
Hematopoietic Progenitors and the Bone Marrow Niche Shape the Inflammatory Response and Contribute to Chronic Disease


{kind=link}
Abstract
:1. Introduction
Advances in Our Understanding of Hematopoiesis and the Role of the Bone Marrow Niche
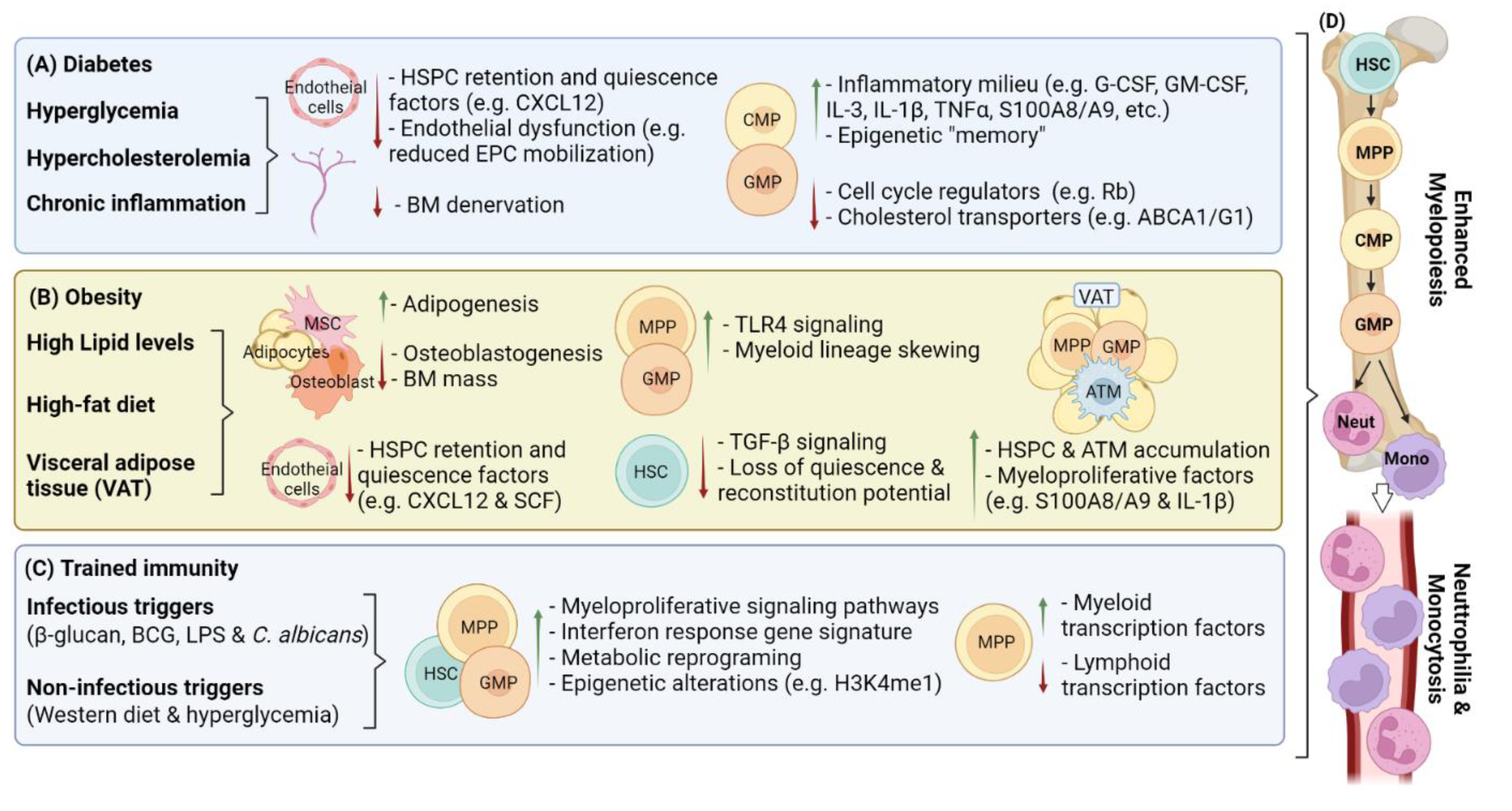
2. Hematopoietic Adaptations in Chronic Inflammatory Disease
2.1. Diabetes-Mediated Changes to Hematopoiesis

2.2. Diabetes-Mediated Changes to the Bone Marrow Niche
2.3. Obesity-Mediated Changes to Hematopoiesis
2.4. Obesity-Mediated Changes to the Bone Marrow Niche
2.5. Hematopoietic Alterations in Trained Immunity
2.6. β-Glucan as a Modulator of Hematopoiesis and a Trigger of Trained Immunity
2.7. BCG Vaccine as a Modulator of Hematopoiesis and a Trigger of Trained Immunity
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Laurenti, E.; Gottgens, B. From haematopoietic stem cells to complex differentiation landscapes. Nature 2018, 553, 418–426. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, E.A.; Till, J.E. The radiation sensitivity of normal mouse bone marrow cells, determined by quantitative marrow transplantation into irradiated mice. Radiat. Res. 1960, 13, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catlin, S.N.; Busque, L.; Gale, R.E.; Guttorp, P.; Abkowitz, J.L. The replication rate of human hematopoietic stem cells in vivo. Blood 2011, 117, 4460–4466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busch, K.; Klapproth, K.; Barile, M.; Flossdorf, M.; Holland-Letz, T.; Schlenner, S.M.; Reth, M.; Hofer, T.; Rodewald, H.R. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature 2015, 518, 542–546. [Google Scholar] [CrossRef]
- Sawai, C.M.; Babovic, S.; Upadhaya, S.; Knapp, D.; Lavin, Y.; Lau, C.M.; Goloborodko, A.; Feng, J.; Fujisaki, J.; Ding, L.; et al. Hematopoietic Stem Cells Are the Major Source of Multilineage Hematopoiesis in Adult Animals. Immunity 2016, 45, 597–609. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Ramos, A.; Chapman, B.; Johnnidis, J.B.; Le, L.; Ho, Y.J.; Klein, A.; Hofmann, O.; Camargo, F.D. Clonal dynamics of native haematopoiesis. Nature 2014, 514, 322–327. [Google Scholar] [CrossRef]
- Pietras, E.M.; Reynaud, D.; Kang, Y.A.; Carlin, D.; Calero-Nieto, F.J.; Leavitt, A.D.; Stuart, J.M.; Göttgens, B.; Passegué, E. Functionally Distinct Subsets of Lineage-Biased Multipotent Progenitors Control Blood Production in Normal and Regenerative Conditions. Cell Stem Cell 2015, 17, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Haas, S.; Trumpp, A.; Milsom, M.D. Causes and Consequences of Hematopoietic Stem Cell Heterogeneity. Cell Stem Cell 2018, 22, 627–638. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, R.; Morita, Y.; Ooehara, J.; Hamanaka, S.; Onodera, M.; Rudolph, K.L.; Ema, H.; Nakauchi, H. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell 2013, 154, 1112–1126. [Google Scholar] [CrossRef] [Green Version]
- Dykstra, B.; Kent, D.; Bowie, M.; McCaffrey, L.; Hamilton, M.; Lyons, K.; Lee, S.J.; Brinkman, R.; Eaves, C. Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell 2007, 1, 218–229. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Fraticelli, A.E.; Wolock, S.L.; Weinreb, C.S.; Panero, R.; Patel, S.H.; Jankovic, M.; Sun, J.; Calogero, R.A.; Klein, A.M.; Camargo, F.D. Clonal analysis of lineage fate in native haematopoiesis. Nature 2018, 553, 212–216. [Google Scholar] [CrossRef]
- King, K.Y.; Goodell, M.A. Inflammatory modulation of HSCs: Viewing the HSC as a foundation for the immune response. Nat. Rev. Immunol. 2011, 11, 685–692. [Google Scholar] [CrossRef]
- Collins, A.; Mitchell, C.A.; Passegue, E. Inflammatory signaling regulates hematopoietic stem and progenitor cell development and homeostasis. J. Exp. Med. 2021, 218, e20201545. [Google Scholar] [CrossRef]
- Pietras, E.M. Inflammation: A key regulator of hematopoietic stem cell fate in health and disease. Blood 2017, 130, 1693–1698. [Google Scholar] [CrossRef] [Green Version]
- Poller, W.C.; Nahrendorf, M.; Swirski, F.K. Hematopoiesis and Cardiovascular Disease. Circ. Res. 2020, 126, 1061–1085. [Google Scholar] [CrossRef]
- Shizuru, J.A.; Negrin, R.S.; Weissman, I.L. Hematopoietic stem and progenitor cells: Clinical and preclinical regeneration of the hematolymphoid system. Annu. Rev. Med. 2005, 56, 509–538. [Google Scholar] [CrossRef] [Green Version]
- Trumpp, A.; Essers, M.; Wilson, A. Awakening dormant haematopoietic stem cells. Nat. Rev. Immunol. 2010, 10, 201–209. [Google Scholar] [CrossRef]
- Seita, J.; Weissman, I.L. Hematopoietic stem cell: Self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 640–653. [Google Scholar] [CrossRef] [Green Version]
- Notta, F.; Zandi, S.; Takayama, N.; Dobson, S.; Gan, O.I.; Wilson, G.; Kaufmann, K.B.; McLeod, J.; Laurenti, E.; Dunant, C.F.; et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 2016, 351, aab2116. [Google Scholar] [CrossRef] [Green Version]
- Oguro, H.; Ding, L.; Morrison, S.J. SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell 2013, 13, 102–116. [Google Scholar] [CrossRef] [Green Version]
- Chavakis, T.; Mitroulis, I.; Hajishengallis, G. Hematopoietic progenitor cells as integrative hubs for adaptation to and fine-tuning of inflammation. Nat. Immunol. 2019, 20, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Loughran, S.J.; Haas, S.; Wilkinson, A.C.; Klein, A.M.; Brand, M. Lineage commitment of hematopoietic stem cells and progenitors: Insights from recent single cell and lineage tracing technologies. Exp. Hematol. 2020, 88, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Cabezas-Wallscheid, N.; Klimmeck, D.; Hansson, J.; Lipka, D.B.; Reyes, A.; Wang, Q.; Weichenhan, D.; Lier, A.; von Paleske, L.; Renders, S.; et al. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell 2014, 15, 507–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, Y.; Ema, H.; Nakauchi, H. Heterogeneity and hierarchy within the most primitive hematopoietic stem cell compartment. J. Exp. Med. 2010, 207, 1173–1182. [Google Scholar] [CrossRef] [Green Version]
- Wilson, N.K.; Kent, D.G.; Buettner, F.; Shehata, M.; Macaulay, I.C.; Calero-Nieto, F.J.; Sanchez Castillo, M.; Oedekoven, C.A.; Diamanti, E.; Schulte, R.; et al. Combined Single-Cell Functional and Gene Expression Analysis Resolves Heterogeneity within Stem Cell Populations. Cell Stem Cell 2015, 16, 712–724. [Google Scholar] [CrossRef] [Green Version]
- Schofield, R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 1978, 4, 7–25. [Google Scholar]
- Mendelson, A.; Frenette, P.S. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat. Med. 2014, 20, 833–846. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Frenette, P.S. Niches for Hematopoietic Stem Cells and Their Progeny. Immunity 2018, 48, 632–648. [Google Scholar] [CrossRef] [Green Version]
- Bianco, P.; Robey, P.G.; Simmons, P.J. Mesenchymal stem cells: Revisiting history, concepts, and assays. Cell Stem Cell 2008, 2, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Birbrair, A.; Frenette, P.S. Niche heterogeneity in the bone marrow. Ann. N. Y. Acad. Sci. 2016, 1370, 82–96. [Google Scholar] [CrossRef]
- Tikhonova, A.N.; Lasry, A.; Austin, R.; Aifantis, I. Cell-by-Cell Deconstruction of Stem Cell Niches. Cell Stem Cell 2020, 27, 19–34. [Google Scholar] [CrossRef]
- Dolgalev, I.; Tikhonova, A.N. Connecting the Dots: Resolving the Bone Marrow Niche Heterogeneity. Front. Cell Dev. Biol. 2021, 9, 622519. [Google Scholar] [CrossRef]
