
Cerebellar and Striatal Implications in Autism Spectrum Disorders: From Clinical Observations to Animal Models


 and
and 
Abstract
:1. Introduction
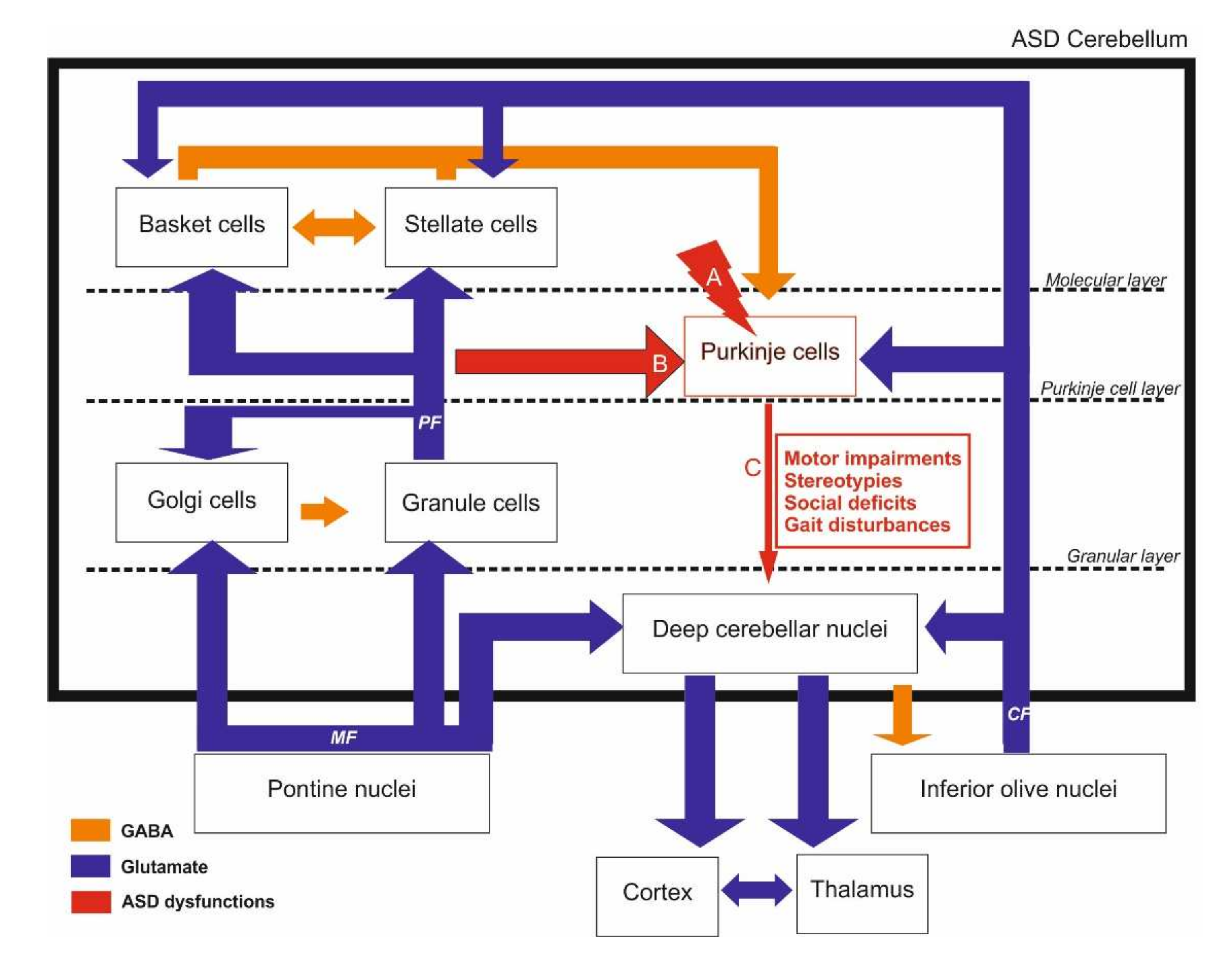
2. Cerebellar Involvement in ASD
2.1. Structure and Function of the Cerebellum
2.2. Anatomical Evidence of Cerebellar Involvement in ASD
2.3. Cellular Correlates
2.4. Neurotransmission Systems Implicated
2.5. Evidence from Our Previous Work
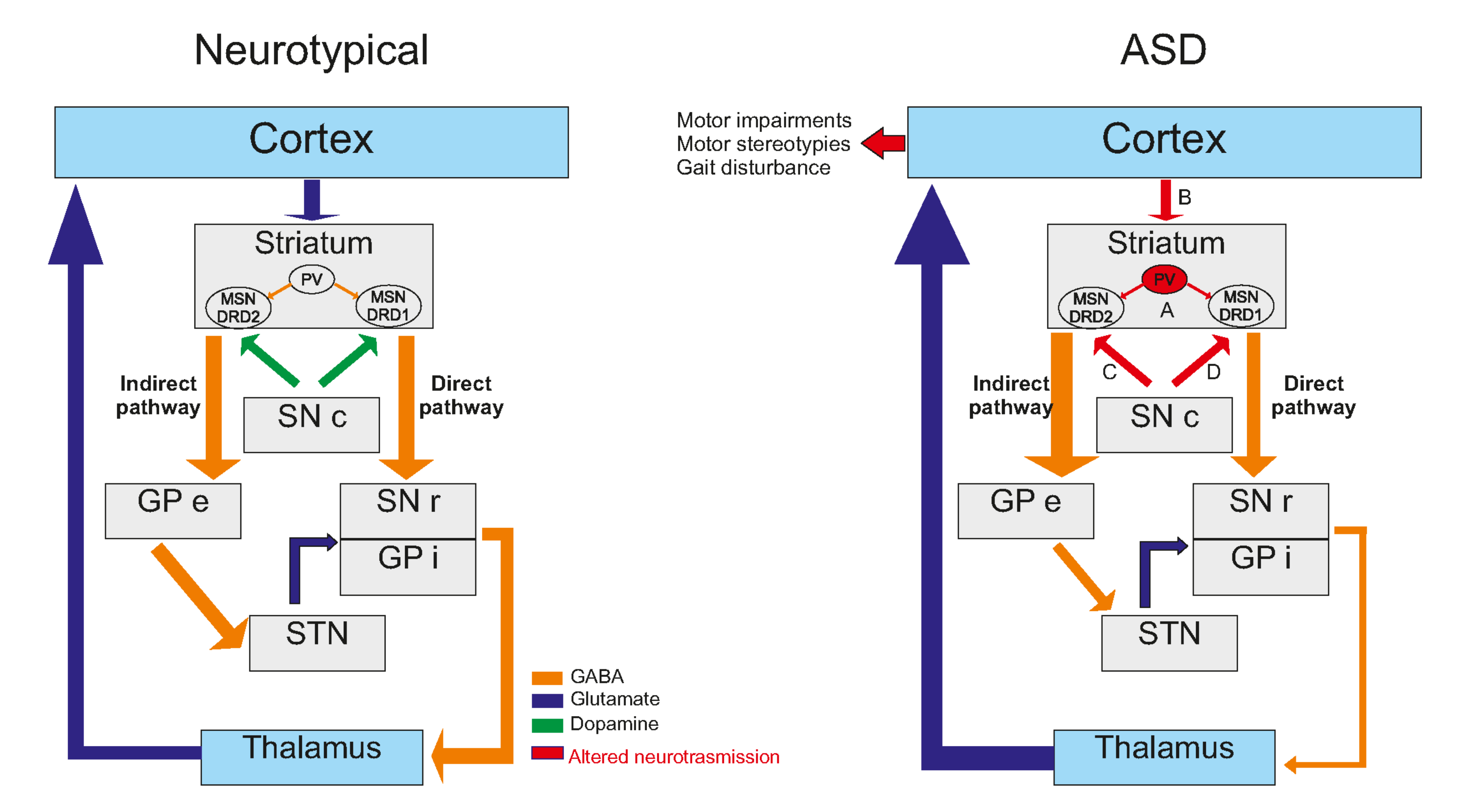
3. Striatal Involvement in ASD
3.1. Anatomical Evidence of Striatal Involvement in ASD
3.2. Cellular Consequences Correlates
3.3. Neurotransmission Systems Implicated
4. Epigenetic Alterations in the Brain of ASD Animal Models
5. Conclusions and Future Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Lai, M.C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef]
- Loomes, R.; Hull, L.; Mandy, W.P.L. What Is the Male-to-Female Ratio in Autism Spectrum Disorder? A Systematic Review and Meta-Analysis. J. Am. Acad. Child Adolesc. Psychiatry 2017, 56, 466–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Nyholt, D.R.; Magnussen, P.; Parano, E.; Pavone, P.; Geschwind, D.; Lord, C.; Iversen, P.; Hoh, J.; Autism Genetic Resource Exchange Consortium; et al. A Genomewide Screen for Autism Susceptibility Loci. Am. J. Hum. Genet. 2001, 69, 327–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folstein, S.E.; Rutter, M.L. Autism: Familial aggregation and genetic implications. J. Autism Dev. Disord. 1988, 18, 3–30. [Google Scholar] [CrossRef] [PubMed]
- Bailey, W.; Popovich, B.; Jones, K.L. Monozygotic twins discordant for Russell-Silver syndrome. Am. J. Med. Genet. 1995, 58, 101–105. [Google Scholar] [CrossRef]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Larsson, H.; Hultman, C.M.; Reichenberg, A. The familial risk of autism. JAMA-J. Am. Med. Assoc. 2014, 311, 1770–1777. [Google Scholar] [CrossRef]
- Constantino, J.N.; Zhang, Y.; Frazier, T.; Abbacchi, A.M.; Law, P. Sibling recurrence and the genetic epidemiology of autism. Am. J. Psychiatry 2010, 167, 1349–1356. [Google Scholar] [CrossRef] [Green Version]
- Risch, N.; Hoffmann, T.J.; Anderson, M.; Croen, L.A.; Grether, J.K.; Windham, G.C. Familial recurrence of autism spectrum disorder: Evaluating genetic and environmental contributions. Am. J. Psychiatry 2014, 171, 1206–1213. [Google Scholar] [CrossRef]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Jiang, H.Y.; Xu, L.L.; Shao, L.; Xia, R.M.; Yu, Z.H.; Ling, Z.X.; Yang, F.; Deng, M.; Ruan, B. Maternal infection during pregnancy and risk of autism spectrum disorders: A systematic review and meta-analysis. Brain Behav. Immun. 2016, 58, 165–172. [Google Scholar] [CrossRef]
- Shigemoto-Mogami, Y.; Hoshikawa, K.; Sato, K. Activated microglia disrupt the blood-brain barrier and induce chemokines and cytokines in a rat in vitro model. Front. Cell. Neurosci. 2018, 12, 494. [Google Scholar] [CrossRef] [PubMed]
- Zawadzka, A.; Cieślik, M.; Adamczyk, A. The Role of Maternal Immune Activation in the Pathogenesis of Autism: A Review of the Evidence, Proposed Mechanisms and Implications for Treatment. Int. J. Mol. Sci. 2021, 22, 11561. [Google Scholar] [CrossRef]
- Phiel, C.J.; Zhang, F.; Huang, E.Y.; Guenther, M.G.; Lazar, M.A.; Klein, P.S. Histone Deacetylase is a Direct Target of Valproic Acid, a Potent Anticonvulsant, Mood Stabilizer, and Teratogen. J. Biol. Chem. 2001, 276, 36734–36741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löscher, W. Basic pharmacology of valproate: A review after 35 years of clinical use for the treatment of epilepsy. CNS Drugs 2002, 16, 669–694. [Google Scholar] [CrossRef] [PubMed]
- Coste, J.; Blotiere, P.O.; Miranda, S.; Mikaeloff, Y.; Peyre, H.; Ramus, F.; Zureik, M.; Weill, A.; Dray-Spira, R. Risk of early neurodevelopmental disorders associated with in utero exposure to valproate and other antiepileptic drugs: A nationwide cohort study in France. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Bailly, Y.; Bossu, J.L. Regional and sex-dependent alterations in Purkinje cell density in the valproate mouse model of autism. Neuroreport 2019, 30, 82–88. [Google Scholar] [CrossRef]
- Morová, M.; Kršková, L. Autistic-Like Traits in Laboratory Rodents Exposed to Phthalic Acid Esters During Early Development – an Animal Model of Autism? Physiol. Res. 2021, 70, 345–361. [Google Scholar] [CrossRef]
- Wöhr, M.; Roullet, F.I.; Hung, A.Y.; Sheng, M.; Crawley, J.N. Communication impairments in mice lacking shank1: Reduced levels of ultrasonic vocalizations and scent marking behavior. PLoS ONE 2011, 6, e0020631. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Bozdagi, O.; Scattoni, M.L.; Wöhr, M.; Roullet, F.I.; Katz, A.M.; Abrams, D.N.; Kalikhman, D.; Simon, H.; Woldeyohannes, L.; et al. Reduced excitatory neurotransmission and mild Autism-Relevant phenotypes in adolescent shank3 null mutant mice. J. Neurosci. 2012, 32, 6525–6541. [Google Scholar] [CrossRef]
- Roy, S.; Watkins, N.; Heck, D. Comprehensive Analysis of Ultrasonic Vocalizations in a Mouse Model of Fragile X Syndrome Reveals Limited, Call Type Specific Deficits. PLoS ONE 2012, 7, e44816. [Google Scholar] [CrossRef] [Green Version]
- Kanner, L. Autistic Disturbances of affective contact. Nerv. Child 1943, 2, 217–250. [Google Scholar]
- Asperger, H. Die „Autistischen Psychopathen” im Kindesalter. Arch. Psychiatr. Nervenkrankh. 1944, 117, 76–136. [Google Scholar] [CrossRef]
- Amaral, D.G.; Schumann, C.M.; Nordahl, C.W. Neuroanatomy of autism. Trends Neurosci. 2008, 31, 137–145. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders; American Psychiatric Association: Washington, DC, USA, 2013; ISBN 0-89042-555-8. [Google Scholar]
- Sarnat, H.B.; Netsky, M.G. When does a ganglion become a brain? Evolutionary origin of the central nervous system. Semin. Pediatr. Neurol. 2002, 9, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Dianne, M. Broussard What Do. In The Cerebellum; Wiley Online Books; Wiley: Hoboken, NJ, USA, 2013; pp. 213–214. ISBN 9781118730133. [Google Scholar]
- Leiner, H.C.; Leiner, A.L.; Dow, R.S. Does the Cerebellum Contribute to Mental Skills? Behav. Neurosci. 1986, 100, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Middleton, F.A.; Strick, P.L. Anatomical evidence for cerebellar and basal ganglia involvement in higher cognitive function. Science 1994, 266, 458–461. [Google Scholar] [CrossRef]
- Allen, G.; Buxton, R.B.; Wong, E.C.; Courchesne, E. Attentional activation of the cerebellum independent of motor involvement. Science 1997, 275, 1940–1943. [Google Scholar] [CrossRef]
