
Role of Mitochondrial Pathways in Cell Apoptosis during He-Patic Ischemia/Reperfusion Injury

 , ,
, ,
Abstract
:1. Introduction
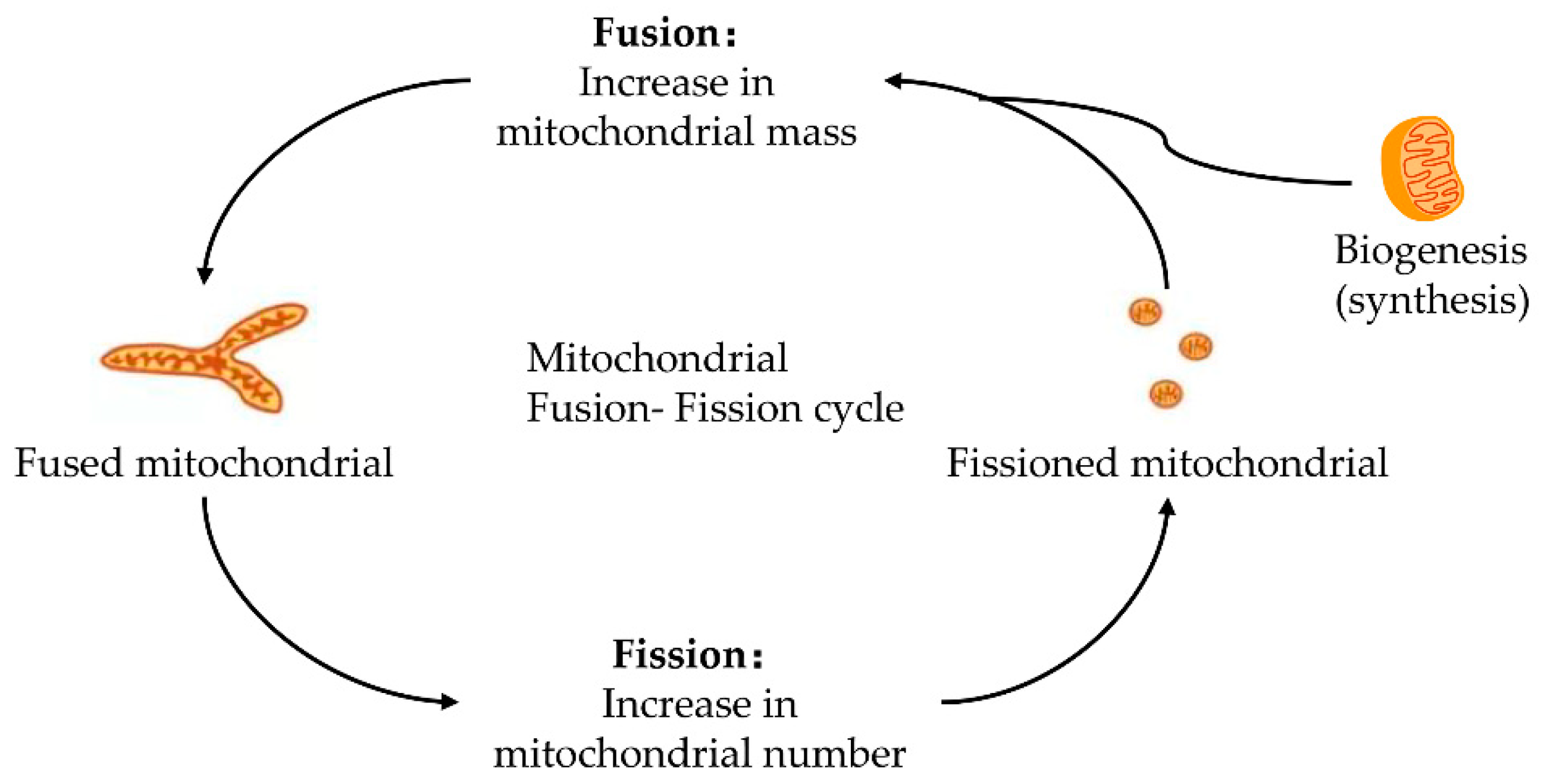
2. The Initiation of Apoptosis: Mitochondrial Fission and Fusion
2.1. Mitochondrial Fission and Fission
2.2. Mitochondrial Fusion
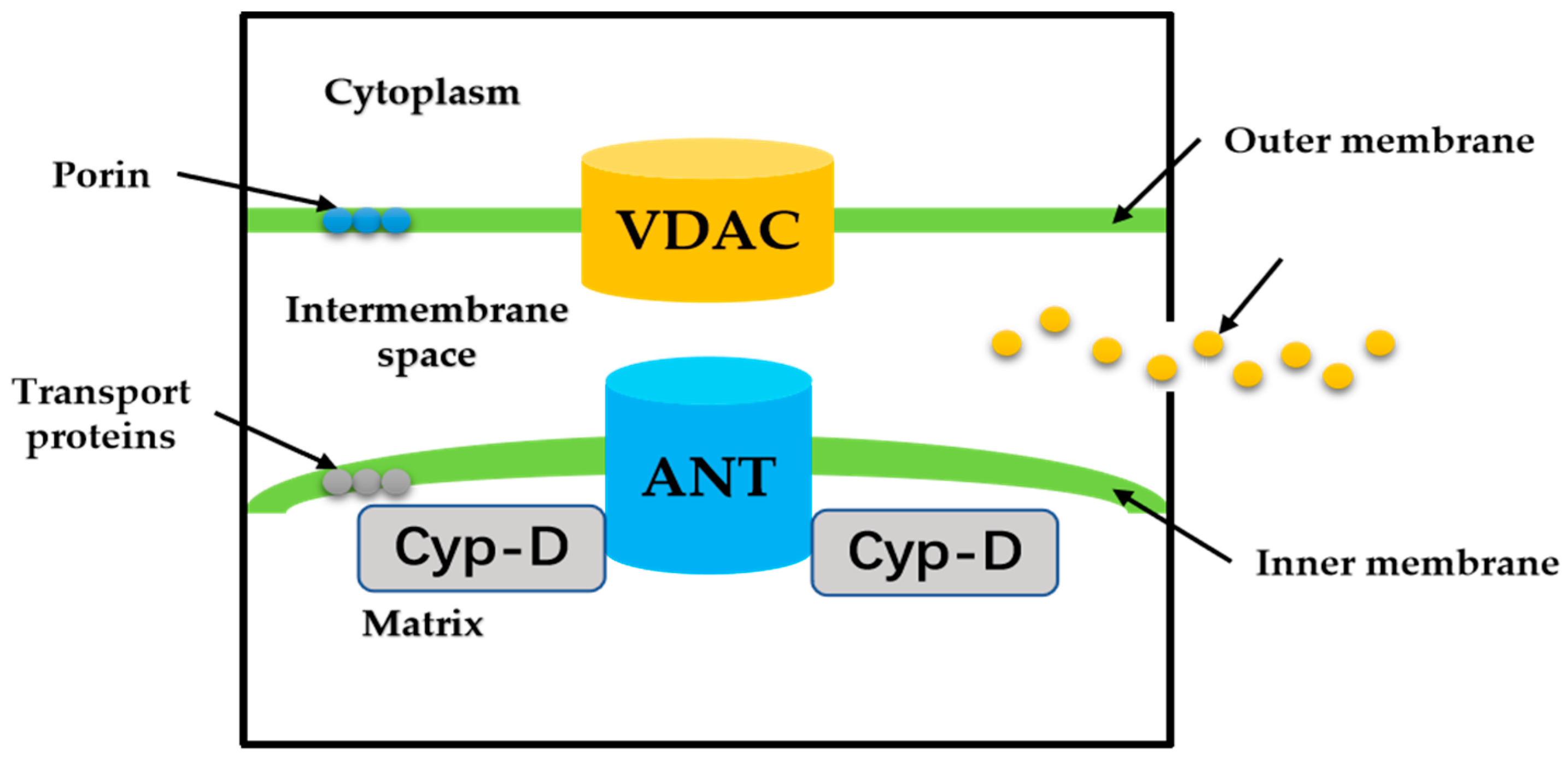
3. “Switch” Role of Mitochondrial Permeability Transition Pore
3.1. ROS Triggered MPTP Opening during Hepatic IRI
3.2. Calcium Overload and MPTP Opening during IRI
3.3. Mitochondrial Membrane Potential Loss and MPTP Opening during IRI
3.4. Regulatory Role of Akt/GSK-3β Pathway on MPTP Opening
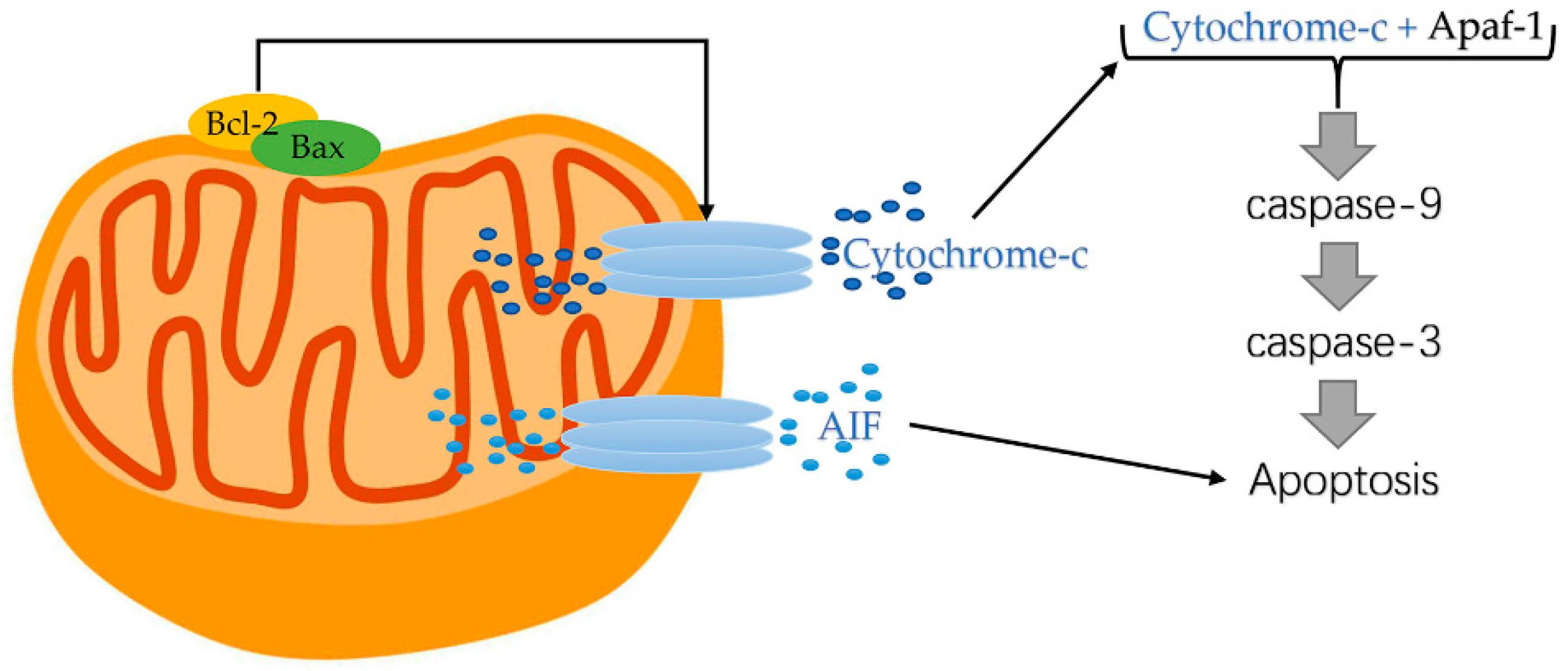
4. The Release of Apoptosis-Related Proteins
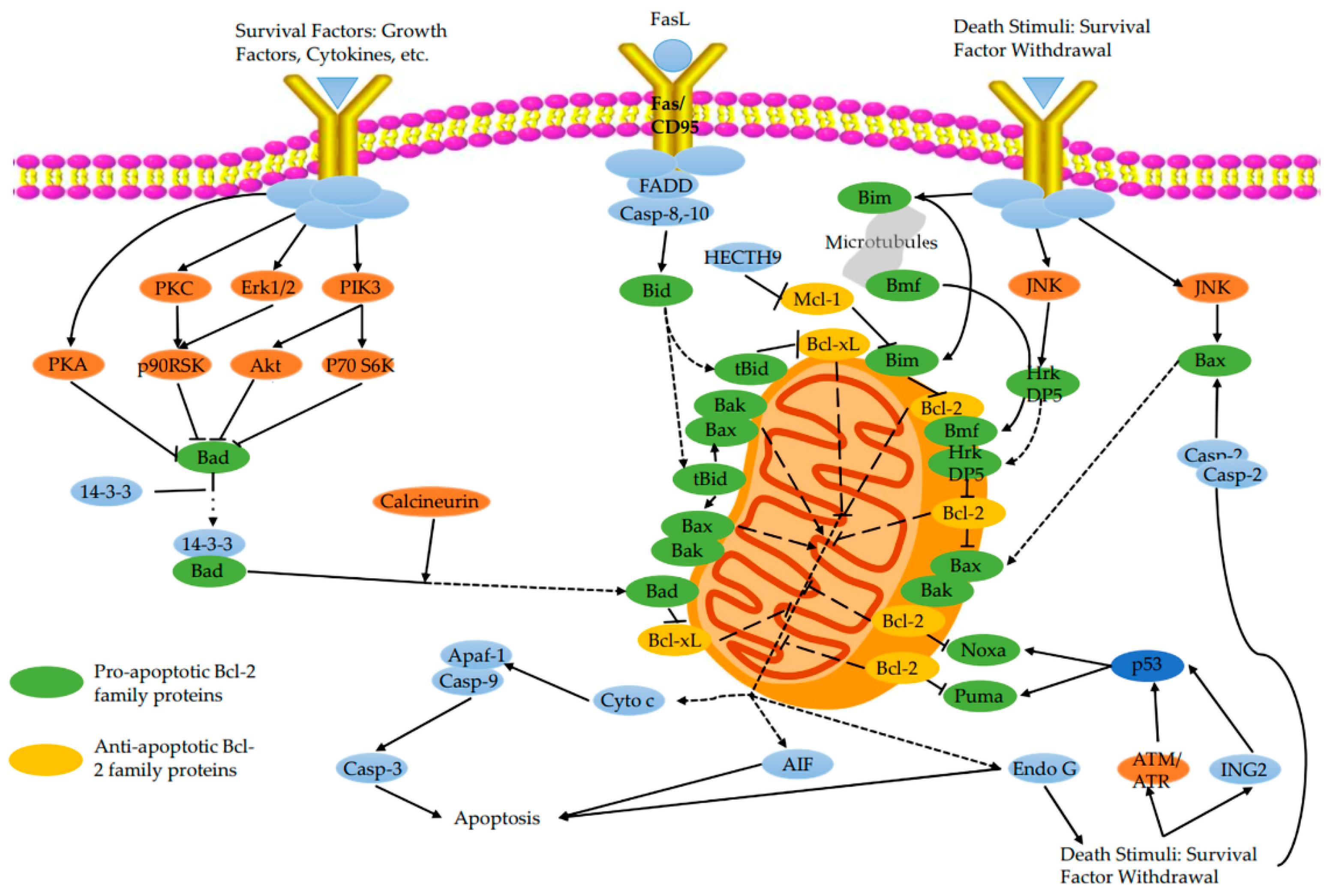
5. Regulatory Role of B Cell Lymphoma-2(Bcl-2) Family Proteins
5.1. Pro-Apoptotic Bcl-2 Family Proteins
5.2. Anti-Apoptotic Bcl-2 Family Proteins
6. ATP Depletion
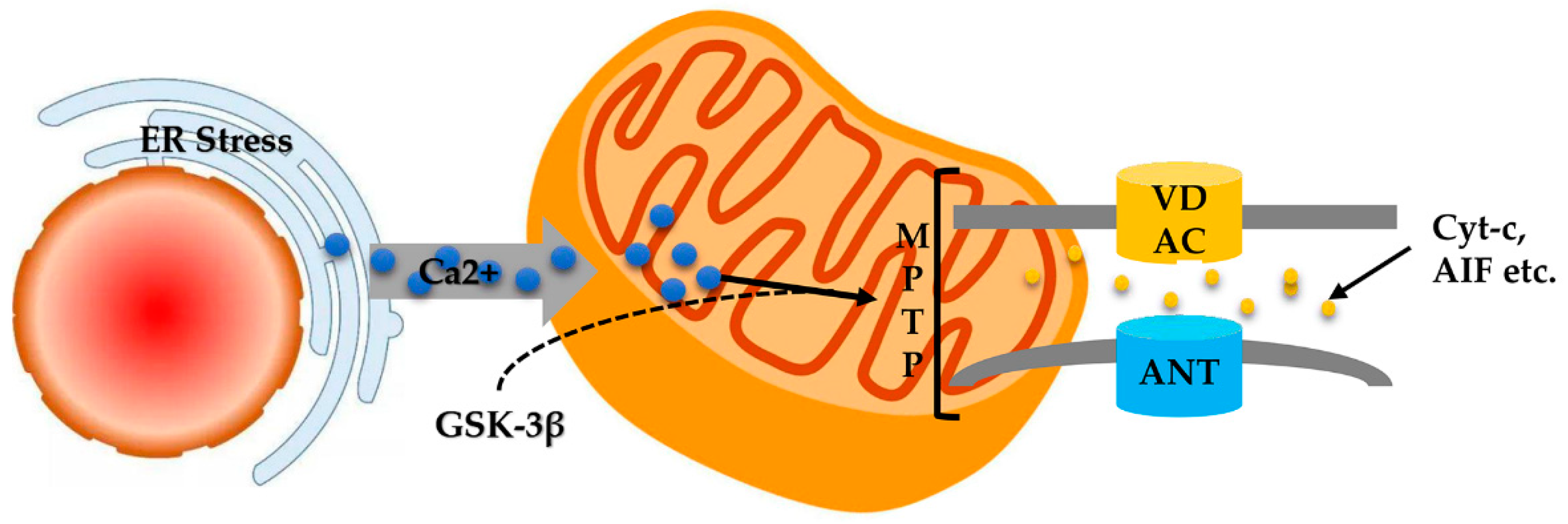
7. Endoplasmic Reticulum Stress
8. The Role of Nitric Oxide in I/R Injury
9. Other Mitochondrial-Mediated Apoptosis Signaling Pathways
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kerr, J.F. History of the events leading to the formulation of the apoptosis concept. Toxicology 2002, 181, 471–474. [Google Scholar] [CrossRef]
- Xu, X.; Lai, Y.; Hua, Z.C. Apoptosis and apoptotic body: Disease message and therapeutic target potentials. Biosci. Rep. 2019, 39, BSR20180992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.L.; Tuli, R.; Manner, P.A.; Sharkey, P.F.; Hall, D.J.; Tuan, R.S. Direct and indirect induction of apoptosis in human mesenchymal stem cells in response to titanium particles. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2003, 21, 697–707. [Google Scholar] [CrossRef]
- Bi, J.; Zhang, J.; Ren, Y.; Du, Z.; Li, Q.; Wang, Y.; Wei, S.; Yang, L.; Zhang, J.; Liu, C.; et al. Irisin alleviates liver ischemia-reperfusion injury by inhibiting excessive mitochondrial fission, promoting mitochondrial biogenesis and decreasing oxidative stress. Redox Biol. 2019, 20, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.W.; He, X.Y.; Yan, A.L.; Wang, S.W.; Li, S.; Nian, S.; Wang, Y.L.; Liang, F.L. Ischemic postconditioning lightening ischemia/reperfusion apoptosis of rats via mitochondria pathway. Eur. Rev. Med. Pharm. Sci. 2019, 23, 6307–6314. [Google Scholar]
- Guicciardi, M.E.; Malhi, H.; Mott, J.L.; Gores, G.J. Apoptosis and Necrosis in the Liver. Compr. Physiol. 2013, 3, 977–1010. [Google Scholar] [PubMed] [Green Version]
- Zhang, L.; Zhang, S.N.; Li, L.; Zhang, X.B.; Wu, R.C.; Liu, J.H. Prolonged warm ischemia aggravates hepatic mitochondria damage and apoptosis in DCD liver by regulating Ca(2+)/CaM/CaMKII signaling pathway. Int. J. Clin. Exp. Pathol. 2019, 12, 217–228. [Google Scholar]
- Serviddio, G.; Bellanti, F.; Sastre, J.; Vendemiale, G.; Altomare, E. Targeting mitochondria: A new promising approach for the treatment of liver diseases. Curr. Med. Chem. 2010, 17, 2325–2337. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.J.; Li, W.; An, W. Adenoviral gene transfer of hepatic stimulator substance confers resistance against hepatic ischemia-reperfusion injury by improving mitochondrial function. Hum. Gene Ther. 2013, 24, 443–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, L.Y.; Fu, P.Y.; Li, P.D.; Li, Z.N.; Liu, H.Y.; Xin, M.G.; Li, W. Mechanisms of hepatic ischemia-reperfusion injury and protective effects of nitric oxide. World J. Gastrointest. Surg. 2014, 6, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lemasters, J.J. Translocation of iron from lysosomes to mitochondria during ischemia predisposes to injury after reperfusion in rat hepatocytes. Free Radic. Biol. Med. 2013, 63, 243–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brekke, E.; Berger, H.R.; Wideroe, M.; Sonnewald, U.; Morken, T.S. Glucose and Intermediary Metabolism and Astrocyte-Neuron Interactions Following Neonatal Hypoxia-Ischemia in Rat. Neurochem. Res. 2017, 42, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yan, Q.; Wang, X.; Chen, X.; Chen, Y.; Du, J.; Chen, L. The Role of Mitochondria in Liver Ischemia-Reperfusion Injury: From Aspects of Mitochondrial Oxidative Stress, Mitochondrial Fission, Mitochondrial Membrane Permeable Transport Pore Formation, Mitophagy, and Mitochondria-Related Protective Measures. Oxid. Med. Cell. Longev. 2021, 2021, 6670579. [Google Scholar] [CrossRef]