- Tikhonova, A.N.; Dolgalev, I.; Hu, H.; Sivaraj, K.K.; Hoxha, E.; Cuesta-Dominguez, A.; Pinho, S.; Akhmetzyanova, I.; Gao, J.; Witkowski, M.; et al. The bone marrow microenvironment at single-cell resolution. Nature 2019, 569, 222–228. [Google Scholar] [CrossRef]
- Itkin, T.; Gur-Cohen, S.; Spencer, J.A.; Schajnovitz, A.; Ramasamy, S.K.; Kusumbe, A.P.; Ledergor, G.; Jung, Y.; Milo, I.; Poulos, M.G.; et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature 2016, 532, 323–328. [Google Scholar] [CrossRef]
- Pinho, S.; Marchand, T.; Yang, E.; Wei, Q.; Nerlov, C.; Frenette, P.S. Lineage-Biased Hematopoietic Stem Cells Are Regulated by Distinct Niches. Dev. Cell 2018, 44, 634–641. [Google Scholar] [CrossRef] [Green Version]
- Kunisaki, Y.; Bruns, I.; Scheiermann, C.; Ahmed, J.; Pinho, S.; Zhang, D.; Mizoguchi, T.; Wei, Q.; Lucas, D.; Ito, K.; et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 2013, 502, 637–643. [Google Scholar] [CrossRef] [Green Version]
- Walter, D.; Lier, A.; Geiselhart, A.; Thalheimer, F.B.; Huntscha, S.; Sobotta, M.C.; Moehrle, B.; Brocks, D.; Bayindir, I.; Kaschutnig, P.; et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 2015, 520, 549–552. [Google Scholar] [CrossRef]
- Helbling, P.M.; Pineiro-Yanez, E.; Gerosa, R.; Boettcher, S.; Al-Shahrour, F.; Manz, M.G.; Nombela-Arrieta, C. Global Transcriptomic Profiling of the Bone Marrow Stromal Microenvironment during Postnatal Development, Aging, and Inflammation. Cell Rep. 2019, 29, 3313–3330. [Google Scholar] [CrossRef] [Green Version]
- Boettcher, S.; Gerosa, R.C.; Radpour, R.; Bauer, J.; Ampenberger, F.; Heikenwalder, M.; Kopf, M.; Manz, M.G. Endothelial cells translate pathogen signals into G-CSF-driven emergency granulopoiesis. Blood 2014, 124, 1393–1403. [Google Scholar] [CrossRef] [Green Version]
- Hormaechea-Agulla, D.; Le, D.T.; King, K.Y. Common Sources of Inflammation and Their Impact on Hematopoietic Stem Cell Biology. Curr. Stem Cell Rep. 2020, 6, 96–107. [Google Scholar] [CrossRef]
- Schultze, J.L.; Mass, E.; Schlitzer, A. Emerging Principles in Myelopoiesis at Homeostasis and during Infection and Inflammation. Immunity 2019, 50, 288–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herault, A.; Binnewies, M.; Leong, S.; Calero-Nieto, F.J.; Zhang, S.Y.; Kang, Y.A.; Wang, X.; Pietras, E.M.; Chu, S.H.; Barry-Holson, K.; et al. Myeloid progenitor cluster formation drives emergency and leukaemic myelopoiesis. Nature 2017, 544, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Mitroulis, I.; Kalafati, L.; Hajishengallis, G.; Chavakis, T. Myelopoiesis in the Context of Innate Immunity. J. Innate Immun. 2018, 10, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Bousounis, P.; Bergo, V.; Trompouki, E. Inflammation, Aging and Hematopoiesis: A Complex Relationship. Cells 2021, 10, 1386. [Google Scholar] [CrossRef]
- Bowers, E.; Singer, K. Obesity-induced inflammation: The impact of the hematopoietic stem cell niche. JCI Insight 2021, 6, e145295. [Google Scholar] [CrossRef]
- Murphy, A.J.; Tall, A.R. Disordered haematopoiesis and athero-thrombosis. Eur. Heart J. 2016, 37, 1113–1121. [Google Scholar] [CrossRef] [Green Version]
- Nahrendorf, M. Myeloid cell contributions to cardiovascular health and disease. Nat. Med. 2018, 24, 711–720. [Google Scholar] [CrossRef]
- Christ, A.; Günther, P.; Lauterbach, M.A.R.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Baßler, K.; et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 2018, 172, 162–175. [Google Scholar] [CrossRef] [Green Version]
- Nagareddy, P.R.; Kraakman, M.; Masters, S.L.; Stirzaker, R.A.; Gorman, D.J.; Grant, R.W.; Dragoljevic, D.; Hong, E.S.; Abdel-Latif, A.; Smyth, S.S.; et al. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab. 2014, 19, 821–835. [Google Scholar] [CrossRef] [Green Version]
- Murphy, A.J.; Akhtari, M.; Tolani, S.; Pagler, T.; Bijl, N.; Kuo, C.L.; Wang, M.; Sanson, M.; Abramowicz, S.; Welch, C.; et al. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J. Clin. Investig. 2011, 121, 4138–4149. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, E.; Sanz, J.; Dunn, J.L.; Khan, N.; Mendonca, L.E.; Pacis, A.; Tzelepis, F.; Pernet, E.; Dumaine, A.; Grenier, J.C.; et al. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell 2018, 172, 176–190. [Google Scholar] [CrossRef] [Green Version]
- Mitroulis, I.; Ruppova, K.; Wang, B.; Chen, L.S.; Grzybek, M.; Grinenko, T.; Eugster, A.; Troullinaki, M.; Palladini, A.; Kourtzelis, I.; et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 2018, 172, 147–161. [Google Scholar] [CrossRef] [Green Version]
- Mellitus, D. Diagnosis and classification of diabetes mellitus. Diabetes Care 2005, 28, S5–S10. [Google Scholar]
- Barrett, T.J.; Murphy, A.J.; Goldberg, I.J.; Fisher, E.A. Diabetes-mediated myelopoiesis and the relationship to cardiovascular risk. Ann. N. Y. Acad. Sci. 2017, 1402, 31–42. [Google Scholar] [CrossRef]
- Miller, R.G.; Mahajan, H.D.; Costacou, T.; Sekikawa, A.; Anderson, S.J.; Orchard, T.J. A Contemporary Estimate of Total Mortality and Cardiovascular Disease Risk in Young Adults with Type 1 Diabetes: The Pittsburgh Epidemiology of Diabetes Complications Study. Diabetes Care 2016, 39, 2296–2303. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.J.; Distel, E.; Murphy, A.J.; Hu, J.; Garshick, M.S.; Ogando, Y.; Liu, J.; Vaisar, T.; Heinecke, J.W.; Berger, J.S.; et al. Apolipoprotein AI Promotes Atherosclerosis Regression in Diabetic Mice by Suppressing Myelopoiesis and Plaque Inflammation. Circulation 2019, 140, 1170–1184. [Google Scholar] [CrossRef]
- Nagareddy, P.R.; Murphy, A.J.; Stirzaker, R.A.; Hu, Y.; Yu, S.; Miller, R.G.; Ramkhelawon, B.; Distel, E.; Westerterp, M.; Huang, L.S.; et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013, 17, 695–708. [Google Scholar] [CrossRef] [Green Version]
- Soro-Paavonen, A.; Watson, A.M.; Li, J.; Paavonen, K.; Koitka, A.; Calkin, A.C.; Barit, D.; Coughlan, M.T.; Drew, B.G.; Lancaster, G.I.; et al. Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes 2008, 57, 2461–2469. [Google Scholar] [CrossRef] [Green Version]
- Averill, M.M.; Barnhart, S.; Becker, L.; Li, X.; Heinecke, J.W.; Leboeuf, R.C.; Hamerman, J.A.; Sorg, C.; Kerkhoff, C.; Bornfeldt, K.E. S100A9 differentially modifies phenotypic states of neutrophils, macrophages, and dendritic cells: Implications for atherosclerosis and adipose tissue inflammation. Circulation 2011, 123, 1216–1226. [Google Scholar] [CrossRef]
- Healy, A.M.; Pickard, M.D.; Pradhan, A.D.; Wang, Y.; Chen, Z.; Croce, K.; Sakuma, M.; Shi, C.; Zago, A.C.; Garasic, J.; et al. Platelet expression profiling and clinical validation of myeloid-related protein-14 as a novel determinant of cardiovascular events. Circulation 2006, 113, 2278–2284. [Google Scholar] [CrossRef] [Green Version]
- Orchard, T.J.; Olson, J.C.; Erbey, J.R.; Williams, K.; Forrest, K.Y.; Smithline Kinder, L.; Ellis, D.; Becker, D.J. Insulin resistance-related factors, but not glycemia, predict coronary artery disease in type 1 diabetes: 10-year follow-up data from the Pittsburgh Epidemiology of Diabetes Complications Study. Diabetes Care 2003, 26, 1374–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Group, A.S.; Gerstein, H.C.; Miller, M.E.; Genuth, S.; Ismail-Beigi, F.; Buse, J.B.; Goff, D.C., Jr.; Probstfield, J.L.; Cushman, W.C.; Ginsberg, H.N.; et al. Long-term effects of intensive glucose lowering on cardiovascular outcomes. N. Engl. J. Med. 2011, 364, 818–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiem, K.; Keating, S.T.; Netea, M.G.; Riksen, N.P.; Tack, C.J.; van Diepen, J.; Stienstra, R. Hyperglycemic Memory of Innate Immune Cells Promotes In Vitro Proinflammatory Responses of Human Monocytes and Murine Macrophages. J. Immunol. 2021, 206, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Edgar, L.; Akbar, N.; Braithwaite, A.T.; Krausgruber, T.; Gallart-Ayala, H.; Bailey, J.; Corbin, A.L.; Khoyratty, T.E.; Chai, J.T.; Alkhalil, M.; et al. Hyperglycemia Induces Trained Immunity in Macrophages and Their Precursors and Promotes Atherosclerosis. Circulation 2021, 144, 961–982. [Google Scholar] [CrossRef]