- Leroi, I.; O’Hearn, E.; Marsh, L.; Lyketsos, C.G.; Rosenblatt, A.; Ross, C.A.; Brandt, J.; Margolis, R.L. Psychopathology in Patients With Degenerative Cerebellar Diseases: A Comparison to Huntington’s Disease. Am. J. Psychiatry 2002, 159, 1306–1314. [Google Scholar] [CrossRef]
- Ramnani, N.; Behrens, T.E.J.; Johansen-Berg, H.; Richter, M.C.; Pinsk, M.A.; Andersson, J.L.R.; Rudebeck, P.; Ciccarelli, O.; Richter, W.; Thompson, A.J.; et al. The Evolution of Prefrontal Inputs to the Cortico-pontine System: Diffusion Imaging Evidence from Macaque Monkeys and Humans. Cereb. Cortex 2006, 16, 811–818. [Google Scholar] [CrossRef] [Green Version]
- Sugihara, I. Crus I in the Rodent Cerebellum: Its Homology to Crus I and II in the Primate Cerebellum and Its Anatomical Uniqueness among Neighboring Lobules. Cerebellum 2018, 17, 49–55. [Google Scholar] [CrossRef]
- Braitenberg, V.; Atwood, R.P. Morphological observations on the cerebellar cortex. J. Comp. Neurol. 1958, 109, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Eccles, J.C. An instruction-selection theory of learning in the cerebellar cortex. Brain Res. 1977, 127, 327–352. [Google Scholar] [CrossRef]
- Ellegood, J.; Pacey, L.K.; Hampson, D.R.; Lerch, J.P.; Henkelman, R.M. Anatomical phenotyping in a mouse model of fragile X syndrome with magnetic resonance imaging. Neuroimage 2010, 53, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Reith, R.M.; McKenna, J.; Wu, H.; Hashmi, S.S.; Cho, S.H.; Dash, P.K.; Gambello, M.J. Loss of Tsc2 in Purkinje cells is associated with autistic-like behavior in a mouse model of tuberous sclerosis complex. Neurobiol. Dis. 2013, 51, 93–103. [Google Scholar] [CrossRef]
- Varghese, M.; Keshav, N.; Jacot-Descombes, S.; Warda, T.; Wicinski, B.; Dickstein, D.L.; Harony-Nicolas, H.; De Rubeis, S.; Drapeau, E.; Buxbaum, J.D.; et al. Autism spectrum disorder: Neuropathology and animal models. Acta Neuropathol. 2017, 134, 537–566. [Google Scholar] [CrossRef]
- Al Sagheer, T.; Haida, O.; Balbous, A.; Francheteau, M.; Matas, E.; Fernagut, P.O.; Jaber, M. Motor impairments correlate with social deficits and restricted neuronal loss in an environmental model of autism. Int. J. Neuropsychopharmacol. 2018, 21, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Haida, O.; Al Sagheer, T.; Balbous, A.; Francheteau, M.; Matas, E.; Soria, F.; Fernagut, P.O.; Jaber, M. Sex-dependent behavioral deficits and neuropathology in a maternal immune activation model of autism. Transl. Psychiatry 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Matas, E.; Maisterrena, A.; Thabault, M.; Balado, E.; Francheteau, M.; Balbous, A.; Galvan, L.; Jaber, M. Major motor and gait deficits with sexual dimorphism in a Shank3 mutant mouse model. Mol. Autism 2021, 12. [Google Scholar] [CrossRef]
- Main, S.L.; Kulesza, R.J. Repeated prenatal exposure to valproic acid results in cerebellar hypoplasia and ataxia. Neuroscience 2017, 340, 34–47. [Google Scholar] [CrossRef]
- Mejias, R.; Chiu, S.L.; Han, M.; Rose, R.; Gil-Infante, A.; Zhao, Y.; Huganir, R.L.; Wang, T. Purkinje cell-specific Grip1/2 knockout mice show increased repetitive self-grooming and enhanced mGluR5 signaling in cerebellum. Neurobiol. Dis. 2019, 132. [Google Scholar] [CrossRef]
- Hassan, T.H.; Abdelrahman, H.M.; Abdel Fattah, N.R.; El-Masry, N.M.; Hashim, H.M.; El-Gerby, K.M.; Abdel Fattah, N.R. Blood and brain glutamate levels in children with autistic disorder. Res. Autism Spectr. Disord. 2013, 7, 541–548. [Google Scholar] [CrossRef]
- Ingram, J.L.; Peckham, S.M.; Tisdale, B.; Rodier, P.M. Prenatal exposure of rats to valproic acid reproduces the cerebellar anomalies associated with autism. Neurotoxicol. Teratol. 2000, 22, 319–324. [Google Scholar] [CrossRef]
- Mychasiuk, R.; Richards, S.; Nakahashi, A.; Kolb, B.; Gibb, R. Effects of rat prenatal exposure to valproic acid on behaviour and neuro-anatomy. Dev. Neurosci. 2012, 34, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Servais, L.; Bearzatto, B.; Schwaller, B.; Dumont, M.; De Saedeleer, C.; Dan, B.; Barski, J.J.; Schiffmann, S.N.; Cheron, G. Mono- and dual-frequency fast cerebellar oscillation in mice lacking parvalbumin and/or calbindin D-28k. Eur. J. Neurosci. 2005, 22, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Piochon, C.; Kloth, A.D.; Grasselli, G.; Titley, H.K.; Nakayama, H.; Hashimoto, K.; Wan, V.; Simmons, D.H.; Eissa, T.; Nakatani, J.; et al. Cerebellar plasticity and motor learning deficits in a copy-number variation mouse model of autism. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albus, J.S. A theory of cerebellar function. Math. Biosci. 1971, 10, 25–61. [Google Scholar] [CrossRef]
- Kemper, T.L.; Bauman, M.L. The contribution of neuropathologic studies to the understanding of autism. Neurol. Clin. 1993, 11, 175–187. [Google Scholar] [CrossRef]
- Ghaziuddin, M.; Butler, E.; Tsai, L.; Ghaziuddin, N. Is clumsiness a marker for Asperger syndrome? J. Intellect. Disabil. Res. 2008, 38, 519–527. [Google Scholar] [CrossRef] [Green Version]
- Gowen, E.; Hamilton, A. Motor abilities in autism: A review using a computational context. J. Autism Dev. Disord. 2013, 43, 323–344. [Google Scholar] [CrossRef]
- Maudsley, H. Reviews and Notices. BMJ 1867, 1, 540–541. [Google Scholar] [CrossRef]
- Limperopoulos, C.; Bassan, H.; Gauvreau, K.; Robertson, R.L.; Sullivan, N.R.; Benson, C.B.; Avery, L.; Stewart, J.; Soul, J.S.; Ringer, S.A.; et al. Does cerebellar injury in premature infants contribute to the high prevalence of long-term cognitive, learning, and behavioral disability in survivors? Pediatrics 2007, 120, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Courchesne, E. Brainstem, cerebellar and limbic neuroanatomical abnormalities in autism. Curr. Opin. Neurobiol. 1997, 7, 269–278. [Google Scholar] [CrossRef]
- Bailey, A.; Luthert, P.; Dean, A.; Harding, B.; Janota, I.; Montgomery, M.; Rutter, M.; Lantos, P. A clinicopathological study of autism. In The Science of Mental Health: Volume 2: Autism; Routledge: New York, NY, USA, 2013; Volume 121, pp. 141–157. ISBN 9781136800818. [Google Scholar]
- Baron-Cohen, S.; Ashwin, E.; Ashwin, C.; Tavassoli, T.; Chakrabarti, B. Talent in autism: Hyper-systemizing, hyper-attention to detail and sensory hypersensitivity. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 1377–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.R.; Hewedi, D.H.; Eissa, A.M.; Moustafa, A.A. The Cerebellum and Psychiatric Disorders. Front. Public Health 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- Arnett, A.B.; Wang, T.; Eichler, E.E.; Bernier, R.A. Reflections on the genetics-first approach to advancements in molecular genetic and neurobiological research on neurodevelopmental disorders. J. Neurodev. Disord. 2021, 13, 24. [Google Scholar] [CrossRef] [PubMed]