- Masior, L.; Grat, M. Methods of Attenuating Ischemia-Reperfusion Injury in Liver Transplantation for Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 8229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, X.; Piao, C.; Jiao, Z.; Ma, Y.; Wang, Y.; Liu, T.; Xu, J.; Wang, H. Effect of conditioned medium from adipose derived mesenchymal stem cells on endoplasmic reticulum stress and lipid metabolism after hepatic ischemia reperfusion injury and hepatectomy in swine. Life Sci. 2022, 289, 120212. [Google Scholar] [CrossRef]
- Qiu, F.; Hu, M.; Tang, B.; Liu, X.; Zhuang, H.; Yang, J.; Hua, Z.C. Annexin V-TRAIL fusion protein is a more sensitive and potent apoptotic inducer for cancer therapy. Sci. Rep. 2013, 3, 3565. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef]
- Batandier, C.; Leverve, X.; Fontaine, E. Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex I. J. Biol. Chem. 2004, 279, 17197–17204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Tang, C.; Zhang, Y.J.; Jiang, Y.; Li, X.W.; Wang, S.G.; Bie, P. Diazoxide suppresses hepatic ischemia/reperfusion injury after mouse liver transplantation by a BCL-2-dependent mechanism. J. Surg. Res. 2011, 169, e155–e166. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, A.; Togliatto, G.; Rolo, A.P.; Teodoro, J.S.; Granata, R.; Ghigo, E.; Columbano, A.; Palmeira, C.M.; Brizzi, M.F. Unacylated ghrelin prevents mitochondrial dysfunction in a model of ischemia/reperfusion liver injury. Cell Death Discov. 2017, 3, 17077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, R.T.; Peng, J.B.; Huang, L.L.; Jiang, G.P.; Liao, Y.J.; Sun, H.; Hu, Y.D.; Liao, X.H. Augmenter of Liver Regeneration Alleviates Renal Hypoxia-Reoxygenation Injury by Regulating Mitochondrial Dynamics in Renal Tubular Epithelial Cells. Mol. Cells 2019, 42, 893–905. [Google Scholar] [CrossRef]
- Andrieux, P.; Chevillard, C.; Cunha-Neto, E.; Nunes, J.P.S. Mitochondria as a Cellular Hub in Infection and Inflammation. Int. J. Mol. Sci. 2021, 22, 1338. [Google Scholar] [CrossRef]
- Lechado Terradas, A.; Zittlau, K.I.; Macek, B.; Fraiberg, M.; Elazar, Z.; Kahle, P.J. Regulation of mitochondrial cargo-selective autophagy by posttranslational modifications. J. Biol. Chem. 2021, 297, 101339. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Qi, Y.; Tsang, S.Y. Mitochondrial Biogenesis, Mitochondrial Dynamics, and Mitophagy in the Maturation of Cardiomyocytes. Cells 2021, 10, 2463. [Google Scholar] [CrossRef]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Zhang, T.; Guo, J.; Chen, K.; Wang, G.; Li, H.; Wang, J. Ursodeoxycholyl lysophosphatidylethanolamide protects against hepatic ischemia/reperfusion injury via phospholipid metabolism-mediated mitochondrial quality control. FASEB J. 2020, 34, 6198–6214. [Google Scholar] [CrossRef] [Green Version]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Jia, L.; Yu, W.; Du, H. Dephosphorylation by calcineurin regulates translocation of dynamin-related protein 1 to mitochondria in hepatic ischemia reperfusion induced hippocampus injury in young mice. Brain Res. 2019, 1711, 68–76. [Google Scholar] [CrossRef]
- Zhang, C.; Huang, J.; An, W. Hepatic stimulator substance resists hepatic ischemia/reperfusion injury by regulating Drp1 translocation and activation. Hepatology 2017, 66, 1989–2001. [Google Scholar] [CrossRef] [Green Version]
