Bacon County | Xfm | Xfm |
| FS4 | Rebel | 2019 | Site 1 | Bacon County | Xfm | Xfm |
| FS5 | Rebel | 2019 | Site 1 | Bacon County | Xfm | Xfm |
| FS6 | Rebel | 2019 | Site 2 | Bacon County | Xff | Xff |
| FS7 | Rebel | 2019 | Site 2 | Bacon County | Xfm | Xfm |
| FS8 | Rebel | 2019 | Site 2 | Bacon County | Xfm | Xfm |
| FS9 | Rebel | 2019 | Site 2 | Bacon County | Xfm | Xfm |
| FS10 | Rebel | 2017 | Site 2 | Bacon County | Xfm | Xfm |
| FS11 | Rebel | 2018 | Site 2 | Bacon County | Xff | Xff |
| FS12 | Rebel | 2018 | Site 1 | Bacon County | Xfm | Xfm |
| FS13 | Rebel | 2018 | Site 1 | Bacon County | Xfm | Xfm |
| FS14 | Rebel | 2018 | Site 1 | Bacon County | Xfm | Xfm |
| FS15 | Rebel | 2017 | Site 2 | Bacon County | Xff | Xff |
| FS16 | Rebel | 2017 | Site 2 | Bacon County | Xff | Xff |
| FS17 | Rebel | 2017 | Site 2 | Bacon County | Xfm | Xfm |
| FS18 | Star | 2017 | Site 3 | Bacon County | Xfm | Xfm |
| FS19 | Meadowlark | 2017 | Site 4 | Pierce County | Xff | Xff |
| FS20 | Sweet Crisp | 2018 | Site 5 | Ware County | Xfm | Xfm |
| FS21 | Rebel | 2019 | Site 1 | Bacon County | Xfm | Xfm |
| FS22 | Rebel | 2019 | Site 1 | Bacon County | Xfm | Xfm |
| FS23 | Rebel | 2019 | Site 1 | Bacon County | Xfm | Xfm |
| FS24 | Rebel | 2019 | Site 1 | Bacon County | Xfm | Xfm |
| FS25 | Rebel | 2019 | Site 1 | Bacon County | Xfm | Xfm |
| FS26 | Rebel | 2019 | Site 1 | Bacon County | Xfm | Xfm |
| Isolate Name | SHB Cultivar | Isolation Location | Subsp ID a | Used for b | Reference | RST Accession Number |
|---|---|---|---|---|---|---|
| PierceMed1 | Meadowlark | Pierce County, GA, USA | Xff | in silico analyses and LAMP | Di Genova et al. [17] | MN590439 |
| AlmaReb1 | Rebel | Bacon County, GA, USA | Xff | in silico analyses, LAMP, and CAPS | Di Genova et al. [17] | MN590433 |
| AlmaReb2 | Rebel | Bacon County, GA, USA | Xff | in silico analyses, LAMP, and CAPS | Di Genova et al. [17] | MN590434 |
| AlmaReb3 | Rebel | Bacon County, GA, USA | Xfm | in silico analyses, LAMP, and CAPS | Di Genova et al. [17] | MN590435 |
| AlmaStar1 | Star | Bacon County, GA, USA | Xfm | in silico analyses, LAMP, and CAPS | Di Genova et al. [17] | MN590436 |
| AlmaStar2 | Star | Bacon County, GA, USA | Xfm | in silico analyses and LAMP | Di Genova et al. [17] | MN590437 |
| AlmaStar3 | Star | Bacon County, GA, USA | Xfm | in silico analyses and LAMP | Di Genova et al. [17] | MN590438 |
| AlmaEm3 | Emerald | Bacon County, GA, USA | Xfm | in silico analyses only | Oliver et al. 2014 [22] | PUIY01000010 |
| BB01 | V1 | Brantley County, GA, USA | Xfm | in silico analyses only | Van Horn et al. 2017 [56] | MPAZ01000016 |
| BB08-1 | Windsor | Putnam County, FL, USA | Xfm | in silico analyses only | Oliver et al. 2014 [22] | PUIZ01000048 |
| BBI64 | V1 | Brantley County, GA, USA | Xfm | in silico analyses only | Oliver et al. 2014 [22] | PUJA01000073 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waliullah, S.; Di Genova, D.; Oliver, J.E.; Ali, M.E. Development of a CAPS Marker and a LAMP Assay for Rapid Detection of Xylella fastidiosa Subsp. multiplex and Differentiation from X. fastidiosa Subsp. fastidiosa on Blueberry. Int. J. Mol. Sci. 2022, 23, 1937. https://doi.org/10.3390/ijms23041937
Waliullah S, Di Genova D, Oliver JE, Ali ME. Development of a CAPS Marker and a LAMP Assay for Rapid Detection of Xylella fastidiosa Subsp. multiplex and Differentiation from X. fastidiosa Subsp. fastidiosa on Blueberry. International Journal of Molecular Sciences. 2022; 23(4):1937. https://doi.org/10.3390/ijms23041937
Chicago/Turabian StyleWaliullah, Sumyya, Dario Di Genova, Jonathan E. Oliver, and Md Emran Ali. 2022. "Development of a CAPS Marker and a LAMP Assay for Rapid Detection of Xylella fastidiosa Subsp. multiplex and Differentiation from X. fastidiosa Subsp. fastidiosa on Blueberry" International Journal of Molecular Sciences 23, no. 4: 1937. https://doi.org/10.3390/ijms23041937
APA StyleWaliullah, S., Di Genova, D., Oliver, J. E., & Ali, M. E. (2022). Development of a CAPS Marker and a LAMP Assay for Rapid Detection of Xylella fastidiosa Subsp. multiplex and Differentiation from X. fastidiosa Subsp. fastidiosa on Blueberry. International Journal of Molecular Sciences, 23(4), 1937. https://doi.org/10.3390/ijms23041937