- Murakami, J.W.; Courchesne, E.; Press, G.A.; Yeung-Courchesne, R.; Hesselink, J.R. Reduced Cerebellar Hemisphere Size and Its Relationship to Vermal Hypoplasia in Autism. Arch. Neurol. 1989, 46, 689–694. [Google Scholar] [CrossRef]
- Allen, G.; Müller, R.A.; Courchesne, E. Cerebellar function in autism: Functional magnetic resonance image activation during a simple motor task. Biol. Psychiatry 2004, 56, 269–278. [Google Scholar] [CrossRef]
- Ellegood, J.; Crawley, J.N. Behavioral and Neuroanatomical Phenotypes in Mouse Models of Autism. Neurotherapeutics 2015, 12, 521–533. [Google Scholar] [CrossRef]
- Dum, R.P.; Strick, P.L. An unfolded map of the cerebellar dentate nucleus and its projections to the cerebral cortex. J. Neurophysiol. 2003, 89, 634–639. [Google Scholar] [CrossRef]
- Peter, S.; De Zeeuw, C.I.; Boeckers, T.M.; Schmeisser, M.J. Cerebellar and striatal pathologies in mouse models of autism spectrum disorder. In Advances in Anatomy Embryology and Cell Biology; Springer: New York, NY, USA, 2017; Volume 224, pp. 103–119. [Google Scholar]
- Belichenko, N.P.; Belichenko, P.V.; Hong, H.L.; Mobley, W.C.; Francke, U. Comparative Study of Brain Morphology in Mecp2 Mutant Mouse Models of Rett Syndrome. J. Comp. Neurol. 2008, 508, 184–195. [Google Scholar] [CrossRef]
- Aldinger, K.A.; Kogan, J.; Kimonis, V.; Fernandez, B.; Horn, D.; Klopocki, E.; Chung, B.; Toutain, A.; Weksberg, R.; Millen, K.J.; et al. Cerebellar and posterior fossa malformations in patients with autism-associated chromosome 22q13 terminal deletion. Am. J. Med. Genet. Part A 2013, 161, 131–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.; Scherrer, B.; Prohl, A.K.; Filip-Dhima, R.; Kapur, K.; Kolevzon, A.; Buxbaum, J.D.; Berry-Kravis, E.; Soorya, L.; Thurm, A.; et al. Volumetric Analysis of the Basal Ganglia and Cerebellar Structures in Patients with Phelan-McDermid Syndrome. Pediatr. Neurol. 2019, 90, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Jamain, S.; Radyushkin, K.; Hammerschmidt, K.; Granon, S.; Boretius, S.; Varoqueaux, F.; Ramanantsoa, N.; Gallego, J.; Ronnenberg, A.; Winter, D.; et al. Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc. Natl. Acad. Sci. USA 2008, 105, 1710–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akshoomoff, N.; Lord, C.; Lincoln, A.J.; Courchesne, R.Y.; Carper, R.A.; Townsend, J.; Courchesne, E. Outcome classification of preschool children with autism spectrum disorders using MRI brain measures. J. Am. Acad. Child Adolesc. Psychiatry 2004, 43, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Courchesne, E.; Karns, C.M.; Davis, H.R.; Ziccardi, R.; Carper, R.A.; Tigue, Z.D.; Chisum, H.J.; Moses, P.; Pierce, K.; Lord, C.; et al. Unusual brain growth patterns in early life in patients with autistic disorder: An MRI study. Neurology 2001, 57, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Brito, A.R.; Vasconcelos, M.M.; Domingues, R.C.; Hygino Da Cruz, L.C.; Rodrigues, L.D.S.; Gasparetto, E.L.; Calçada, C.A.B.P. Diffusion tensor imaging findings in school-aged autistic children. J. Neuroimaging 2009, 19, 337–343. [Google Scholar] [CrossRef]
- Hanaie, R.; Mohri, I.; Kagitani-Shimono, K.; Tachibana, M.; Azuma, J.; Matsuzaki, J.; Watanabe, Y.; Fujita, N.; Taniike, M. Altered Microstructural Connectivity of the Superior Cerebellar Peduncle is Related to Motor Dysfunction in Children with Autistic Spectrum Disorders. Cerebellum 2013, 12, 645–656. [Google Scholar] [CrossRef]
- Noonan, S.K.; Haist, F.; Müller, R.A. Aberrant functional connectivity in autism: Evidence from low-frequency BOLD signal fluctuations. Brain Res. 2009, 1262, 48–63. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.J.; Nair, A.; Keown, C.L.; Datko, M.C.; Lincoln, A.J.; Müller, R.A. Cerebro-cerebellar resting-state functional connectivity in children and adolescents with autism spectrum disorder. Biol. Psychiatry 2015, 78, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Pierce, K.; Courchesne, E. Evidence for a cerebellar role in reduced exploration and stereotyped behavior in autism. Biol. Psychiatry 2001, 49, 655–664. [Google Scholar] [CrossRef]
- Stoodley, C.J.; Valera, E.M.; Schmahmann, J.D. Functional topography of the cerebellum for motor and cognitive tasks: An fMRI study. Neuroimage 2012, 59, 1560–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, S.J.; Bird, G.; Brindley, R.; Frith, C.D.; Burgess, P.W. Atypical recruitment of medial prefrontal cortex in autism spectrum disorders: An fMRI study of two executive function tasks. Neuropsychologia 2008, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, E.; Meng, F.; Fujita, H.; Morgado, F.; Kazemi, Y.; Rice, L.C.; Ren, C.; Escamilla, C.O.; Gibson, J.M.; Sajadi, S.; et al. Regulation of autism-relevant behaviors by cerebellar–prefrontal cortical circuits. Nat. Neurosci. 2020, 23, 1102–1110. [Google Scholar] [CrossRef]
- Stoodley, C.J.; D’Mello, A.M.; Ellegood, J.; Jakkamsetti, V.; Liu, P.; Nebel, M.B.; Gibson, J.M.; Kelly, E.; Meng, F.; Cano, C.A.; et al. Altered cerebellar connectivity in autism and cerebellar-mediated rescue of autism-related behaviors in mice. Nat. Neurosci. 2017, 20, 1744–1751. [Google Scholar] [CrossRef]
- Vilensky, J.A. Gait Disturbances in Patients with Autistic Behavior. Arch. Neurol. 1981, 38. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, M.W.; Wang, Z.; Schmitt, L.M.; Tsai, P.; Sweeney, J.A. The role of cerebellar circuitry alterations in the pathophysiology of autism spectrum disorders. Front. Neurosci. 2015, 9, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegiel, J.; Flory, M.; Kuchna, I.; Nowicki, K.; Ma, S.Y.; Imaki, H.; Wegiel, J.; Cohen, I.L.; London, E.; Wisniewski, T.; et al. Stereological study of the neuronal number and volume of 38 brain subdivisions of subjects diagnosed with autism reveals significant alterations restricted to the striatum, amygdala and cerebellum. Acta Neuropathol. Commun. 2014, 2. [Google Scholar] [CrossRef] [Green Version]
- Fatemi, S.H.; Halt, A.R.; Realmuto, G.; Earle, J.; Kist, D.A.; Thuras, P.; Merz, A. Purkinje Cell Size Is Reduced in Cerebellum of Patients with Autism. Cell. Mol. Neurobiol. 2002, 22, 171–175. [Google Scholar] [CrossRef]
- Bailey, A. A clinicopathological study of autism. Brain 1998, 121, 889–905. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.-W.; Tiwari, V.N.; Behen, M.E.; Chugani, H.T.; Chugani, D.C. In vivo detection of reduced Purkinje cell fibers with diffusion MRI tractography in children with autistic spectrum disorders. Front. Hum. Neurosci. 2014, 8, 110. [Google Scholar] [CrossRef] [Green Version]