- Kraus, F.; Roy, K.; Pucadyil, T.J.; Ryan, M.T. Function and regulation of the divisome for mitochondrial fission. Nature 2021, 590, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Qajari, N.M.; Shafaroudi, M.M.; Gholami, M.; Khonakdar-Tarsi, A. Silibinin treatment results in reducing OPA1&MFN1 genes expression in a rat model hepatic ischemia–reperfusion. Mol. Biol. Rep. 2020, 47, 3271–3280. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xu, J.; Li, X.; Liu, Z.; Han, Y.; Xu, X.; Li, X.; Tang, Y.; Liu, Y.; Yu, T.; et al. Sirt3 modulate renal ischemia-reperfusion injury through enhancing mitochondrial fusion and activating the ERK-OPA1 signaling pathway. J. Cell. Physiol. 2019, 234, 23495–23506. [Google Scholar] [CrossRef]
- Jaeschke, H.; Woolbright, B.L. Current strategies to minimize hepatic ischemia-reperfusion injury by targeting reactive oxygen species. Transpl. Rev. 2012, 26, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Martins, R.M.; Teodoro, J.S.; Furtado, E.; Rolo, A.P.; Palmeira, C.M.; Tralhao, J.G. Recent insights into mitochondrial targeting strategies in liver transplantation. Int. J. Med. Sci. 2018, 15, 248–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, E.M.; Pennington, E.R.; Green, W.D.; Beck, M.A.; Brown, D.A.; Shaikh, S.R. Mechanisms by Which Dietary Fatty Acids Regulate Mitochondrial Structure-Function in Health and Disease. Adv. Nutr. 2018, 9, 247–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crompton, M.; Ellinger, H.; Costi, A. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem. J. 1988, 255, 357–360. [Google Scholar]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Basso, E.; Fante, L.; Fowlkes, J.; Petronilli, V.; Forte, M.A.; Bernardi, P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J. Biol. Chem. 2005, 280, 18558–18561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, A.W.; Varanyuwatana, P.; Halestrap, A.P. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J. Biol. Chem. 2008, 283, 26312–26323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzo, I.; Brenner, C.; Zamzami, N.; Susin, S.A.; Beutner, G.; Brdiczka, D.; Rémy, R.; Xie, Z.H.; Reed, J.C.; Kroemer, G. The permeability transition pore complex: A target for apoptosis regulation by caspases and bcl-2-related proteins. J. Exp. Med. 1998, 187, 1261–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pebay-Peyroula, E.; Dahout-Gonzalez, C.; Kahn, R.; Trézéguet, V.; Lauquin, G.J.; Brandolin, G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 2003, 426, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Wallimann, T.; Riek, U.; Moddel, M. Intradialytic creatine supplementation: A scientific rationale for improving the health and quality of life of dialysis patients. Med. Hypotheses 2017, 99, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zaouali, M.A.; Panisello, A.; Lopez, A.; Castro, C.; Folch, E.; Carbonell, T.; Rolo, A.; Palmeira, C.M.; Garcia-Gil, A.; Adam, R.; et al. GSK3β and VDAC Involvement in ER Stress and Apoptosis Modulation during Orthotopic Liver Transplantation. Int. J. Mol. Sci. 2017, 18, 591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, H.A.; Ahmad, M.Z.; Khan, J.A.; Arshad, M.I. Crosstalk of liver immune cells and cell death mechanisms in different murine models of liver injury and its clinical relevance. Hepatobiliary Pancreat. Dis. Int. 2017, 16, 245–256. [Google Scholar] [CrossRef]
- Zhang, Q.; Fu, H.; Zhang, H.; Xu, F.; Zou, Z.; Liu, M.; Wang, Q.; Miao, M.; Shi, X. Hydrogen sulfide preconditioning protects rat liver against ischemia/reperfusion injury by activating Akt-GSK-3beta signaling and inhibiting mitochondrial permeability transition. PLoS ONE 2013, 8, e74422. [Google Scholar] [CrossRef] [Green Version]
- Brown-Suedel, A.N.; Bouchier-Hayes, L. Caspase-2 Substrates: To Apoptosis, Cell Cycle Control, and Beyond. Front Cell Dev. Biol. 2020, 8, 610022. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Lin, H.-C.; Lee, T.-K.; Tsai, C.-C.; Lai, I.R.; Lu, K.-S. Ischemic Postconditioning Protects Liver From Ischemia-Reperfusion Injury by Modulating Mitochondrial Permeability Transition. Transplantation 2012, 93, 265–271. [Google Scholar] [CrossRef]