- Westerterp, M.; Gourion-Arsiquaud, S.; Murphy, A.J.; Shih, A.; Cremers, S.; Levine, R.L.; Tall, A.R.; Yvan-Charvet, L. Regulation of hematopoietic stem and progenitor cell mobilization by cholesterol efflux pathways. Cell Stem Cell 2012, 11, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Swirski, F.K.; Libby, P.; Aikawa, E.; Alcaide, P.; Luscinskas, F.W.; Weissleder, R.; Pittet, M.J. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Investig. 2007, 117, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Westerterp, M.; Bochem, A.E.; Yvan-Charvet, L.; Murphy, A.J.; Wang, N.; Tall, A.R. ATP-binding cassette transporters, atherosclerosis, and inflammation. Circ. Res. 2014, 114, 157–170. [Google Scholar] [CrossRef] [Green Version]
- Tall, A.R.; Yvan-Charvet, L.; Terasaka, N.; Pagler, T.; Wang, N. HDL, ABC transporters, and cholesterol efflux: Implications for the treatment of atherosclerosis. Cell Metab. 2008, 7, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Yvan-Charvet, L.; Pagler, T.; Gautier, E.L.; Avagyan, S.; Siry, R.L.; Han, S.; Welch, C.L.; Wang, N.; Randolph, G.J.; Snoeck, H.W.; et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 2010, 328, 1689–1693. [Google Scholar] [CrossRef] [Green Version]
- Murphy, A.J.; Bijl, N.; Yvan-Charvet, L.; Welch, C.B.; Bhagwat, N.; Reheman, A.; Wang, Y.; Shaw, J.A.; Levine, R.L.; Ni, H.; et al. Cholesterol efflux in megakaryocyte progenitors suppresses platelet production and thrombocytosis. Nat. Med. 2013, 19, 586–594. [Google Scholar] [CrossRef] [Green Version]
- Distel, E.; Barrett, T.J.; Chung, K.; Girgis, N.M.; Parathath, S.; Essau, C.C.; Murphy, A.J.; Moore, K.J.; Fisher, E.A. miR33 inhibition overcomes deleterious effects of diabetes mellitus on atherosclerosis plaque regression in mice. Circ. Res. 2014, 115, 759–769. [Google Scholar] [CrossRef] [Green Version]
- Mauldin, J.P.; Srinivasan, S.; Mulya, A.; Gebre, A.; Parks, J.S.; Daugherty, A.; Hedrick, C.C. Reduction in ABCG1 in Type 2 diabetic mice increases macrophage foam cell formation. J. Biol. Chem. 2006, 281, 21216–21224. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Kanter, J.E.; Bornfeldt, K.E.; Leboeuf, R.C.; Oram, J.F. Diabetes reduces the cholesterol exporter ABCA1 in mouse macrophages and kidneys. J. Lipid Res. 2010, 51, 1719–1728. [Google Scholar] [CrossRef] [Green Version]
- Passarelli, M.; Tang, C.; McDonald, T.O.; O’Brien, K.D.; Gerrity, R.G.; Heinecke, J.W.; Oram, J.F. Advanced glycation end product precursors impair ABCA1-dependent cholesterol removal from cells. Diabetes 2005, 54, 2198–2205. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Zhao, D.; Schouteden, S.; Sorci-Thomas, M.G.; Van Veldhoven, P.P.; Eggermont, K.; Liu, G.; Verfaillie, C.M.; Feng, Y. Regulation of high-density lipoprotein on hematopoietic stem/progenitor cells in atherosclerosis requires scavenger receptor type BI expression. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1900–1909. [Google Scholar] [CrossRef] [Green Version]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef]
- Hermetet, F.; Buffiere, A.; Aznague, A.; Pais de Barros, J.P.; Bastie, J.N.; Delva, L.; Quere, R. High-fat diet disturbs lipid raft/TGF-beta signaling-mediated maintenance of hematopoietic stem cells in mouse bone marrow. Nat. Commun. 2019, 10, 523. [Google Scholar] [CrossRef]
- Rayner, K.J.; Sheedy, F.J.; Esau, C.C.; Hussain, F.N.; Temel, R.E.; Parathath, S.; van Gils, J.M.; Rayner, A.J.; Chang, A.N.; Suarez, Y.; et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J. Clin. Investig. 2011, 121, 2921–2931. [Google Scholar] [CrossRef] [Green Version]
- Seijkens, T.; Hoeksema, M.A.; Beckers, L.; Smeets, E.; Meiler, S.; Levels, J.; Tjwa, M.; de Winther, M.P.; Lutgens, E. Hypercholesterolemia-induced priming of hematopoietic stem and progenitor cells aggravates atherosclerosis. FASEB J. 2014, 28, 2202–2213. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef]
- Tie, G.; Messina, K.E.; Yan, J.; Messina, J.A.; Messina, L.M. Hypercholesterolemia induces oxidant stress that accelerates the ageing of hematopoietic stem cells. J. Am. Heart Assoc. 2014, 3, e000241. [Google Scholar] [CrossRef] [Green Version]
- Patel, D.C.; Albrecht, C.; Pavitt, D.; Paul, V.; Pourreyron, C.; Newman, S.P.; Godsland, I.F.; Valabhji, J.; Johnston, D.G. Type 2 diabetes is associated with reduced ATP-binding cassette transporter A1 gene expression, protein and function. PLoS ONE 2011, 6, e22142. [Google Scholar] [CrossRef] [Green Version]
- Asleh, R.; Levy, A.P. Divergent effects of alpha-tocopherol and vitamin C on the generation of dysfunctional HDL associated with diabetes and the Hp 2-2 genotype. Antioxid. Redox Signal. 2010, 12, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Robbins, C.S.; Chudnovskiy, A.; Rauch, P.J.; Figueiredo, J.L.; Iwamoto, Y.; Gorbatov, R.; Etzrodt, M.; Weber, G.F.; Ueno, T.; van Rooijen, N.; et al. Extramedullary hematopoiesis generates Ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation 2012, 125, 364–374. [Google Scholar] [CrossRef] [Green Version]
- Tacke, F.; Alvarez, D.; Kaplan, T.J.; Jakubzick, C.; Spanbroek, R.; Llodra, J.; Garin, A.; Liu, J.; Mack, M.; van Rooijen, N.; et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J. Clin. Investig. 2007, 117, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Spinetti, G.; Cordella, D.; Fortunato, O.; Sangalli, E.; Losa, S.; Gotti, A.; Carnelli, F.; Rosa, F.; Riboldi, S.; Sessa, F.; et al. Global remodeling of the vascular stem cell niche in bone marrow of diabetic patients: Implication of the microRNA-155/FOXO3a signaling pathway. Circ. Res. 2013, 112, 510–522. [Google Scholar] [CrossRef] [Green Version]
- Fadini, G.P.; Ferraro, F.; Quaini, F.; Asahara, T.; Madeddu, P. Concise review: Diabetes, the bone marrow niche, and impaired vascular regeneration. Stem Cells Transl. Med. 2014, 3, 949–957. [Google Scholar] [CrossRef]
- Vinci, M.C.; Gambini, E.; Bassetti, B.; Genovese, S.; Pompilio, G. When Good Guys Turn Bad: Bone Marrow’s and Hematopoietic Stem Cells’ Role in the Pathobiology of Diabetic Complications. Int. J. Mol. Sci. 2020, 21, 3864. [Google Scholar] [CrossRef]
- Avogaro, A.; de Kreutzenberg, S.V.; Fadini, G. Endothelial dysfunction: Causes and consequences in patients with diabetes mellitus. Diabetes Res. Clin. Pract. 2008, 82 (Suppl. 2), S94–S101. [Google Scholar] [CrossRef]
- Kojima, H.; Kim, J.; Chan, L. Emerging roles of hematopoietic cells in the pathobiology of diabetic complications. Trends Endocrinol. Metab. 2014, 25, 178–187. [Google Scholar] [CrossRef] [Green Version]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Zalos, G.; Halcox, J.P.; Schenke, W.H.; Waclawiw, M.A.; Quyyumi, A.A.; Finkel, T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N. Engl. J. Med. 2003, 348, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Miorin, M.; Facco, M.; Bonamico, S.; Baesso, I.; Grego, F.; Menegolo, M.; de Kreutzenberg, S.V.; Tiengo, A.; Agostini, C.; et al. Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J. Am. Coll. Cardiol. 2005, 45, 1449–1457. [Google Scholar] [CrossRef] [PubMed]