- Skefos, J.; Cummings, C.; Enzer, K.; Holiday, J.; Weed, K.; Levy, E.; Yuce, T.; Kemper, T.; Bauman, M. Regional alterations in Purkinje cell density in patients with autism. PLoS ONE 2014, 9, e0081255. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.M.; Wright, C.L. Convergence of Sex Differences and the Neuroimmune System in Autism Spectrum Disorder. Biol. Psychiatry 2017, 81, 402–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haraguchi, S.; Sasahara, K.; Shikimi, H.; Honda, S.I.; Harada, N.; Tsutsui, K. Estradiol promotes purkinje dendritic growth, spinogenesis, and synaptogenesis during neonatal life by inducing the expression of BDNF. Cerebellum 2012, 11, 416–417. [Google Scholar] [CrossRef]
- Hoffman, J.F.; Wright, C.L.; McCarthy, M.M. A critical period in purkinje cell development is mediated by local estradiol synthesis, disrupted by inflammation, and has enduring consequences only for males. J. Neurosci. 2016, 36, 10039–10049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecellio, M.; Schwaller, B.; Meyer, M.; Hunziker, W.; Celio, M.R. Alterations in Purkinje cell spines of calbindin D-28 k and parvalbumin knock-out mice. Eur. J. Neurosci. 2000, 12, 945–954. [Google Scholar] [CrossRef]
- Leblond, C.S.; Nava, C.; Polge, A.; Gauthier, J.; Huguet, G.; Lumbroso, S.; Giuliano, F.; Stordeur, C.; Depienne, C.; Mouzat, K.; et al. Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: A Gradient of Severity in Cognitive Impairments. PLoS Genet. 2014, 10, e1004580. [Google Scholar] [CrossRef] [Green Version]
- Koekkoek, S.K.E.; Yamaguchi, K.; Milojkovic, B.A.; Dortland, B.R.; Ruigrok, T.J.H.; Maex, R.; De Graaf, W.; Smit, A.E.; VanderWerf, F.; Bakker, C.E.; et al. Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X syndrome. Neuron 2005, 47, 339–352. [Google Scholar] [CrossRef] [Green Version]
- Tobia, M.J.; Woodruff-Pak, D.S. Delay Eyeblink Classical Conditioning is Impaired in Fragile X Syndrome. Behav. Neurosci. 2009, 123, 665. [Google Scholar] [CrossRef] [Green Version]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef]
- Sobaniec-Lotowska, M.E. Ultrastructure of Purkinje cell perikarya and their dendritic processes in the rat cerebellar cortex in experimental encephalopathy induced by chronic application of valproate. Int. J. Exp. Pathol. 2001, 82, 337–348. [Google Scholar] [CrossRef]
- Whitney, E.R.; Kemper, T.L.; Rosene, D.L.; Bauman, M.L.; Blatt, G.J. Density of cerebellar basket and stellate cells in autism: Evidence for a late developmental loss of Purkinje cells. J. Neurosci. Res. 2009, 87, 2245–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubenstein, J.L.R.; Merzenich, M.M. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003, 2, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Purcell, A.E.; Jeon, O.H.; Zimmerman, A.W.; Blue, M.E.; Pevsner, J. Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology 2001, 57, 1618–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeVito, T.J.; Drost, D.J.; Neufeld, R.W.J.; Rajakumar, N.; Pavlosky, W.; Williamson, P.; Nicolson, R. Evidence for Cortical Dysfunction in Autism: A Proton Magnetic Resonance Spectroscopic Imaging Study. Biol. Psychiatry 2007, 61, 465–473. [Google Scholar] [CrossRef]
- Neill Epperson, C.; Haga, K.; Mason, G.F.; Sellers, E.; Gueorguieva, R.; Zhang, W.; Weiss, E.; Rothman, D.L.; Krystal, J.H. Cortical gamma-aminobutyric acid levels across the menstrual cycle in healthy women and those with premenstrual dysphoric disorder: A proton magnetic resonance spectroscopy study. Arch. Gen. Psychiatry 2002, 59, 851–858. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, A.; Jones, M.; Milne, E. Measuring neural excitation and inhibition in autism: Different approaches, different findings and different interpretations. Brain Res. 2016, 1648, 277–289. [Google Scholar] [CrossRef]
- Peter, S.; Ten Brinke, M.M.; Stedehouder, J.; Reinelt, C.M.; Wu, B.; Zhou, H.; Zhou, K.; Boele, H.J.; Kushner, S.A.; Lee, M.G.; et al. Dysfunctional cerebellar Purkinje cells contribute to autism-like behaviour in Shank2-deficient mice. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Ha, S.; Lee, D.; Cho, Y.S.; Chung, C.; Yoo, Y.E.; Kim, J.; Lee, J.; Kim, W.; Kim, H.; Bae, Y.; et al. Cerebellar shank2 regulates excitatory synapse density, motor coordination, and specific repetitive and anxiety-like behaviors. J. Neurosci. 2016, 36, 12129–12143. [Google Scholar] [CrossRef] [Green Version]
- Tsai, P.T.; Hull, C.; Chu, Y.; Greene-Colozzi, E.; Sadowski, A.R.; Leech, J.M.; Steinberg, J.; Crawley, J.N.; Regehr, W.G.; Sahin, M. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 2012, 488, 647–651. [Google Scholar] [CrossRef] [Green Version]
- Fatemi, S.H.; Halt, A.R.; Stary, J.M.; Kanodia, R.; Schulz, S.C.; Realmuto, G.R. Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol. Psychiatry 2002, 52, 805–810. [Google Scholar] [CrossRef]
- Yip, J.; Soghomonian, J.-J.; Blatt, G.J. IncreasedGAD67 mRNA expression in cerebellar interneurons in autism: Implications for Purkinje cell dysfunction. J. Neurosci. Res. 2008, 86. [Google Scholar] [CrossRef] [PubMed]
- Dhossche, D.; Applegate, H.; Abraham, A.; Maertens, P.; Bland, L.; Bencsath, A.; Martinez, J. Elevated plasma gamma-aminobutyric acid (GABA) levels in autistic youngsters: Stimulus for a GABA hypothesis of autism. Med. Sci. Monit. 2002, 8, PR1–PR6. [Google Scholar] [PubMed]
- Pétriz, A.; Reyes-Haro, D.; González-González, M.A.; Miledi, R.; Martínez-Torres, A. GABAρ subunits confer a bicuculline-insensitive component to GFAP + cells of cerebellum. Proc. Natl. Acad. Sci. USA 2014, 111, 17522–17527. [Google Scholar] [CrossRef] [Green Version]
- Varman, D.R.; Soria-Ortíz, M.B.; Martínez-Torres, A.; Reyes-Haro, D. GABAρ3 expression in lobule X of the cerebellum is reduced in the valproate model of autism. Neurosci. Lett. 2018, 687, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Hong, A.; Zhang, A.; Ke, Y.; El Idrissi, A.; Shen, C.H. Downregulation of GABA(A) β subunits is transcriptionally controlled by Fmr1p. J. Mol. Neurosci. 2012, 46, 272–275. [Google Scholar] [CrossRef]
- Heulens, I.; D’Hulst, C.; Van Dam, D.; De Deyn, P.P.; Kooy, R.F. Pharmacological treatment of fragile X syndrome with GABAergic drugs in a knockout mouse model. Behav. Brain Res. 2012, 229, 244–249. [Google Scholar] [CrossRef]