- Weigand, K.; Brost, S.; Steinebrunner, N.; Büchler, M.; Schemmer, P.; Müller, M. Ischemia/Reperfusion Injury in Liver Surgery and Transplantation: Pathophysiology. HPB Surg. 2012, 2012, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Lan, X.; Ahsan, A.; Xi, Y.; Liu, S.; Zhang, Z.; Chu, P.; Song, Y.; Piao, F.; Peng, J.; et al. Phosphocreatine protects against LPS-induced human umbilical vein endothelial cell apoptosis by regulating mitochondrial oxidative phosphorylation. Apoptosis 2016, 21, 283–297. [Google Scholar] [CrossRef]
- Lin, J.; Huang, H.F.; Yang, S.K.; Duan, J.; Qu, S.M.; Yuan, B.; Zeng, Z. The effect of Ginsenoside Rg1 in hepatic ischemia reperfusion (I/R) injury ameliorates ischemia-reperfusion-induced liver injury by inhibiting apoptosis. Biomed. Pharm. 2020, 129, 110398. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Li, Y.; Zhang, Q.; Sun, H.; Yan, X.; Hua, T.; Zhu, Q.; Xu, H.; Fu, H. Salidroside protects rat liver against ischemia/reperfusion injury by regulating the GSK-3beta/Nrf2-dependent antioxidant response and mitochondrial permeability transition. Eur. J. Pharm. 2017, 806, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sun, J.J.; Chen, G.Y.; Wang, W.W.; Xie, Z.T.; Tang, G.F.; Wei, S.D. Carnosic acid nanoparticles suppress liver ischemia/reperfusion injury by inhibition of ROS, Caspases and NF-kappaB signaling pathway in mice. Biomed. Pharm. 2016, 82, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Bellanti, F.; Mirabella, L.; Mitarotonda, D.; Blonda, M.; Tamborra, R.; Cinnella, G.; Fersini, A.; Ambrosi, A.; Dambrosio, M.; Vendemiale, G.; et al. Propofol but not sevoflurane prevents mitochondrial dysfunction and oxidative stress by limiting HIF-1alpha activation in hepatic ischemia/reperfusion injury. Free Radic. Biol. Med. 2016, 96, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, A.; Muller, X.; Dutkowski, P. Hypothermic Machine Preservation of the Liver: State of the Art. Curr. Transplant. Rep. 2018, 5, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Weng, J.; Li, W.; Jia, X.; An, W. Alleviation of Ischemia-Reperfusion Injury in Liver Steatosis by Augmenter of Liver Regeneration Is Attributed to Antioxidation and Preservation of Mitochondria. Transplantation 2017, 101, 2340–2348. [Google Scholar] [CrossRef]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [Green Version]
- Lei, Z.; Deng, M.; Yi, Z.; Sun, Q.; Shapiro, R.A.; Xu, H.; Li, T.; Loughran, P.A.; Griepentrog, J.E.; Huang, H.; et al. cGAS-mediated autophagy protects the liver from ischemia-reperfusion injury independently of STING. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G655–G667. [Google Scholar] [CrossRef] [Green Version]
- Ravingerova, T.; Kindernay, L.; Bartekova, M.; Ferko, M.; Adameova, A.; Zohdi, V.; Bernatova, I.; Ferenczyova, K.; Lazou, A. The Molecular Mechanisms of Iron Metabolism and Its Role in Cardiac Dysfunction and Cardioprotection. Int. J. Mol. Sci. 2020, 21, 7889. [Google Scholar] [CrossRef]
- Panisello-Roselló, A.; Alva, N.; Flores, M.; Lopez, A.; Castro Benítez, C.; Folch-Puy, E.; Rolo, A.; Palmeira, C.; Adam, R.; Carbonell, T.; et al. Aldehyde Dehydrogenase 2 (ALDH2) in Rat Fatty Liver Cold Ischemia Injury. Int. J. Mol. Sci. 2018, 19, 2479. [Google Scholar] [CrossRef] [Green Version]
- Panisello-Rosello, A.; Lopez, A.; Folch-Puy, E.; Carbonell, T.; Rolo, A.; Palmeira, C.; Adam, R.; Net, M.; Rosello-Catafau, J. Role of aldehyde dehydrogenase 2 in ischemia reperfusion injury: An update. World J. Gastroenterol. 2018, 24, 2984–2994. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Feng, Z.; Gao, W.; Duan, Y.; Fan, G.; Geng, X.; Wu, B.; Li, K.; Liu, K.; Peng, C. Aucubin Attenuates Liver Ischemia-Reperfusion Injury by Inhibiting the HMGB1/TLR-4/NF-kappaB Signaling Pathway, Oxidative Stress, and Apoptosis. Front. Pharm. 2020, 11, 544124. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W. Therapeutic Potential of Heme Oxygenase-1 and Carbon Monoxide in Acute Organ Injury, Critical Illness, and Inflammatory Disorders. Antioxidants 2020, 9, 1153. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.S.; Moon, J.S.; Xu, J.F.; Ifedigbo, E.; Ryter, S.W.; Choi, A.M.; Nakahira, K. Carbon monoxide negatively regulates NLRP3 inflammasome activation in macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L1058–L1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stucki, D.; Steinhausen, J.; Westhoff, P.; Krahl, H.; Brilhaus, D.; Massenberg, A.; Weber, A.P.M.; Reichert, A.S.; Brenneisen, P.; Stahl, W. Endogenous Carbon Monoxide Signaling Modulates Mitochondrial Function and Intracellular Glucose Utilization: Impact of the Heme Oxygenase Substrate Hemin. Antioxidants 2020, 9, 652. [Google Scholar] [CrossRef]
- Cannistra, M.; Ruggiero, M.; Zullo, A.; Gallelli, G.; Serafini, S.; Maria, M.; Naso, A.; Grande, R.; Serra, R.; Nardo, B. Hepatic ischemia reperfusion injury: A systematic review of literature and the role of current drugs and biomarkers. Int. J. Surg. 2016, 33 (Suppl. 1), S57–S70. [Google Scholar] [CrossRef]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef]
- Lu, H.I.; Huang, T.H.; Sung, P.H.; Chen, Y.L.; Chua, S.; Chai, H.Y.; Chung, S.Y.; Liu, C.F.; Sun, C.K.; Chang, H.W.; et al. Administration of antioxidant peptide SS-31 attenuates transverse aortic constriction-induced pulmonary arterial hypertension in mice. Acta Pharm. Sin. 2016, 37, 589–603. [Google Scholar] [CrossRef] [Green Version]
- Gong, W.-H. Coexistence of hyperlipidemia and acute cerebral ischemia/reperfusion induces severe liver damage in a rat model. World J. Gastroenterol. 2012, 18, 4934. [Google Scholar] [CrossRef]
- Boyman, L.; Greiser, M.; Lederer, W.L. Calcium influx through the mitochondrial calcium uniporter holocomplex, MCUcx. J. Mol. Cell. Cardiol. 2021, 151, 145–154. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 2010, 467, 291–296. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Li, R.J.; Lv, G.Y.; Liu, H.Q. The mechanisms and strategies to protect from hepatic ischemia-reperfusion injury. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 2036–2047. [Google Scholar] [PubMed]
- Chang, W.J.; Chehab, M.; Kink, S.; Toledo-Pereyra, L.H. Intracellular Calcium Signaling Pathways during Liver Ischemia and Reperfusion. J. Investig. Surg. 2010, 23, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Saeed, W.K.; Jun, D.W.; Jang, K.; Chae, Y.J.; Lee, J.S.; Kang, H.T. Does necroptosis have a crucial role in hepatic ischemia-reperfusion injury? PLoS ONE 2017, 12, e0184752. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Zhang, X.B.; Wu, R.C.; Zhang, S.N.; Liu, J.; Gao, Y.; Zheng, K.P.; Ran, J.H. Calcium-calmodulin-dependent protein kinase type 2 induces apoptosis of hepatocytes after liver transplantation. Eur. Rev. Med. Pharm. Sci. 2020, 24, 3331–3343. [Google Scholar] [CrossRef]
- Nakazato, P.C.G.; Victorino, J.P.; Fina, C.F.; Mendes, K.D.S.; Gomes, M.C.J.; Evora, P.R.B.; D’Albuquerque, L.A.C.; Castro, E.S.O. Liver ischemia and reperfusion injury. Pathophysiology and new horizons in preconditioning and therapy. Acta Cir. Bras. 2018, 33, 723–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolder, M.; Walzel, B.; Speer, O.; Schlattner, U.; Wallimann, T. Inhibition of the mitochondrial permeability transition by creatine kinase substrates. Requirement for microcompartmentation. J. Biol. Chem. 2003, 278, 17760–17766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caretti, A.; Bianciardi, P.; Sala, G.; Terruzzi, C.; Lucchina, F.; Samaja, M. Supplementation of creatine and ribose prevents apoptosis in ischemic cardiomyocytes. Cell. Physiol. Biochem. 2010, 26, 831–838. [Google Scholar] [CrossRef]
- Weemhoff, J.L.; Woolbright, B.L.; Jenkins, R.E.; McGill, M.R.; Sharpe, M.R.; Olson, J.C.; Antoine, D.J.; Curry, S.C.; Jaeschke, H. Plasma biomarkers to study mechanisms of liver injury in patients with hypoxic hepatitis. Liver Int. 2017, 37, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Shen, X.; Nan, H.; Yan, L.; Zhao, H.; Yu, J.; Lv, Y. Remifentanil protects liver against ischemia/reperfusion injury through activation of anti-apoptotic pathways. J. Surg. Res. 2013, 183, 827–834. [Google Scholar] [CrossRef]