- Sibal, L.; Aldibbiat, A.; Agarwal, S.C.; Mitchell, G.; Oates, C.; Razvi, S.; Weaver, J.U.; Shaw, J.A.; Home, P.D. Circulating endothelial progenitor cells, endothelial function, carotid intima-media thickness and circulating markers of endothelial dysfunction in people with type 1 diabetes without macrovascular disease or microalbuminuria. Diabetologia 2009, 52, 1464–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tepper, O.M.; Galiano, R.D.; Capla, J.M.; Kalka, C.; Gagne, P.J.; Jacobowitz, G.R.; Levine, J.P.; Gurtner, G.C. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation 2002, 106, 2781–2786. [Google Scholar] [CrossRef] [Green Version]
- Mangialardi, G.; Katare, R.; Oikawa, A.; Meloni, M.; Reni, C.; Emanueli, C.; Madeddu, P. Diabetes causes bone marrow endothelial barrier dysfunction by activation of the RhoA-Rho-associated kinase signaling pathway. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Ferraro, F.; Lymperi, S.; Mendez-Ferrer, S.; Saez, B.; Spencer, J.A.; Yeap, B.Y.; Masselli, E.; Graiani, G.; Prezioso, L.; Rizzini, E.L.; et al. Diabetes impairs hematopoietic stem cell mobilization by altering niche function. Sci. Transl. Med. 2011, 3, 104ra101. [Google Scholar] [CrossRef] [Green Version]
- Hazra, S.; Jarajapu, Y.P.; Stepps, V.; Caballero, S.; Thinschmidt, J.S.; Sautina, L.; Bengtsson, N.; Licalzi, S.; Dominguez, J.; Kern, T.S.; et al. Long-term type 1 diabetes influences haematopoietic stem cells by reducing vascular repair potential and increasing inflammatory monocyte generation in a murine model. Diabetologia 2013, 56, 644–653. [Google Scholar] [CrossRef] [Green Version]
- Busik, J.V.; Tikhonenko, M.; Bhatwadekar, A.; Opreanu, M.; Yakubova, N.; Caballero, S.; Player, D.; Nakagawa, T.; Afzal, A.; Kielczewski, J.; et al. Diabetic retinopathy is associated with bone marrow neuropathy and a depressed peripheral clock. J. Exp. Med. 2009, 206, 2897–2906. [Google Scholar] [CrossRef]
- Tepper, O.M.; Carr, J.; Allen, R.J., Jr.; Chang, C.C.; Lin, C.D.; Tanaka, R.; Gupta, S.M.; Levine, J.P.; Saadeh, P.B.; Warren, S.M. Decreased circulating progenitor cell number and failed mechanisms of stromal cell-derived factor-1alpha mediated bone marrow mobilization impair diabetic tissue repair. Diabetes 2010, 59, 1974–1983. [Google Scholar] [CrossRef] [Green Version]
- Albiero, M.; Poncina, N.; Ciciliot, S.; Cappellari, R.; Menegazzo, L.; Ferraro, F.; Bolego, C.; Cignarella, A.; Avogaro, A.; Fadini, G.P. Bone Marrow Macrophages Contribute to Diabetic Stem Cell Mobilopathy by Producing Oncostatin M. Diabetes 2015, 64, 2957–2968. [Google Scholar] [CrossRef] [Green Version]
- Asahara, T.; Kawamoto, A.; Masuda, H. Concise review: Circulating endothelial progenitor cells for vascular medicine. Stem Cells 2011, 29, 1650–1655. [Google Scholar] [CrossRef]
- Hoyer, F.F.; Zhang, X.; Coppin, E.; Vasamsetti, S.B.; Modugu, G.; Schloss, M.J.; Rohde, D.; McAlpine, C.S.; Iwamoto, Y.; Libby, P.; et al. Bone Marrow Endothelial Cells Regulate Myelopoiesis in Diabetes Mellitus. Circulation 2020, 142, 244–258. [Google Scholar] [CrossRef]
- Oikawa, A.; Siragusa, M.; Quaini, F.; Mangialardi, G.; Katare, R.G.; Caporali, A.; van Buul, J.D.; van Alphen, F.P.; Graiani, G.; Spinetti, G.; et al. Diabetes mellitus induces bone marrow microangiopathy. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 498–508. [Google Scholar] [CrossRef]
- Ferland-McCollough, D.; Maselli, D.; Spinetti, G.; Sambataro, M.; Sullivan, N.; Blom, A.; Madeddu, P. MCP-1 Feedback Loop Between Adipocytes and Mesenchymal Stromal Cells Causes Fat Accumulation and Contributes to Hematopoietic Stem Cell Rarefaction in the Bone Marrow of Patients with Diabetes. Diabetes 2018, 67, 1380–1394. [Google Scholar] [CrossRef] [Green Version]
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef]
- Slater, S.C.; Jover, E.; Martello, A.; Mitic, T.; Rodriguez-Arabaolaza, I.; Vono, R.; Alvino, V.V.; Satchell, S.C.; Spinetti, G.; Caporali, A.; et al. MicroRNA-532-5p Regulates Pericyte Function by Targeting the Transcription Regulator BACH1 and Angiopoietin-1. Mol. Ther. 2018, 26, 2823–2837. [Google Scholar] [CrossRef] [Green Version]
- Urabe, H.; Terashima, T.; Lin, F.; Kojima, H.; Chan, L. Bone marrow-derived TNF-alpha causes diabetic neuropathy in mice. Diabetologia 2015, 58, 402–410. [Google Scholar] [CrossRef] [Green Version]
- Katayama, Y.; Battista, M.; Kao, W.M.; Hidalgo, A.; Peired, A.J.; Thomas, S.A.; Frenette, P.S. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 2006, 124, 407–421. [Google Scholar] [CrossRef] [Green Version]
- Albiero, M.; Poncina, N.; Tjwa, M.; Ciciliot, S.; Menegazzo, L.; Ceolotto, G.; Vigili de Kreutzenberg, S.; Moura, R.; Giorgio, M.; Pelicci, P.; et al. Diabetes causes bone marrow autonomic neuropathy and impairs stem cell mobilization via dysregulated p66Shc and Sirt1. Diabetes 2014, 63, 1353–1365. [Google Scholar] [CrossRef] [Green Version]
- Rota, M.; LeCapitaine, N.; Hosoda, T.; Boni, A.; De Angelis, A.; Padin-Iruegas, M.E.; Esposito, G.; Vitale, S.; Urbanek, K.; Casarsa, C.; et al. Diabetes promotes cardiac stem cell aging and heart failure, which are prevented by deletion of the p66shc gene. Circ. Res. 2006, 99, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menini, S.; Amadio, L.; Oddi, G.; Ricci, C.; Pesce, C.; Pugliese, F.; Giorgio, M.; Migliaccio, E.; Pelicci, P.; Iacobini, C.; et al. Deletion of p66Shc longevity gene protects against experimental diabetic glomerulopathy by preventing diabetes-induced oxidative stress. Diabetes 2006, 55, 1642–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadini, G.P.; Albiero, M.; Menegazzo, L.; Boscaro, E.; Pagnin, E.; Iori, E.; Cosma, C.; Lapolla, A.; Pengo, V.; Stendardo, M.; et al. The redox enzyme p66Shc contributes to diabetes and ischemia-induced delay in cutaneous wound healing. Diabetes 2010, 59, 2306–2314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, Z.; Avolio, E.; Albertario, A.; Sala-Newby, G.B.; Thomas, A.C.; Wang, N.; Emanueli, C.; Madeddu, P. Nerve growth factor gene therapy improves bone marrow sensory innervation and nociceptor-mediated stem cell release in a mouse model of type 1 diabetes with limb ischaemia. Diabetologia 2019, 62, 1297–1311. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.H.; Metharom, P.; Schmeckpeper, J.; Weiss, S.; Martin, K.; Caplice, N.M. Bone marrow-derived CX3CR1 progenitors contribute to neointimal smooth muscle cells via fractalkine CX3CR1 interaction. FASEB J. 2010, 24, 81–92. [Google Scholar] [CrossRef]
- Fadini, G.P.; Albiero, M.; Menegazzo, L.; Boscaro, E.; Agostini, C.; de Kreutzenberg, S.V.; Rattazzi, M.; Avogaro, A. Procalcific phenotypic drift of circulating progenitor cells in type 2 diabetes with coronary artery disease. Exp. Diabetes Res. 2012, 2012, 921685. [Google Scholar] [CrossRef]
- Loomans, C.J.; van Haperen, R.; Duijs, J.M.; Verseyden, C.; de Crom, R.; Leenen, P.J.; Drexhage, H.A.; de Boer, H.C.; de Koning, E.J.; Rabelink, T.J.; et al. Differentiation of bone marrow-derived endothelial progenitor cells is shifted into a proinflammatory phenotype by hyperglycemia. Mol. Med. 2009, 15, 152–159. [Google Scholar] [CrossRef]