- Henderson, C.; Wijetunge, L.; Kinoshita, M.N.; Shumway, M.; Hammond, R.S.; Postma, F.R.; Brynczka, C.; Rush, R.; Thomas, A.; Paylor, R.; et al. Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Folsom, T.D.; Kneeland, R.E.; Liesch, S.B. Metabotropic glutamate Receptor 5 upregulation in children with autism is associated with underexpression of both fragile X mental retardation protein and GABA A receptor beta 3 in adults with autism. Anat. Rec. 2011, 294, 1635–1645. [Google Scholar] [CrossRef] [Green Version]
- Muddashetty, R.S.; Kelić, S.; Gross, C.; Xu, M.; Bassell, G.J. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J. Neurosci. 2007, 27, 5338–5348. [Google Scholar] [CrossRef]
- Bear, M.F.; Huber, K.M.; Warren, S.T. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004, 27, 370–377. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Aldinger, K.A.; Ashwood, P.; Bauman, M.L.; Blaha, C.D.; Blatt, G.J.; Chauhan, A.; Chauhan, V.; Dager, S.R.; Dickson, P.E.; et al. Consensus paper: Pathological role of the cerebellum in Autism. Cerebellum 2012, 11, 777–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicidomini, C.; Ponzoni, L.; Lim, D.; Schmeisser, M.J.; Reim, D.; Morello, N.; Orellana, D.; Tozzi, A.; Durante, V.; Scalmani, P.; et al. Pharmacological enhancement of mGlu5 receptors rescues behavioral deficits in SHANK3 knock-out mice. Mol. Psychiatry 2017, 22, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blatt, G.J. GABAergic Cerebellar System In Autism: A Neuropathological And Developmental Perspective. Int. Rev. Neurobiol. 2005, 71, 167–178. [Google Scholar] [PubMed]
- Ito, M.; Sakurai, M.; Tongroach, P. Climbing fibre induced depression of both mossy fibre responsiveness and glutamate sensitivity of cerebellar Purkinje cells. J. Physiol. 1982, 324, 113–134. [Google Scholar] [CrossRef] [PubMed]
- Kano, M.; Hashimoto, K.; Kurihara, H.; Watanabe, M.; Inoue, Y.; Aiba, A.; Tonegawa, S. Persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking mGluR1. Neuron 1997, 18, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Ichise, T.; Kano, M.; Hashimoto, K.; Yanagihara, D.; Nakao, K.; Shigemoto, R.; Katsuki, M.; Aiba, A. mGluR1 in cerebellar Purkinje cells essential for long-term depression, synapse elimination, and motor coordination. Science 2000, 288, 1832–1835. [Google Scholar] [CrossRef]
- Bannai, H.; Niwa, F.; Sherwood, M.W.; Shrivastava, A.N.; Arizono, M.; Miyamoto, A.; Sugiura, K.; Lévi, S.; Triller, A.; Mikoshiba, K. Bidirectional Control of Synaptic GABAAR Clustering by Glutamate and Calcium. Cell Rep. 2015, 13, 2768. [Google Scholar] [CrossRef]
- Nicolini, C.; Fahnestock, M. The valproic acid-induced rodent model of autism. Exp. Neurol. 2018, 299, 217–227. [Google Scholar] [CrossRef]
- Shi, L.; Smith, S.E.P.; Malkova, N.; Tse, D.; Su, Y.; Patterson, P.H. Activation of the maternal immune system alters cerebellar development in the offspring. Brain Behav. Immun. 2009, 23, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Kouser, M.; Speed, H.E.; Dewey, C.M.; Reimers, J.M.; Widman, A.J.; Gupta, N.; Liu, S.; Jaramillo, T.C.; Bangash, M.; Xiao, B.; et al. Loss of predominant shank3 isoforms results in hippocampus-dependent impairments in behavior and synaptic transmission. J. Neurosci. 2013, 33, 18448–18468. [Google Scholar] [CrossRef]
- Lanciego, J.L.; Luquin, N.; Obeso, J.A. Functional neuroanatomy of the basal ganglia. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Kreitzer, A.C. Physiology and Pharmacology of Striatal Neurons. Annu. Rev. Neurosci. 2009, 32, 127–147. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, K.; Brandenburg, C.; Orsati, F.; Soghomonian, J.J.; Hussman, J.P.; Blatt, G.J. Basal ganglia and autism—A translational perspective. Autism Res. 2017, 10, 1751–1775. [Google Scholar] [CrossRef] [PubMed]
- Creasey, H.; Rumsey, J.M.; Schwartz, M.; Duara, R.; Rapoport, J.L.; Rapoport, S.I. Brain Morphometry in Autistic Men as Measured by Volumetric Computed Tomography. Arch. Neurol. 1986, 43, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Aylward, E.H.; Schwartz, J.; Machlin, S.; Pearl, G. Bicaudate Ratio as a Measure of Caudate Volume on MR Images. Am. J. Neuroradiol. 1991, 12, 1217–1222. [Google Scholar]
- Reiss, A.L.; Faruque, F.; Naidu, S.; Abrams, M.; Beaty, T.; Bryan, R.N.; Moser, H. Neuroanatomy of Rett syndrome: A volumetric imaging study. Ann. Neurol. 1993, 34, 227–234. [Google Scholar] [CrossRef]
- Hollander, E.; Anagnostou, E.; Chaplin, W.; Esposito, K.; Haznedar, M.M.; Licalzi, E.; Wasserman, S.; Soorya, L.; Buchsbaum, M. Striatal volume on magnetic resonance imaging and repetitive behaviors in autism. Biol. Psychiatry 2005, 58, 226–232. [Google Scholar] [CrossRef]
- Langen, M.; Schnack, H.G.; Nederveen, H.; Bos, D.; Lahuis, B.E.; de Jonge, M.V.; van Engeland, H.; Durston, S. Changes in the Developmental Trajectories of Striatum in Autism. Biol. Psychiatry 2009, 66, 327–333. [Google Scholar] [CrossRef]
- Rojas, D.C.; Peterson, E.; Winterrowd, E.; Reite, M.L.; Rogers, S.J.; Tregellas, J.R. Regional gray matter volumetric changes in autism associated with social and repetitive behavior symptoms. BMC Psychiatry 2006, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Portmann, T.; Yang, M.; Mao, R.; Panagiotakos, G.; Ellegood, J.; Dolen, G.; Bader, P.L.; Grueter, B.A.; Goold, C.; Fisher, E.; et al. Behavioral abnormalities and circuit defects in the basal ganglia of a mouse model of 16p11.2 deletion syndrome. Cell Rep. 2014, 7, 1077–1092. [Google Scholar] [CrossRef] [Green Version]
- Ellegood, J.; Anagnostou, E.; Babineau, B.A.; Crawley, J.N.; Lin, L.; Genestine, M.; Dicicco-Bloom, E.; Lai, J.K.Y.; Foster, J.A.; Peñagarikano, O.; et al. Clustering autism: Using neuroanatomical differences in 26 mouse models to gain insight into the heterogeneity. Mol. Psychiatry 2015, 20, 118–125. [Google Scholar] [CrossRef]