- Kang, J.H.; Lee, H.S.; Park, D.; Kang, Y.W.; Kim, S.M.; Gong, J.R.; Cho, K.H. Context-independent essential regulatory interactions for apoptosis and hypertrophy in the cardiac signaling network. Sci. Rep. 2017, 7, 34. [Google Scholar] [CrossRef] [Green Version]
- Joiner, M.L.; Koval, O.M.; Li, J.; He, B.J.; Allamargot, C.; Gao, Z.; Luczak, E.D.; Hall, D.D.; Fink, B.D.; Chen, B.; et al. CaMKII determines mitochondrial stress responses in heart. Nature 2012, 491, 269–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Meng, W.; Li, Y.; Liu, W.; Fu, B.; Yang, Y.; Zhang, Q.; Chen, G. The protective effects of CHIR99021 against oxidative injury in LO2 cells. Pharmazie 2016, 71, 629–635. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Casillas-Ramírez, A.; Peralta, C. Molecular pathways in protecting the liver from ischaemia/reperfusion injury: A 2015 update. Clin. Sci. 2015, 129, 345–362. [Google Scholar] [CrossRef]
- Calvert, J.W.; Jha, S.; Gundewar, S.; Elrod, J.W.; Ramachandran, A.; Pattillo, C.B.; Kevil, C.G.; Lefer, D.J. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 2009, 105, 365–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, H.; Chen, H.; Wang, C.; Xu, H.; Liu, F.; Guo, M.; Wang, Q.; Shi, X. Flurbiprofen, a cyclooxygenase inhibitor, protects mice from hepatic ischemia/reperfusion injury by inhibiting GSK-3beta signaling and mitochondrial permeability transition. Mol. Med. 2012, 18, 1128–1135. [Google Scholar] [CrossRef]
- Zhao, G.; Ma, H.; Shen, X.; Xu, G.F.; Zhu, Y.L.; Chen, B.; Tie, R.; Qu, P.; Lv, Y.; Zhang, H.; et al. Role of glycogen synthase kinase 3beta in protective effect of propofol against hepatic ischemia-reperfusion injury. J. Surg. Res. 2013, 185, 388–398. [Google Scholar] [CrossRef]
- Han, S.J.; Choi, H.S.; Kim, J.I.; Park, J.W.; Park, K.M. IDH2 deficiency increases the liver susceptibility to ischemia-reperfusion injury via increased mitochondrial oxidative injury. Redox Biol. 2018, 14, 142–153. [Google Scholar] [CrossRef]
- Hadj Abdallah, N.; Baulies, A.; Bouhlel, A.; Bejaoui, M.; Zaouali, M.A.; Ben Mimouna, S.; Messaoudi, I.; Fernandez-Checa, J.C.; Garcia Ruiz, C.; Ben Abdennebi, H. The effect of zinc acexamate on oxidative stress, inflammation and mitochondria induced apoptosis in rat model of renal warm ischemia. Biomed. Pharm. 2018, 105, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Sun, W.; Gu, C.; Yang, Z.; Quan, N.; Yang, J.; Shi, Z.; Yu, L.; Ma, H. Targeting ALDH2 for Therapeutic Interventions in Chronic Pain-Related Myocardial Ischemic Susceptibility. Theranostics 2018, 8, 1027–1041. [Google Scholar] [CrossRef] [PubMed]
- Kalpage, H.A.; Vaishnav, A.; Liu, J.; Varughese, A.; Wan, J.; Turner, A.A.; Ji, Q.; Zurek, M.P.; Kapralov, A.A.; Kagan, V.E.; et al. Serine-47 phosphorylation of cytochrome c in the mammalian brain regulates cytochrome c oxidase and caspase-3 activity. FASEB J. 2019, 33, 13503–13514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakshmi Devi, S.; Anuradha, C.V. Mitochondrial damage, cytotoxicity and apoptosis in iron-potentiated alcoholic liver fibrosis: Amelioration by taurine. Amino Acids 2009, 38, 869–879. [Google Scholar] [CrossRef]
- He, Q.; Pu, J.; Yuan, A.; Lau, W.B.; Gao, E.; Koch, W.J.; Ma, X.L.; He, B. Activation of liver-X-receptor alpha but not liver-X-receptor beta protects against myocardial ischemia/reperfusion injury. Circ. Heart Fail. 2014, 7, 1032–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Chen, Z.; Zhang, F.; Jing, H.; Xu, W.; Ning, S.; Li, Z.; Liu, K.; Yao, J.; Tian, X. Blockade of PKCbeta protects against remote organ injury induced by intestinal ischemia and reperfusion via a p66shc-mediated mitochondrial apoptotic pathway. Apoptosis 2014, 19, 1342–1353. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Chen, M.-T.; Hong, H.-M.; Wang, Y.; Li, Q.; Liu, H.; Yang, M.-W.; Hong, F.-F.; Yang, S.-L. Role of Reduced Nitric Oxide in Liver Cell Apoptosis Inhibition During Liver Damage. Arch. Med. Res. 2018, 49, 219–225. [Google Scholar] [CrossRef]