- Gallagher, K.A.; Joshi, A.; Carson, W.F.; Schaller, M.; Allen, R.; Mukerjee, S.; Kittan, N.; Feldman, E.L.; Henke, P.K.; Hogaboam, C.; et al. Epigenetic changes in bone marrow progenitor cells influence the inflammatory phenotype and alter wound healing in type 2 diabetes. Diabetes 2015, 64, 1420–1430. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Tie, G.; Wang, S.; Tutto, A.; DeMarco, N.; Khair, L.; Fazzio, T.G.; Messina, L.M. Diabetes impairs wound healing by Dnmt1-dependent dysregulation of hematopoietic stem cells differentiation towards macrophages. Nat. Commun. 2018, 9, 33. [Google Scholar] [CrossRef] [Green Version]
- van Diepen, J.A.; Thiem, K.; Stienstra, R.; Riksen, N.P.; Tack, C.J.; Netea, M.G. Diabetes propels the risk for cardiovascular disease: Sweet monocytes becoming aggressive? Cell Mol. Life Sci. 2016, 73, 4675–4684. [Google Scholar] [CrossRef] [Green Version]
- Arnold, M.; Leitzmann, M.; Freisling, H.; Bray, F.; Romieu, I.; Renehan, A.; Soerjomataram, I. Obesity and cancer: An update of the global impact. Cancer Epidemiol. 2016, 41, 8–15. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef]
- Kopelman, P.G. Obesity as a medical problem. Nature 2000, 404, 635–643. [Google Scholar] [CrossRef]
- Swinburn, B.A.; Sacks, G.; Hall, K.D.; McPherson, K.; Finegood, D.T.; Moodie, M.L.; Gortmaker, S.L. The global obesity pandemic: Shaped by global drivers and local environments. Lancet 2011, 378, 804–814. [Google Scholar] [CrossRef]
- Berg, A.H.; Scherer, P.E. Adipose tissue, inflammation, and cardiovascular disease. Circ. Res. 2005, 96, 939–949. [Google Scholar] [CrossRef] [Green Version]
- Trottier, M.D.; Naaz, A.; Li, Y.; Fraker, P.J. Enhancement of hematopoiesis and lymphopoiesis in diet-induced obese mice. Proc. Natl. Acad. Sci. USA 2012, 109, 7622–7629. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Chen, G.L.; Hannemann, N.; Ipseiz, N.; Krönke, G.; Bäuerle, T.; Munos, L.; Wirtz, S.; Schett, G.; Bozec, A. Microbiota from Obese Mice Regulate Hematopoietic Stem Cell Differentiation by Altering the Bone Niche. Cell Metab. 2015, 22, 886–894. [Google Scholar] [CrossRef] [Green Version]
- Wouters, K.; Gaens, K.; Bijnen, M.; Verboven, K.; Jocken, J.; Wetzels, S.; Wijnands, E.; Hansen, D.; van Greevenbroek, M.; Duijvestijn, A.; et al. Circulating classical monocytes are associated with CD11c(+) macrophages in human visceral adipose tissue. Sci. Rep. 2017, 7, 42665. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Hudis, C.A.; Dannenberg, A.J. Obesity and cancer: Local and systemic mechanisms. Annu. Rev. Med. 2015, 66, 297–309. [Google Scholar] [CrossRef]
- Kamei, N.; Tobe, K.; Suzuki, R.; Ohsugi, M.; Watanabe, T.; Kubota, N.; Ohtsuka-Kowatari, N.; Kumagai, K.; Sakamoto, K.; Kobayashi, M.; et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J. Biol. Chem. 2006, 281, 26602–26614. [Google Scholar] [CrossRef] [Green Version]
- Talukdar, S.; Oh, D.Y.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2012, 18, 1407–1412. [Google Scholar] [CrossRef] [Green Version]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef]
- Adler, B.J.; Kaushansky, K.; Rubin, C.T. Obesity-driven disruption of haematopoiesis and the bone marrow niche. Nat. Rev. Endocrinol. 2014, 10, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Claycombe, K.; King, L.E.; Fraker, P.J. A role for leptin in sustaining lymphopoiesis and myelopoiesis. Proc. Natl. Acad. Sci. USA 2008, 105, 2017–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.; Chen, M.; Kumar, R.; Stefanovic-Racic, M.; O’Doherty, R.M.; Ding, Y.; Jahnen-Dechent, W.; Borghesi, L. Bone marrow lympho-myeloid malfunction in obesity requires precursor cell-autonomous TLR4. Nat. Commun. 2018, 9, 708. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, S.M.; Seijkens, T.T.; Kusters, P.J.; Beckers, L.; den Toom, M.; Smeets, E.; Levels, J.; de Winther, M.P.; Lutgens, E. Diet-induced obesity in mice diminishes hematopoietic stem and progenitor cells in the bone marrow. FASEB J. 2016, 30, 1779–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, K.; DelProposto, J.; Morris, D.L.; Zamarron, B.; Mergian, T.; Maley, N.; Cho, K.W.; Geletka, L.; Subbaiah, P.; Muir, L.; et al. Diet-induced obesity promotes myelopoiesis in hematopoietic stem cells. Mol. Metab. 2014, 3, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Griffin, C.; Eter, L.; Lanzetta, N.; Abrishami, S.; Varghese, M.; McKernan, K.; Muir, L.; Lane, J.; Lumeng, C.N.; Singer, K. TLR4, TRIF, and MyD88 are essential for myelopoiesis and CD11c(+) adipose tissue macrophage production in obese mice. J. Biol. Chem. 2018, 293, 8775–8786. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Govindarajah, V.; Goddard, B.; Hinge, A.; Muench, D.E.; Filippi, M.D.; Aronow, B.; Cancelas, J.A.; Salomonis, N.; Grimes, H.L.; et al. Obesity alters the long-term fitness of the hematopoietic stem cell compartment through modulation of Gfi1 expression. J. Exp. Med. 2018, 215, 627–644. [Google Scholar] [CrossRef]
- Brotfain, E.; Hadad, N.; Shapira, Y.; Avinoah, E.; Zlotnik, A.; Raichel, L.; Levy, R. Neutrophil functions in morbidly obese subjects. Clin. Exp. Immunol. 2015, 181, 156–163. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, K.; Sommer, M.; Strobel, S.; Thrum, S.; Bluher, M.; Wagner, U.; Rossol, M. Perturbation of the Monocyte Compartment in Human Obesity. Front. Immunol. 2019, 10, 1874. [Google Scholar] [CrossRef] [Green Version]
- Kullo, I.J.; Hensrud, D.D.; Allison, T.G. Comparison of numbers of circulating blood monocytes in men grouped by body mass index (<25, 25 to <30, > or = 30). Am. J. Cardiol. 2002, 89, 1441–1443. [Google Scholar] [CrossRef]
- Yang, H.; Youm, Y.H.; Vandanmagsar, B.; Rood, J.; Kumar, K.G.; Butler, A.A.; Dixit, V.D. Obesity accelerates thymic aging. Blood 2009, 114, 3803–3812. [Google Scholar] [CrossRef] [Green Version]
- Chan, M.E.; Adler, B.J.; Green, D.E.; Rubin, C.T. Bone structure and B-cell populations, crippled by obesity, are partially rescued by brief daily exposure to low-magnitude mechanical signals. FASEB J. 2012, 26, 4855–4863. [Google Scholar] [CrossRef] [Green Version]
- Calixto, M.C.; Lintomen, L.; Schenka, A.; Saad, M.J.; Zanesco, A.; Antunes, E. Obesity enhances eosinophilic inflammation in a murine model of allergic asthma. Br. J. Pharmacol. 2010, 159, 617–625. [Google Scholar] [CrossRef] [Green Version]
- Kraakman, M.J.; Lee, M.K.; Al-Sharea, A.; Dragoljevic, D.; Barrett, T.J.; Montenont, E.; Basu, D.; Heywood, S.; Kammoun, H.L.; Flynn, M.; et al. Neutrophil-derived S100 calcium-binding proteins A8/A9 promote reticulated thrombocytosis and atherogenesis in diabetes. J. Clin. Investig. 2017, 127, 2133–2147. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, C.; Eberle, J.U.; Voehringer, D. Basophils in inflammation. Eur. J. Pharmacol. 2016, 778, 90–95. [Google Scholar] [CrossRef]
- Bolus, W.R.; Peterson, K.R.; Hubler, M.J.; Kennedy, A.J.; Gruen, M.L.; Hasty, A.H. Elevating adipose eosinophils in obese mice to physiologically normal levels does not rescue metabolic impairments. Mol. Metab. 2018, 8, 86–95. [Google Scholar] [CrossRef]
- Luche, E.; Robert, V.; Cuminetti, V.; Pomie, C.; Sastourne-Arrey, Q.; Waget, A.; Arnaud, E.; Varin, A.; Labit, E.; Laharrague, P.; et al. Corrupted adipose tissue endogenous myelopoiesis initiates diet-induced metabolic disease. eLife 2017, 6, e23194. [Google Scholar] [CrossRef] [Green Version]