- Kuo, H.Y.; Liu, F.C. Valproic acid induces aberrant development of striatal compartments and corticostriatal pathways in a mouse model of autism spectrum disorder. FASEB J. 2017, 31, 4458–4471. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, S.L.; Polis, B.; David, O.; Morris, G. Striatal cholinergic interneurons exert inhibition on competing default behaviours controlled by the nucleus accumbens and dorsolateral striatum. Eur. J. Neurosci. 2021, 53, 2078–2089. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Zhao, H.; Li, B.; Sen, Y.; Lu, W.; Yongjin, T.; Zhujun, L.; Dazhang, B.; Caijuan, L.; Yingqi, L.; et al. CRISPR/Cas9-mediated disruption of SHANK3 in monkey leads to drug-treatable autism-like symptoms. Hum. Mol. Genet. 2019, 28, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Lingawi, N.W.; Balleine, B.W. Amygdala central nucleus interacts with dorsolateral striatum to regulate the acquisition of habits. J. Neurosci. 2012, 32, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuccillo, M.V. Striatal circuits as a common node for autism pathophysiology. Front. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavi, S.; Iezzi, D.; Manduca, A.; Leone, S.; Melancia, F.; Carbone, C.; Petrella, M.; Mannaioni, G.; Masi, A.; Trezza, V. Reward-Related Behavioral, Neurochemical and Electrophysiological Changes in a Rat Model of Autism Based on Prenatal Exposure to Valproic Acid. Front. Cell. Neurosci. 2019, 13, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauber, E.; Filice, F.; Schwaller, B. Prenatal valproate exposure differentially affects parvalbumin-expressing neurons and related circuits in the cortex and striatum of mice. Front. Mol. Neurosci. 2016, 9, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauber, E.; Filice, F.; Schwaller, B. Dysregulation of Parvalbumin Expression in the Cntnap2−/− Mouse Model of Autism Spectrum Disorder. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaber, M.; Robinson, S.W.; Missale, C.; Caron, M.G. Dopamine receptors and brain function. Neuropharmacology 1996, 35, 1503–1519. [Google Scholar] [CrossRef]
- Kreitzer, A.C.; Malenka, R.C. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature 2007, 445, 643–647. [Google Scholar] [CrossRef]
- Peça, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Li, C.; Chen, Q.; Van Der Goes, M.S.; Hawrot, J.; Yao, A.Y.; Gao, X.; Lu, C.; Zang, Y.; Zhang, Q.; et al. Striatopallidal dysfunction underlies repetitive behavior in Shank3-deficient model of autism. J. Clin. Investig. 2017, 127, 1978–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandenburg, C.; Soghomonian, J.J.; Zhang, K.; Sulkaj, I.; Randolph, B.; Kachadoorian, M.; Blatt, G.J. Increased Dopamine Type 2 Gene Expression in the Dorsal Striatum in Individuals with Autism Spectrum Disorder Suggests Alterations in Indirect Pathway Signaling and Circuitry. Front. Cell. Neurosci. 2020, 14, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Filice, F.; Janickova, L.; Henzi, T.; Bilella, A.; Schwaller, B. The Parvalbumin Hypothesis of Autism Spectrum Disorder. Front. Cell. Neurosci. 2020, 14, 1–24. [Google Scholar] [CrossRef]
- Li, W.; Pozzo-Miller, L. Dysfunction of the corticostriatal pathway in autism spectrum disorders. J. Neurosci. Res. 2020, 98, 2130–2147. [Google Scholar] [CrossRef]
- Modi, M.E.; Brooks, J.M.; Guilmette, E.R.; Beyna, M.; Graf, R.; Reim, D.; Schmeisser, M.J.; Boeckers, T.M.; O’Donnell, P.; Buhl, D.L. Hyperactivity and hypermotivation associated with increased striatal mglur1 signaling in a Shank2 rat model of autism. Front. Mol. Neurosci. 2018, 11, 1–17. [Google Scholar] [CrossRef]
- Provenzano, G.; Zunino, G.; Genovesi, S.; Sgadó, P.; Bozzi, Y. Mutant mouse models of autism spectrum disorders. Dis. Markers 2012, 33, 225–239. [Google Scholar] [CrossRef]
- Rothwell, P.E.; Fuccillo, M.V.; Maxeiner, S.; Hayton, S.J.; Gokce, O.; Lim, B.K.; Fowler, S.C.; Malenka, R.C.; Südhof, T.C. Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell 2014, 158, 198–212. [Google Scholar] [CrossRef] [Green Version]
- Espinosa, F.; Xuan, Z.; Liu, S.; Powell, C.M. Neuroligin 1 modulates striatal glutamatergic neurotransmission in a pathway and NMDAR subunit-specific manner. Front. Synaptic Neurosci. 2015, 7, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Rapanelli, M.; Frick, L.R.; Xu, M.; Groman, S.M.; Jindachomthong, K.; Tamamaki, N.; Tanahira, C.; Taylor, J.R.; Pittenger, C. Targeted Interneuron Depletion in the Dorsal Striatum Produces Autism-like Behavioral Abnormalities in Male but Not Female Mice. Biol. Psychiatry 2017, 82, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Poppi, L.A.; Ho-Nguyen, K.T.; Shi, A.; Daut, C.T.; Tischfield, M.A. Recurrent Implication of Striatal Cholinergic Interneurons in a Range of Neurodevelopmental, Neurodegenerative, and Neuropsychiatric Disorders. Cells 2021, 10, 907. [Google Scholar] [CrossRef]
- Aliane, V.; Pérez, S.; Bohren, Y.; Deniau, J.M.; Kemel, M.L. Key role of striatal cholinergic interneurons in processes leading to arrest of motor stereotypies. Brain 2011, 134, 110–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Kobets, A.; Du, J.C.; Lennington, J.; Li, L.; Banasr, M.; Duman, R.S.; Vaccarino, F.M.; DiLeone, R.J.; Pittenger, C. Targeted ablation of cholinergic interneurons in the dorsolateral striatum produces behavioral manifestations of tourette syndrome. Proc. Natl. Acad. Sci. USA 2015, 112, 893–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito-Ishida, A.; Ure, K.; Chen, H.; Swann, J.W.; Zoghbi, H.Y. Loss of MeCP2 in Parvalbumin-and Somatostatin-Expressing Neurons in Mice Leads to Distinct Rett Syndrome-like Phenotypes. Neuron 2015, 88, 651–658. [Google Scholar] [CrossRef] [Green Version]
- Owen, S.F.; Berke, J.D.; Kreitzer, A.C. Fast-Spiking Interneurons Supply Feedforward Control of Bursting, Calcium, and Plasticity for Efficient Learning. Cell 2018, 172, 683–695.e15. [Google Scholar] [CrossRef] [Green Version]
- Wöhr, M.; Orduz, D.; Gregory, P.; Moreno, H.; Khan, U.; Vörckel, K.J.; Wolfer, D.P.; Welzl, H.; Gall, D.; Schiffmann, S.N.; et al. Lack of parvalbumin in mice leads to behavioral deficits relevant to all human autism core symptoms and related neural morphofunctional abnormalities. Transl. Psychiatry 2015, 5, e525. [Google Scholar] [CrossRef]
- Filice, F.; Vörckel, K.J.; Sungur, A.Ö.; Wöhr, M.; Schwaller, B. Reduction in parvalbumin expression not loss of the parvalbumin-expressing GABA interneuron subpopulation in genetic parvalbumin and shank mouse models of autism. Mol. Brain 2016, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Petrelli, F.; Pucci, L.; Bezzi, P. Astrocytes and microglia and their potential link with autism spectrum disorders. Front. Cell. Neurosci. 2016, 10, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Lioy, D.T.; Garg, S.K.; Monaghan, C.E.; Raber, J.; Foust, K.D.; Kaspar, B.K.; Hirrlinger, P.G.; Kirchhoff, F.; Bissonnette, J.M.; Ballas, N.; et al. A role for glia in the progression of Rett-syndrome. Nature 2011, 475, 497–500. [Google Scholar] [CrossRef]