- Zhuang, Z.; Lian, P.; Wu, X.; Shi, B.; Zhuang, M.; Zhou, R.; Zhao, R.; Zhao, Z.; Guo, S.; Ji, Z.; et al. Abate Cytochrome C induced apoptosome to protect donor liver against ischemia reperfusion injury on rat liver transplantation model. Am. J. Transl. Res. 2016, 8, 1738–1747. [Google Scholar] [PubMed]
- Chen, Q.; Paillard, M.; Gomez, L.; Ross, T.; Hu, Y.; Xu, A.; Lesnefsky, E.J. Activation of mitochondrial mu-calpain increases AIF cleavage in cardiac mitochondria during ischemia-reperfusion. Biochem. Biophys. Res. Commun. 2011, 415, 533–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, P.L.; Chen, K.H.; Chang, T.C.; Chien, C.T. Repetitively hypoxic preconditioning attenuates ischemia/reperfusion-induced liver dysfunction through upregulation of hypoxia-induced factor-1 alpha-dependent mitochondrial Bcl-xl in rat. Chin. J. Physiol. 2020, 63, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.P.; Yuan, F.; Xu, J.; Sai, K.; Chen, J.; Guan, S. Cryptotanshinone Ameliorates Hepatic Normothermic Ischemia and Reperfusion Injury in Rats by Anti-mitochondrial Apoptosis. Biol. Pharm. Bull. 2014, 37, 1758–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunenfelder, J.; Miniati, D.N.; Murata, S.; Falk, V.; Hoyt, E.G.; Kown, M.; Koransky, M.L.; Robbins, R.C. Upregulation of Bcl-2 through caspase-3 inhibition ameliorates ischemia/reperfusion injury in rat cardiac allografts. Circulation 2001, 104, I202–I206. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xiong, X.; Guo, H.; Wu, M.; Li, X.; Hu, Y.; Xie, G.; Shen, J.; Tian, Q. ZnPP reduces autophagy and induces apoptosis, thus aggravating liver ischemia/reperfusion injury in vitro. Int. J. Mol. Med. 2014, 34, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Eum, H.A.; Billiar, T.R.; Lee, S.M. Role of heme oxygenase 1 in TNF/TNF receptor-mediated apoptosis after hepatic ischemia/reperfusion in rats. Shock 2013, 39, 380–388. [Google Scholar] [CrossRef]
- Mahmoud, A.R.; Ali, F.E.M.; Abd-Elhamid, T.H.; Hassanein, E.H.M. Coenzyme Q10 protects hepatocytes from ischemia reperfusion-induced apoptosis and oxidative stress via regulation of Bax/Bcl-2/PUMA and Nrf-2/FOXO-3/Sirt-1 signaling pathways. Tissue Cell 2019, 60, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Xing, W.J.; Kong, F.J.; Li, G.W.; Qiao, K.; Zhang, W.H.; Zhang, L.; Bai, S.Z.; Xi, Y.H.; Li, H.X.; Tian, Y.; et al. Calcium-sensing receptors induce apoptosis during simulated ischaemia-reperfusion in Buffalo rat liver cells. Clin. Exp. Pharmacol. Physiol. 2011, 38, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, P.; Chaudhury, P.; Wahi, A.K. Bcl-2 expression alters the mitochondrial tri carboxyl Acid pathway in hepatic ischemic and reperfusion induced necrosis and apoptosis in rat liver. Indian J. Pharm. Sci. 2010, 72, 437–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abogresha, N.M.; Greish, S.M.; Abdelaziz, E.Z.; Khalil, W.F. Remote effect of kidney ischemia-reperfusion injury on pancreas: Role of oxidative stress and mitochondrial apoptosis. Arch. Med. Sci. 2016, 12, 252–262. [Google Scholar] [CrossRef]
- Chattopadhyay, P.; Chaudhury, P.; Wahi, A.K. Ca2+ concentrations are key determinants of ischemia-reperfusion-induced apoptosis: Significance for the molecular mechanism of Bcl-2 action. Appl. Biochem. Biotechnol. 2010, 160, 1968–1977. [Google Scholar] [CrossRef] [PubMed]
- Parente, A.; Osei-Bordom, D.C.; Ronca, V.; Perera, M.; Mirza, D. Organ Restoration With Normothermic Machine Perfusion and Immune Reaction. Front. Immunol. 2020, 11, 565616. [Google Scholar] [CrossRef] [PubMed]