- Amano, S.U.; Cohen, J.L.; Vangala, P.; Tencerova, M.; Nicoloro, S.M.; Yawe, J.C.; Shen, Y.; Czech, M.P.; Aouadi, M. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab. 2014, 19, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Arkan, M.C.; Hevener, A.L.; Greten, F.R.; Maeda, S.; Li, Z.W.; Long, J.M.; Wynshaw-Boris, A.; Poli, G.; Olefsky, J.; Karin, M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat. Med. 2005, 11, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Han, M.S.; Jung, D.Y.; Morel, C.; Lakhani, S.A.; Kim, J.K.; Flavell, R.A.; Davis, R.J. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 2013, 339, 218–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Oh, D.Y.; Bandyopadhyay, G.; Lagakos, W.S.; Talukdar, S.; Osborn, O.; Johnson, A.; Chung, H.; Mayoral, R.; Maris, M.; et al. LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat. Med. 2015, 21, 239–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsouris, D.; Li, P.P.; Thapar, D.; Chapman, J.; Olefsky, J.M.; Neels, J.G. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. 2008, 8, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Saberi, M.; Woods, N.B.; de Luca, C.; Schenk, S.; Lu, J.C.; Bandyopadhyay, G.; Verma, I.M.; Olefsky, J.M. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009, 10, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Luche, E.; Sengenes, C.; Arnaud, E.; Laharrague, P.; Casteilla, L.; Cousin, B. Differential Hematopoietic Activity in White Adipose Tissue Depending on its Localization. J. Cell. Physiol. 2015, 230, 3076–3083. [Google Scholar] [CrossRef]
- Huang, J.Y.; Zhou, Q.L.; Huang, C.H.; Song, Y.; Sharma, A.G.; Liao, Z.; Zhu, K.; Massidda, M.W.; Jamieson, R.R.; Zhang, J.Y.; et al. Neutrophil Elastase Regulates Emergency Myelopoiesis Preceding Systemic Inflammation in Diet-induced Obesity. J. Biol. Chem. 2017, 292, 4770–4776. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, G.I.; Langley, K.G.; Berglund, N.A.; Kammoun, H.L.; Reibe, S.; Estevez, E.; Weir, J.; Mellett, N.A.; Pernes, G.; Conway, J.R.W.; et al. Evidence that TLR4 Is Not a Receptor for Saturated Fatty Acids but Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell Metab. 2018, 27, 1096–1110. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.S.; Nakahira, K.; Chung, K.P.; DeNicola, G.M.; Koo, M.J.; Pabón, M.A.; Rooney, K.T.; Yoon, J.H.; Ryter, S.W.; Stout-Delgado, H.; et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat. Med. 2016, 22, 1002–1012. [Google Scholar] [CrossRef]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Vila, I.K.; Badin, P.M.; Marques, M.A.; Monbrun, L.; Lefort, C.; Mir, L.; Louche, K.; Bourlier, V.; Roussel, B.; Gui, P.; et al. Immune cell Toll-like receptor 4 mediates the development of obesity- and endotoxemia-associated adipose tissue fibrosis. Cell Rep. 2014, 7, 1116–1129. [Google Scholar] [CrossRef]
- Orr, J.S.; Puglisi, M.J.; Ellacott, K.L.; Lumeng, C.N.; Wasserman, D.H.; Hasty, A.H. Toll-like receptor 4 deficiency promotes the alternative activation of adipose tissue macrophages. Diabetes 2012, 61, 2718–2727. [Google Scholar] [CrossRef] [Green Version]
- Luck, H.; Tsai, S.; Chung, J.; Clemente-Casares, X.; Ghazarian, M.; Revelo, X.S.; Lei, H.; Luk, C.T.; Shi, S.Y.; Surendra, A.; et al. Regulation of obesity-related insulin resistance with gut anti-inflammatory agents. Cell Metab. 2015, 21, 527–542. [Google Scholar] [CrossRef] [Green Version]
- Nagai, Y.; Garrett, K.P.; Ohta, S.; Bahrun, U.; Kouro, T.; Akira, S.; Takatsu, K.; Kincade, P.W. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 2006, 24, 801–812. [Google Scholar] [CrossRef] [Green Version]
- Khosravi, A.; Yanez, A.; Price, J.G.; Chow, A.; Merad, M.; Goodridge, H.S.; Mazmanian, S.K. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe 2014, 15, 374–381. [Google Scholar] [CrossRef] [Green Version]
- Tencerova, M.; Figeac, F.; Ditzel, N.; Taipaleenmaki, H.; Nielsen, T.K.; Kassem, M. High-Fat Diet-Induced Obesity Promotes Expansion of Bone Marrow Adipose Tissue and Impairs Skeletal Stem Cell Functions in Mice. J. Bone Miner. Res. 2018, 33, 1154–1165. [Google Scholar] [CrossRef] [Green Version]
- Ambrosi, T.H.; Scialdone, A.; Graja, A.; Gohlke, S.; Jank, A.M.; Bocian, C.; Woelk, L.; Fan, H.; Logan, D.W.; Schürmann, A.; et al. Adipocyte Accumulation in the Bone Marrow during Obesity and Aging Impairs Stem Cell-Based Hematopoietic and Bone Regeneration. Cell Stem Cell 2017, 20, 771–784. [Google Scholar] [CrossRef] [Green Version]
- Tencerova, M.; Frost, M.; Figeac, F.; Nielsen, T.K.; Ali, D.; Lauterlein, J.L.; Andersen, T.L.; Haakonsson, A.K.; Rauch, A.; Madsen, J.S.; et al. Obesity-Associated Hypermetabolism and Accelerated Senescence of Bone Marrow Stromal Stem Cells Suggest a Potential Mechanism for Bone Fragility. Cell Rep. 2019, 27, 2050–2062. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.O.; Yu, H.; Yue, R.; Zhao, Z.; Rios, J.J.; Naveiras, O.; Morrison, S.J. Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat. Cell Biol. 2017, 19, 891–903. [Google Scholar] [CrossRef]
- Cawthorn, W.P.; Scheller, E.L.; Learman, B.S.; Parlee, S.D.; Simon, B.R.; Mori, H.; Ning, X.; Bree, A.J.; Schell, B.; Broome, D.T.; et al. Bone marrow adipose tissue is an endocrine organ that contributes to increased circulating adiponectin during caloric restriction. Cell Metab. 2014, 20, 368–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, N.; Han, S.J.; Enamorado, M.; Link, V.M.; Huang, B.; Moseman, E.A.; Kishton, R.J.; Shannon, J.P.; Dixit, D.; Schwab, S.R.; et al. The Bone Marrow Protects and Optimizes Immunological Memory during Dietary Restriction. Cell 2019, 178, 1088–1101. [Google Scholar] [CrossRef] [PubMed]
- Mattiucci, D.; Maurizi, G.; Izzi, V.; Cenci, L.; Ciarlantini, M.; Mancini, S.; Mensà, E.; Pascarella, R.; Vivarelli, M.; Olivieri, A.; et al. Bone marrow adipocytes support hematopoietic stem cell survival. J. Cell. Physiol. 2018, 233, 1500–1511. [Google Scholar] [CrossRef] [PubMed]
- Masamoto, Y.; Arai, S.; Sato, T.; Kubota, N.; Takamoto, I.; Kadowaki, T.; Kurokawa, M. Adiponectin Enhances Quiescence Exit of Murine Hematopoietic Stem Cells and Hematopoietic Recovery Through mTORC1 Potentiation. Stem Cells 2017, 35, 1835–1848. [Google Scholar] [CrossRef] [Green Version]
- Dias, C.C.; Nogueira-Pedro, A.; Tokuyama, P.Y.; Martins, M.N.; Segreto, H.R.; Buri, M.V.; Miranda, A.; Paredes-Gamero, E.J. A synthetic fragment of leptin increase hematopoietic stem cell population and improve its engraftment ability. J. Cell. Biochem. 2015, 116, 1334–1340. [Google Scholar] [CrossRef]
- Janssens, R.; Struyf, S.; Proost, P. The unique structural and functional features of CXCL12. Cell. Mol. Immunol. 2018, 15, 299–311. [Google Scholar] [CrossRef]
- Crewe, C.; Joffin, N.; Rutkowski, J.M.; Kim, M.; Zhang, F.; Towler, D.A.; Gordillo, R.; Scherer, P.E. An Endothelial-to-Adipocyte Extracellular Vesicle Axis Governed by Metabolic State. Cell 2018, 175, 695–708. [Google Scholar] [CrossRef] [Green Version]
- Asada, N.; Kunisaki, Y.; Pierce, H.; Wang, Z.; Fernandez, N.F.; Birbrair, A.; Ma’ayan, A.; Frenette, P.S. Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat. Cell Biol. 2017, 19, 214–223. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Saunders, T.L.; Enikolopov, G.; Morrison, S.J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef] [Green Version]
- Comazzetto, S.; Murphy, M.M.; Berto, S.; Jeffery, E.; Zhao, Z.; Morrison, S.J. Restricted Hematopoietic Progenitors and Erythropoiesis Require SCF from Leptin Receptor+ Niche Cells in the Bone Marrow. Cell Stem Cell 2019, 24, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Frodermann, V.; Rohde, D.; Courties, G.; Severe, N.; Schloss, M.J.; Amatullah, H.; McAlpine, C.S.; Cremer, S.; Hoyer, F.F.; Ji, F.; et al. Exercise reduces inflammatory cell production and cardiovascular inflammation via instruction of hematopoietic progenitor cells. Nat. Med. 2019, 25, 1761–1771. [Google Scholar] [CrossRef]