- Cope, E.C.; Briones, B.A.; Brockett, A.T.; Martinez, S.; Vigneron, P.A.; Opendak, M.; Wang, S.S.H.; Gould, E. Immature neurons and radial glia, but not astrocytes or microglia, are altered in adult Cntnap2 and Shank3 mice, models of autism. eNeuro 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.X.; Kim, G.H.; Tan, J.W.; Riso, A.E.; Sun, Y.; Xu, E.Y.; Liao, G.Y.; Xu, H.; Lee, S.H.; Do, N.Y.; et al. Elevated protein synthesis in microglia causes autism-like synaptic and behavioral aberrations. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Mineur, Y.S.; Huynh, L.X.; Crusio, W.E. Social behavior deficits in the Fmr1 mutant mouse. Behav. Brain Res. 2006, 168, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Clipperton-Allen, A.E.; Page, D.T. Pten haploinsufficient mice show broad brain overgrowth but selective impairments in autism-relevant behavioral tests. Hum. Mol. Genet. 2014, 23, 3490–3505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clipperton-Allen, A.E.; Page, D.T. Decreased aggression and increased repetitive behavior in Pten haploinsufficient mice. Genes Brain Behav. 2015, 14, 145–157. [Google Scholar] [CrossRef]
- Kazdoba, T.M.; Leach, P.T.; Silverman, J.L.; Crawley, J.N. Modeling fragile X syndrome in the Fmr1 knockout mouse. Intractable Rare Dis. Res. 2014, 3, 118–133. [Google Scholar] [CrossRef] [Green Version]
- Ravenscroft, P.; Brotchie, J. NMDA receptors in the basal ganglia. J. Anat. 2000, 196, 577–585. [Google Scholar] [CrossRef]
- Stefani, A.; Chen, Q.; Flores-Hernandez, J.; Jiao, Y.; Reiner, A.; Surmeier, D.J. Physiological and molecular properties of AMPA/kainate receptors expressed by striatal medium spiny neurons. Dev. Neurosci. 1998, 20, 242–252. [Google Scholar] [CrossRef]
- Blundell, J.; Blaiss, C.A.; Etherton, M.R.; Espinosa, F.; Tabuchi, K.; Walz, C.; Bolliger, M.F.; Südhof, T.C.; Powell, C.M. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J. Neurosci. 2010, 30, 2115–2129. [Google Scholar] [CrossRef] [Green Version]
- Benthall, K.N.; Cording, K.R.; Agopyan-Miu, A.H.C.W.; Wong, C.D.; Chen, E.Y.; Bateup, H.S. Loss of Tsc1 from striatal direct pathway neurons impairs endocannabinoid-LTD and enhances motor routine learning. Cell Rep. 2021, 36. [Google Scholar] [CrossRef]
- Jung, K.M.; Sepers, M.; Henstridge, C.M.; Lassalle, O.; Neuhofer, D.; Martin, H.; Ginger, M.; Frick, A.; Dipatrizio, N.V.; MacKie, K.; et al. Uncoupling of the endocannabinoid signalling complex in a mouse model of fragile X syndrome. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martella, G.; Meringolo, M.; Trobiani, L.; De Jaco, A.; Pisani, A.; Bonsi, P. The R451C-Neuroligin 3 mutation hampers the expression of long-term synaptic depression in the dorsal striatum. Eur. J. Neurosci. 2017, 47, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, T.C.; Speed, H.E.; Xuan, Z.; Reimers, J.M.; Liu, S.; Powell, C.M. Altered Striatal Synaptic Function and Abnormal Behaviour in Shank3 Exon4-9 Deletion Mouse Model of Autism. Autism Res. 2016, 9, 350–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, E.; Huynh, T.N.; MacAskill, A.F.; Carter, A.G.; Pierre, P.; Ruggero, D.; Kaphzan, H.; Klann, E. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 2013, 493, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Zurek, A.A.; Kemp, S.W.P.; Aga, Z.; Walker, S.; Milenkovic, M.; Ramsey, A.J.; Sibille, E.; Scherer, S.W.; Orser, B.A. α5GABAA receptor deficiency causes autism-like behaviors. Ann. Clin. Transl. Neurol. 2016, 3, 392–398. [Google Scholar] [CrossRef] [PubMed]
- DeLorey, T.M. GABRB3 Gene Deficient Mice: A Potential Model of Autism Spectrum Disorder. Int. Rev. Neurobiol. 2005, 71, 359–382. [Google Scholar] [CrossRef]
- Platt, R.J.; Zhou, Y.; Slaymaker, I.M.; Shetty, A.S.; Weisbach, N.R.; Kim, J.A.; Sharma, J.; Desai, M.; Sood, S.; Kempton, H.R.; et al. Chd8 Mutation Leads to Autistic-like Behaviors and Impaired Striatal Circuits. Cell Rep. 2017, 19, 335–350. [Google Scholar] [CrossRef] [Green Version]
- Centonze, D.; Rossi, S.; Mercaldo, V.; Napoli, I.; Ciotti, M.T.; De Chiara, V.; Musella, A.; Prosperetti, C.; Calabresi, P.; Bernardi, G.; et al. Abnormal Striatal GABA Transmission in the Mouse Model for the Fragile X Syndrome. Biol. Psychiatry 2008, 63, 963–973. [Google Scholar] [CrossRef]
- Aman, M.; Rettiganti, M.; Nagaraja, H.N.; Hollway, J.A.; Mccracken, J.; Mcdougle, C.J.; Tierney, E.; Scahill, L.; Arnold, L.E.; Hellings, J.; et al. Tolerability, Safety, and Benefits of Risperidone in Children and Adolescents with Autism: 21-Month Follow-up after 8-Week Placebo-Controlled Trial. J. Child Adolesc. Psychopharmacol. 2015, 25, 482. [Google Scholar] [CrossRef]
- Aman, M.G.; Kasper, W.; Manos, G.; Mathew, S.; Marcus, R.; Owen, R.; Mankoski, R. Line-item analysis of the Aberrant Behavior Checklist: Results from two studies of aripiprazole in the treatment of irritability associated with autistic disorder. J. Child Adolesc. Psychopharmacol. 2010, 20, 415–422. [Google Scholar] [CrossRef]
- Zürcher, N.R.; Walsh, E.C.; Phillips, R.D.; Cernasov, P.M.; Tseng, C.E.J.; Dharanikota, A.; Smith, E.; Li, Z.; Kinard, J.L.; Bizzell, J.C.; et al. A simultaneous [11C]raclopride positron emission tomography and functional magnetic resonance imaging investigation of striatal dopamine binding in autism. Transl. Psychiatry 2021, 11, 33. [Google Scholar] [CrossRef] [PubMed]
- Kosillo, P.; Bateup, H.S. Dopaminergic Dysregulation in Syndromic Autism Spectrum Disorders: Insights from Genetic Mouse Models. Front. Neural Circuits 2021, 15. [Google Scholar] [CrossRef] [PubMed]
- Shonesy, B.C.; Parrish, W.P.; Haddad, H.K.; Stephenson, J.R.; Báldi, R.; Bluett, R.J.; Marks, C.R.; Centanni, S.W.; Folkes, O.M.; Spiess, K.; et al. Role of Striatal Direct Pathway 2-Arachidonoylglycerol Signaling in Sociability and Repetitive Behavior. Biol. Psychiatry 2018, 84, 304–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Han, P.-L. Early-Life Stress in D2 Heterozygous Mice Promotes Autistic-like Behaviors through the Downregulation of the BDNF-TrkB Pathway in the Dorsal Striatum. Exp. Neurobiol. 2019, 28, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Morel, L.; Higashimori, H.; Tolman, M.; Yang, Y. VGluT1+ Neuronal Glutamatergic signaling regulates postnatal developmental maturation of cortical protoplasmic astroglia. J. Neurosci. 2014, 34, 10950–10962. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, Y.; Park, J.Y.; Kim, J.E.; Kim, T.K.; Choi, J.; Lee, J.E.; Lee, E.H.; Kim, D.; Kim, K.S.; et al. Loss of Adenylyl Cyclase Type-5 in the Dorsal Striatum Produces Autistic-Like Behaviors. Mol. Neurobiol. 2017, 54, 7994–8008. [Google Scholar] [CrossRef]
- Herbert, M.R. Contributions of the environment and environmentally vulnerable physiology to autism spectrum disorders. Curr. Opin. Neurol. 2010, 23, 103–110. [Google Scholar] [CrossRef]
- Perera, F.; Herbstman, J. Prenatal environmental exposures, epigenetics, and disease. Reprod. Toxicol. 2011, 31, 363–373. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Jiang, Y.; Tsai, T.F.; Bressler, J.; Beaudet, A.L. Imprinting in Angelman and Prader-Willi syndromes. Curr. Opin. Genet. Dev. 1998, 8, 334–342. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremblay, M.W.; Jiang, Y. DNA Methylation and Susceptibility to Autism Spectrum Disorder. Annu. Rev. Med. 2019, 70, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Matthew, D.; et al. Global Epigenomic Reconfiguration During Mammalian Brain Development. Science 2013, 341, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Nardone, S.; Sharan Sams, D.; Reuveni, E.; Getselter, D.; Oron, O.; Karpuj, M.; Elliott, E. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl. Psychiatry 2014, 4, e433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardone, S.; Sams, D.S.; Zito, A.; Reuveni, E.; Elliott, E. Dysregulation of cortical neuron DNA methylation profile in autism spectrum disorder. Cereb. Cortex 2017, 27, 5739–5754. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Poschmann, J.; Cruz-Herrera del Rosario, R.; Parikshak, N.N.; Hajan, H.S.; Kumar, V.; Ramasamy, R.; Belgard, T.G.; Elanggovan, B.; Wong, C.C.Y.; et al. Histone Acetylome-wide Association Study of Autism Spectrum Disorder. Cell 2016, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, H.; de la Torre-Ubieta, L.; Stein, J.L.; Parikshak, N.N.; Huang, J.; Opland, C.K.; Gandal, M.J.; Sutton, G.J.; Hormozdiari, F.; Lu, D.; et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 2016, 538. [Google Scholar] [CrossRef] [Green Version]
- Steullet, P.; Cabungcal, J.H.; Monin, A.; Dwir, D.; O’Donnell, P.; Cuenod, M.; Do, K.Q. Redox dysregulation, neuroinflammation, and NMDA receptor hypofunction: A “central hub” in schizophrenia pathophysiology? Schizophr. Res. 2016, 176, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Richetto, J.; Massart, R.; Weber-Stadlbauer, U.; Szyf, M.; Riva, M.A.; Meyer, U. Genome-wide DNA Methylation Changes in a Mouse Model of Infection-Mediated Neurodevelopmental Disorders. Biol. Psychiatry 2017, 81, 265–276. [Google Scholar] [CrossRef]
- Basil, P.; Li, Q.; Dempster, E.L.; Mill, J.; Sham, P.C.; Wong, C.C.Y.; McAlonan, G.M. Prenatal maternal immune activation causes epigenetic differences in adolescent mouse brain. Transl. Psychiatry 2014, 4. [Google Scholar] [CrossRef]
- Dinan, T.G.; Cryan, J.F. Brain–gut–microbiota axis—Mood, metabolism and behaviour. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 69–70. [Google Scholar] [CrossRef] [PubMed]
- Sabit, H.; Tombuloglu, H.; Rehman, S.; Almandil, N.B.; Cevik, E.; Abdel-Ghany, S.; Rashwan, S.; Abasiyanik, M.F.; Yee Waye, M.M. Gut microbiota metabolites in autistic children: An epigenetic perspective. Heliyon 2021, 7, e06105. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Species | Treated Animal | Periodicity | Dose | Age of Treatment | Phenotype |
|---|---|---|---|---|---|
| Xenopus [13] | Embryo | 24 h exposition of the eggs |
| Stage 8 embryo |
|
| C57BL/6J mice [25] | Pregnant female | One IP injection | 450 mg/kg | E12.5 |
|
| C57BL/6J mice [16] | Pregnant female | One IP injection | 600 mg/kg | E12 |
|
| C57BL/6J mice [26] | Pregnant female | One oral administration | 600 mg/kg | E12 |
|
| CD-1 and GFAP-eGFP mice [27] | Pregnant female | One IP injection | 500 mg/kg | E12.5 |
|
| FVB/NJ mice [28] | Pregnant female | One IP injection | 400 mg/kg | E11.5 or E12.75 | E11.5
E12.75
|
| Long Evans rats [29] | Pregnant female | One IP injection | 600 mg/kg | E12.5 |
|
| Long Evans rats [30] | Pregnant female | One oral administration | 800 mg/kg | E12 |
|
| Sprague-Dawley rats [31] | Pregnant female | Two oral administrations | 800 mg/kg | E10 and E12 |
|
| Winstar rats [32] | Young rat | Daily intragastric administration | 200 mg/kg | 1 month 3 months 6 months 9 months 12 months |
|
| Winstar rats [33] | Pregnant female | One IP injection | 500 mg/kg | E12.5 |
|
| Children | Teenagers | Adults | Animal Models | |||
|---|---|---|---|---|---|---|
| <5 years old | 5 to 14 years old | 14 to 21 years old | >21 years old | |||
| Anatomical impairment | Global cerebellar volume |
|
|
|
 Fmr1KO [64] and Nlgn4KO− [65], Fmr1KO [64] and Nlgn4KO− [65],= Shank3∆C/∆C, VPA and Poly I:C models [25,66,67] | |
| White matter (WM) changes |
|
|
|
| ||
| Connectivity |
|
|
| |||
 in the left supplementary motor areas [72] in the left supplementary motor areas [72] Global right overconnectivity [73] | ||||||
| Cellular correlates | Purkinje cell (PC) |
|
|
| PC density [63] PC arborization in LPS rats [84] Dendritic spine density in VPA rats [30] | |
| Bergmann cells |
|
|
| |||
| Microglia |
|
|
| |||
| Neurotransmission | Glutamate |
|
glutamate & glutamate metabolite levels [87] | mGluR5 expression in Shank3∆c/∆c [66] | ||
| GABA |
|
|
|
|
GABA-A ρ3 levels in VPA model [27] GABA-A β1 & β2 levels in Fmr1 KO [99] | |
| Children | Teenagers | Adults | Animal Models | |||
|---|---|---|---|---|---|---|
| <5 yo | 5 to 14 yo | 14 to 21 yo | >21 yo | |||
| Anatomical impairment | Volume |
|
| |||
| ||||||
| Matrice/striosome organization |
|
|
|
| ||
| Cellular correlates | Medium spiny neurons |
|
|
| ||
| PV interneurons |
|
| ||||
| Astrocyte |
|
|
| |||
| Microglia |
|
|
| |||
| Neurotransmission | Glutamate |
|
|
|
| |
| GABA |
|
|
|
|
mIPSCs frequency in ventral DRD1-expressing MSNs only [174] sIPSCs & mIPSCs frequency in Fmr1 KO mice [175] | |
| Dopamine |
|
|
|
|
| |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thabault, M.; Turpin, V.; Maisterrena, A.; Jaber, M.; Egloff, M.; Galvan, L. Cerebellar and Striatal Implications in Autism Spectrum Disorders: From Clinical Observations to Animal Models. Int. J. Mol. Sci. 2022, 23, 2294. https://doi.org/10.3390/ijms23042294
Thabault M, Turpin V, Maisterrena A, Jaber M, Egloff M, Galvan L. Cerebellar and Striatal Implications in Autism Spectrum Disorders: From Clinical Observations to Animal Models. International Journal of Molecular Sciences. 2022; 23(4):2294. https://doi.org/10.3390/ijms23042294
Chicago/Turabian StyleThabault, Mathieu, Valentine Turpin, Alexandre Maisterrena, Mohamed Jaber, Matthieu Egloff, and Laurie Galvan. 2022. "Cerebellar and Striatal Implications in Autism Spectrum Disorders: From Clinical Observations to Animal Models" International Journal of Molecular Sciences 23, no. 4: 2294. https://doi.org/10.3390/ijms23042294
APA StyleThabault, M., Turpin, V., Maisterrena, A., Jaber, M., Egloff, M., & Galvan, L. (2022). Cerebellar and Striatal Implications in Autism Spectrum Disorders: From Clinical Observations to Animal Models. International Journal of Molecular Sciences, 23(4), 2294. https://doi.org/10.3390/ijms23042294