- Agha, N.H.; Baker, F.L.; Kunz, H.E.; Graff, R.; Azadan, R.; Dolan, C.; Laughlin, M.S.; Hosing, C.; Markofski, M.M.; Bond, R.A.; et al. Vigorous exercise mobilizes CD34+ hematopoietic stem cells to peripheral blood via the beta2-adrenergic receptor. Brain Behav. Immun. 2018, 68, 66–75. [Google Scholar] [CrossRef]
- Baker, J.M.; De Lisio, M.; Parise, G. Endurance exercise training promotes medullary hematopoiesis. FASEB J. 2011, 25, 4348–4357. [Google Scholar] [CrossRef]
- Gerbaix, M.; Metz, L.; Mac-Way, F.; Lavet, C.; Guillet, C.; Walrand, S.; Masgrau, A.; Vico, L.; Courteix, D. A well-balanced diet combined or not with exercise induces fat mass loss without any decrease of bone mass despite bone micro-architecture alterations in obese rat. Bone 2013, 53, 382–390. [Google Scholar] [CrossRef]
- Netea, M.G.; Quintin, J.; van der Meer, J.W. Trained immunity: A memory for innate host defense. Cell Host Microbe 2011, 9, 355–361. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Dominguez-Andres, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef] [Green Version]
- Benn, C.S.; Netea, M.G.; Selin, L.K.; Aaby, P. A small jab—A big effect: Nonspecific immunomodulation by vaccines. Trends Immunol. 2013, 34, 431–439. [Google Scholar] [CrossRef]
- Nankabirwa, V.; Tumwine, J.K.; Mugaba, P.M.; Tylleskär, T.; Sommerfelt, H.; for the PROMISE-EBF Study Group. Child survival and BCG vaccination: A community based prospective cohort study in Uganda. BMC Public Health 2015, 15, 175. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Joosten, L.A.; Latz, E.; Mills, K.H.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [Green Version]
- Katzmarski, N.; Domínguez-Andrés, J.; Cirovic, B.; Renieris, G.; Ciarlo, E.; Le Roy, D.; Lepikhov, K.; Kattler, K.; Gasparoni, G.; Händler, K.; et al. Transmission of trained immunity and heterologous resistance to infections across generations. Nat. Immunol. 2021, 22, 1382–1390. [Google Scholar] [CrossRef]
- Belicard, T.; Jareosettasin, P.; Sarkies, P. The piRNA pathway responds to environmental signals to establish intergenerational adaptation to stress. BMC Biol. 2018, 16, 103. [Google Scholar] [CrossRef]
- Cubas, P.; Vincent, C.; Coen, E. An epigenetic mutation responsible for natural variation in floral symmetry. Nature 1999, 401, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.A.; Ginhoux, F.; Yona, S. Monocytes, macrophages, dendritic cells and neutrophils: An update on lifespan kinetics in health and disease. Immunology 2021, 163, 250–261. [Google Scholar] [CrossRef]
- Manz, M.G.; Boettcher, S. Emergency granulopoiesis. Nat. Rev. Immunol. 2014, 14, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, S.; Manz, M.G. Sensing and translation of pathogen signals into demand-adapted myelopoiesis. Curr. Opin. Hematol. 2016, 23, 5–10. [Google Scholar] [CrossRef]
- Kleinnijenhuis, J.; Quintin, J.; Preijers, F.; Joosten, L.A.; Ifrim, D.C.; Saeed, S.; Jacobs, C.; van Loenhout, J.; de Jong, D.; Stunnenberg, H.G.; et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 17537–17542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, N.; Andersen, A.; Hansen, A.S.; Jepsen, F.S.; Barbosa, A.; Biering-Sørensen, S.; Rodrigues, A.; Ravn, H.; Aaby, P.; Benn, C.S. The Effect of Oral Polio Vaccine at Birth on Infant Mortality: A Randomized Trial. Clin. Infect. Dis. 2015, 61, 1504–1511. [Google Scholar] [CrossRef] [Green Version]
- Higgins, J.P.; Soares-Weiser, K.; Lopez-Lopez, J.A.; Kakourou, A.; Chaplin, K.; Christensen, H.; Martin, N.K.; Sterne, J.A.; Reingold, A.L. Association of BCG, DTP, and measles containing vaccines with childhood mortality: Systematic review. BMJ 2016, 355, i5170. [Google Scholar] [CrossRef] [Green Version]
- Quintin, J.; Saeed, S.; Martens, J.H.A.; Giamarellos-Bourboulis, E.J.; Ifrim, D.C.; Logie, C.; Jacobs, L.; Jansen, T.; Kullberg, B.J.; Wijmenga, C.; et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 2012, 12, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Schrum, J.E.; Crabtree, J.N.; Dobbs, K.R.; Kiritsy, M.C.; Reed, G.W.; Gazzinelli, R.T.; Netea, M.G.; Kazura, J.W.; Dent, A.E.; Fitzgerald, K.A.; et al. Cutting Edge: Plasmodium falciparum Induces Trained Innate Immunity. J. Immunol. 2018, 200, 1243–1248. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Sandalova, E.; Low, D.; Gehring, A.J.; Fieni, S.; Amadei, B.; Urbani, S.; Chong, Y.S.; Guccione, E.; Bertoletti, A. Trained immunity in newborn infants of HBV-infected mothers. Nat. Commun. 2015, 6, 6588. [Google Scholar] [CrossRef] [Green Version]
- Bekkering, S.; Quintin, J.; Joosten, L.A.; van der Meer, J.W.; Netea, M.G.; Riksen, N.P. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1731–1738. [Google Scholar] [CrossRef]
- van der Valk, F.M.; Bekkering, S.; Kroon, J.; Yeang, C.; Van den Bossche, J.; van Buul, J.D.; Ravandi, A.; Nederveen, A.J.; Verberne, H.J.; Scipione, C.; et al. Oxidized Phospholipids on Lipoprotein(a) Elicit Arterial Wall Inflammation and an Inflammatory Monocyte Response in Humans. Circulation 2016, 134, 611–624. [Google Scholar] [CrossRef]
- van der Heijden, C.; Keating, S.T.; Groh, L.; Joosten, L.A.B.; Netea, M.G.; Riksen, N.P. Aldosterone induces trained immunity: The role of fatty acid synthesis. Cardiovasc. Res. 2020, 116, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Crisan, T.O.; Cleophas, M.C.P.; Novakovic, B.; Erler, K.; van de Veerdonk, F.L.; Stunnenberg, H.G.; Netea, M.G.; Dinarello, C.A.; Joosten, L.A.B. Uric acid priming in human monocytes is driven by the AKT-PRAS40 autophagy pathway. Proc. Natl. Acad. Sci. USA 2017, 114, 5485–5490. [Google Scholar] [CrossRef] [Green Version]
- Vetvicka, V. Glucan-immunostimulant, adjuvant, potential drug. World J. Clin. Oncol. 2011, 2, 115–119. [Google Scholar] [CrossRef]
- Brown, G.D.; Gordon, S. Fungal beta-glucans and mammalian immunity. Immunity 2003, 19, 311–315. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.D.; Gordon, S. Immune recognition of fungal beta-glucans. Cell Microbiol. 2005, 7, 471–479. [Google Scholar] [CrossRef]
- van Bruggen, R.; Drewniak, A.; Jansen, M.; van Houdt, M.; Roos, D.; Chapel, H.; Verhoeven, A.J.; Kuijpers, T.W. Complement receptor 3, not Dectin-1, is the major receptor on human neutrophils for beta-glucan-bearing particles. Mol. Immunol. 2009, 47, 575–581. [Google Scholar] [CrossRef]
- Moorlag, S.; Khan, N.; Novakovic, B.; Kaufmann, E.; Jansen, T.; van Crevel, R.; Divangahi, M.; Netea, M.G. β-Glucan Induces Protective Trained Immunity against Mycobacterium tuberculosis Infection: A Key Role for IL-1. Cell Rep. 2020, 31, 107634. [Google Scholar] [CrossRef]
- Kalafati, L.; Kourtzelis, I.; Schulte-Schrepping, J.; Li, X.; Hatzioannou, A.; Grinenko, T.; Hagag, E.; Sinha, A.; Has, C.; Dietz, S.; et al. Innate Immune Training of Granulopoiesis Promotes Anti-tumor Activity. Cell 2020, 183, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Saeed, S.; Quintin, J.; Kerstens, H.H.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Cheng, S.C.; Ratter, J.; Berentsen, K.; van der Ent, M.A.; et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014, 345, 1251086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saz-Leal, P.; Del Fresno, C.; Brandi, P.; Martinez-Cano, S.; Dungan, O.M.; Chisholm, J.D.; Kerr, W.G.; Sancho, D. Targeting SHIP-1 in Myeloid Cells Enhances Trained Immunity and Boosts Response to Infection. Cell Rep. 2018, 25, 1118–1126. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, J.C.; Barroso de Figueiredo, A.M.; Teodoro Silva, M.V.; Cirovic, B.; de Bree, L.C.J.; Damen, M.; Moorlag, S.; Gomes, R.S.; Helsen, M.M.; Oosting, M.; et al. beta-Glucan-Induced Trained Immunity Protects against Leishmania braziliensis Infection: A Crucial Role for IL-32. Cell Rep. 2019, 28, 2659–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, N.; Downey, J.; Sanz, J.; Kaufmann, E.; Blankenhaus, B.; Pacis, A.; Pernet, E.; Ahmed, E.; Cardoso, S.; Nijnik, A.; et al. M. tuberculosis Reprograms Hematopoietic Stem Cells to Limit Myelopoiesis and Impair Trained Immunity. Cell 2020, 183, 752–770. [Google Scholar] [CrossRef]
- Arts, R.J.; Novakovic, B.; Ter Horst, R.; Carvalho, A.; Bekkering, S.; Lachmandas, E.; Rodrigues, F.; Silvestre, R.; Cheng, S.C.; Wang, S.Y.; et al. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab. 2016, 24, 807–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.; Rao, N.A.; Aghajanirefah, A.; et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef] [Green Version]
- Bekkering, S.; Arts, R.J.W.; Novakovic, B.; Kourtzelis, I.; van der Heijden, C.; Li, Y.; Popa, C.D.; Ter Horst, R.; van Tuijl, J.; Netea-Maier, R.T.; et al. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell 2018, 172, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Noma, K.; Allis, C.D.; Grewal, S.I. Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science 2001, 293, 1150–1155. [Google Scholar] [CrossRef]
- Wang, H.; Cao, R.; Xia, L.; Erdjument-Bromage, H.; Borchers, C.; Tempst, P.; Zhang, Y. Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol. Cell 2001, 8, 1207–1217. [Google Scholar] [CrossRef]
- Keating, S.T.; Groh, L.; van der Heijden, C.; Rodriguez, H.; Dos Santos, J.C.; Fanucchi, S.; Okabe, J.; Kaipananickal, H.; van Puffelen, J.H.; Helder, L.; et al. The Set7 Lysine Methyltransferase Regulates Plasticity in Oxidative Phosphorylation Necessary for Trained Immunity Induced by beta-Glucan. Cell Rep. 2020, 31, 107548. [Google Scholar] [CrossRef]
- Kleinnijenhuis, J.; van Crevel, R.; Netea, M.G. Trained immunity: Consequences for the heterologous effects of BCG vaccination. Trans. R. Soc. Trop. Med. Hyg. 2015, 109, 29–35. [Google Scholar] [CrossRef]
- Freyne, B.; Marchant, A.; Curtis, N. BCG-associated heterologous immunity, a historical perspective: Intervention studies in animal models of infectious diseases. Trans. R. Soc. Trop. Med. Hyg. 2015, 109, 52–61. [Google Scholar] [CrossRef]
- Aaby, P.; Roth, A.; Ravn, H.; Napirna, B.M.; Rodrigues, A.; Lisse, I.M.; Stensballe, L.; Diness, B.R.; Lausch, K.R.; Lund, N.; et al. Randomized trial of BCG vaccination at birth to low-birth-weight children: Beneficial nonspecific effects in the neonatal period? J. Infect. Dis. 2011, 204, 245–252. [Google Scholar] [CrossRef]
- Biering-Sørensen, S.; Aaby, P.; Lund, N.; Monteiro, I.; Jensen, K.J.; Eriksen, H.B.; Schaltz-Buchholzer, F.; Jørgensen, A.S.P.; Rodrigues, A.; Fisker, A.B.; et al. Early BCG-Denmark and Neonatal Mortality Among Infants Weighing <2500 g: A Randomized Controlled Trial. Clin. Infect. Dis. 2017, 65, 1183–1190. [Google Scholar] [CrossRef]
- Biering-Sørensen, S.; Aaby, P.; Napirna, B.M.; Roth, A.; Ravn, H.; Rodrigues, A.; Whittle, H.; Benn, C.S. Small randomized trial among low-birth-weight children receiving bacillus Calmette-Guérin vaccination at first health center contact. Pediatr. Infect. Dis. J. 2012, 31, 306–308. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, I.; Aaby, P.; Jensen, H. Routine vaccinations and child survival: Follow up study in Guinea-Bissau, West Africa. BMJ 2000, 321, 1435–1438. [Google Scholar] [CrossRef] [Green Version]
- Rieckmann, A.; Villumsen, M.; Sørup, S.; Haugaard, L.K.; Ravn, H.; Roth, A.; Baker, J.L.; Benn, C.S.; Aaby, P. Vaccinations against smallpox and tuberculosis are associated with better long-term survival: A Danish case-cohort study 1971–2010. Int. J. Epidemiol. 2017, 46, 695–705. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, P.; Finn, A.; Curtis, N. Does BCG Vaccination Protect Against Nontuberculous Mycobacterial Infection? A Systematic Review and Meta-Analysis. J. Infect. Dis. 2018, 218, 679–687. [Google Scholar] [CrossRef]
- Cirovic, B.; de Bree, L.C.J.; Groh, L.; Blok, B.A.; Chan, J.; van der Velden, W.; Bremmers, M.E.J.; van Crevel, R.; Händler, K.; Picelli, S.; et al. BCG Vaccination in Humans Elicits Trained Immunity via the Hematopoietic Progenitor Compartment. Cell Host Microbe 2020, 28, 322–334. [Google Scholar] [CrossRef]
- Baldridge, M.T.; King, K.Y.; Boles, N.C.; Weksberg, D.C.; Goodell, M.A. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 2010, 465, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Burberry, A.; Zeng, M.Y.; Ding, L.; Wicks, I.; Inohara, N.; Morrison, S.J.; Nunez, G. Infection mobilizes hematopoietic stem cells through cooperative NOD-like receptor and Toll-like receptor signaling. Cell Host Microbe 2014, 15, 779–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Laval, B.; Maurizio, J.; Kandalla, P.K.; Brisou, G.; Simonnet, L.; Huber, C.; Gimenez, G.; Matcovitch-Natan, O.; Reinhardt, S.; David, E.; et al. C/EBPβ-Dependent Epigenetic Memory Induces Trained Immunity in Hematopoietic Stem Cells. Cell Stem Cell 2020, 26, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Frobel, J.; Landspersky, T.; Percin, G.; Schreck, C.; Rahmig, S.; Ori, A.; Nowak, D.; Essers, M.; Waskow, C.; Oostendorp, R.A.J. The Hematopoietic Bone Marrow Niche Ecosystem. Front. Cell Dev. Biol. 2021, 9, 705410. [Google Scholar] [CrossRef]
- Kamada, R.; Yang, W.; Zhang, Y.; Patel, M.C.; Yang, Y.; Ouda, R.; Dey, A.; Wakabayashi, Y.; Sakaguchi, K.; Fujita, T.; et al. Interferon stimulation creates chromatin marks and establishes transcriptional memory. Proc. Natl. Acad. Sci. USA 2018, 115, E9162–E9171. [Google Scholar] [CrossRef] [Green Version]
- Schnitzler, J.G.; Hoogeveen, R.M.; Ali, L.; Prange, K.H.M.; Waissi, F.; van Weeghel, M.; Bachmann, J.C.; Versloot, M.; Borrelli, M.J.; Yeang, C.; et al. Atherogenic Lipoprotein(a) Increases Vascular Glycolysis, Thereby Facilitating Inflammation and Leukocyte Extravasation. Circ. Res. 2020, 126, 1346–1359. [Google Scholar] [CrossRef]
- Mitroulis, I.; Chen, L.S.; Singh, R.P.; Kourtzelis, I.; Economopoulou, M.; Kajikawa, T.; Troullinaki, M.; Ziogas, A.; Ruppova, K.; Hosur, K.; et al. Secreted protein Del-1 regulates myelopoiesis in the hematopoietic stem cell niche. J. Clin. Investig. 2017, 127, 3624–3639. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Murphy, A.J.; Fleetwood, A.J. Hematopoietic Progenitors and the Bone Marrow Niche Shape the Inflammatory Response and Contribute to Chronic Disease. Int. J. Mol. Sci. 2022, 23, 2234. https://doi.org/10.3390/ijms23042234
Xu Y, Murphy AJ, Fleetwood AJ. Hematopoietic Progenitors and the Bone Marrow Niche Shape the Inflammatory Response and Contribute to Chronic Disease. International Journal of Molecular Sciences. 2022; 23(4):2234. https://doi.org/10.3390/ijms23042234
Chicago/Turabian StyleXu, Yangsong, Andrew J. Murphy, and Andrew J. Fleetwood. 2022. "Hematopoietic Progenitors and the Bone Marrow Niche Shape the Inflammatory Response and Contribute to Chronic Disease" International Journal of Molecular Sciences 23, no. 4: 2234. https://doi.org/10.3390/ijms23042234
APA StyleXu, Y., Murphy, A. J., & Fleetwood, A. J. (2022). Hematopoietic Progenitors and the Bone Marrow Niche Shape the Inflammatory Response and Contribute to Chronic Disease. International Journal of Molecular Sciences, 23(4), 2234. https://doi.org/10.3390/ijms23042234