- Khader, A.; Yang, W.L.; Godwin, A.; Prince, J.M.; Nicastro, J.M.; Coppa, G.F.; Wang, P. Sirtuin 1 Stimulation Attenuates Ischemic Liver Injury and Enhances Mitochondrial Recovery and Autophagy. Crit. Care Med. 2016, 44, e651–e663. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Yoshidome, H.; Kimura, F.; Shimizu, H.; Ohtsuka, M.; Takeuchi, D.; Kato, A.; Furukawa, K.; Yoshitomi, H.; Iida, A.; et al. Hepatocyte apoptosis is enhanced after ischemia/reperfusion in the steatotic liver. J. Clin. Biochem. Nutr. 2011, 48, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.; Zhang, R.; Shen, N.; Jin, Y.; Alina, A.; Yang, S.; Lin, S. Sulforaphane reduces apoptosis and oncosis along with protecting liver injury-induced ischemic reperfusion by activating the Nrf2/ARE pathway. Hepatol. Int. 2015, 9, 321–329. [Google Scholar] [CrossRef]
- Yang, J.; Sun, H.; Guan, R.; Liu, W.; Xia, Y.; Zhao, J.; Liu, J. Hepatocellular protein profiles after hepatic ischemia/reperfusion injury with or without octreotide preconditioning in a rabbit model. Transpl. Proc. 2014, 46, 3282–3288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compagnon, P.; Levesque, E.; Hentati, H.; Disabato, M.; Calderaro, J.; Feray, C.; Corlu, A.; Cohen, J.L.; Ben Mosbah, I.; Azoulay, D. An Oxygenated and Transportable Machine Perfusion System Fully Rescues Liver Grafts Exposed to Lethal Ischemic Damage in a Pig Model of DCD Liver Transplantation. Transplantation 2017, 101, e205–e213. [Google Scholar] [CrossRef] [PubMed]
- Bouhlel, A.; Bejaoui, M.; Ben Mosbah, I.; Hadj Abdallah, N.; Ribault, C.; Viel, R.; Hentati, H.; Corlu, A.; Ben Abdennebi, H. Thymoquinone protects rat liver after partial hepatectomy under ischaemia/reperfusion through oxidative stress and endoplasmic reticulum stress prevention. Clin. Exp. Pharmacol. Physiol. 2018, 45, 943–951. [Google Scholar] [CrossRef]
- Mosbah, I.B.; Zaouali, M.A.; Martel, C.; Bjaoui, M.; Abdennebi, H.B.; Hotter, G.; Brenner, C.; Rosello-Catafau, J. IGL-1 solution reduces endoplasmic reticulum stress and apoptosis in rat liver transplantation. Cell Death Dis. 2012, 3, e279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Mosbah, I.; Alfany-Fernandez, I.; Martel, C.; Zaouali, M.A.; Bintanel-Morcillo, M.; Rimola, A.; Rodes, J.; Brenner, C.; Rosello-Catafau, J.; Peralta, C. Endoplasmic reticulum stress inhibition protects steatotic and non-steatotic livers in partial hepatectomy under ischemia-reperfusion. Cell Death Dis. 2010, 1, e52. [Google Scholar] [CrossRef] [Green Version]
- Oshiro, T.; Shiraishi, M.; Muto, Y. Adenovirus mediated gene transfer of antiapoptotic protein in hepatic ischemia-reperfusion injury: The paradoxical effect of Bcl-2 expression in the reperfused liver. J. Surg. Res. 2002, 103, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Z.; Zhang, S.J.; Chen, Y.X.; Chen, Z.X.; Huang, Y.H.; Zhang, L.J. Effects of platelet-derived growth factor and interleukin-10 on Fas/Fas-ligand and Bcl-2/Bax mRNA expression in rat hepatic stellate cells in vitro. World J. Gastroenterol. 2004, 10, 2706–2710. [Google Scholar] [CrossRef] [PubMed]
- Weber, L.W.; Boll, M.; Stampfl, A. Hepatotoxicity and mechanism of action of haloalkanes: Carbon tetrachloride as a toxicological model. Crit. Rev. Toxicol. 2003, 33, 105–136. [Google Scholar] [CrossRef] [PubMed]
- Varadarajan, R.; Golden-Mason, L.; Young, L.; McLoughlin, P.; Nolan, N.; McEntee, G.; Traynor, O.; Geoghegan, J.; Hegarty, J.E.; O’Farrelly, C. Nitric oxide in early ischaemia reperfusion injury during human orthotopic liver transplantation. Transplantation 2004, 78, 250–256. [Google Scholar] [CrossRef]
- Hines, I.N.; Hoffman, J.M.; Scheerens, H.; Day, B.J.; Harada, H.; Pavlick, K.P.; Bharwani, S.; Wolf, R.; Gao, B.; Flores, S.; et al. Regulation of postischemic liver injury following different durations of ischemia. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G536–G545. [Google Scholar] [CrossRef] [Green Version]
- Pannen, B.H.; Al-Adili, F.; Bauer, M.; Clemens, M.G.; Geiger, K.K. Role of endothelins and nitric oxide in hepatic reperfusion injury in the rat. Hepatology 1998, 27, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Zhai, C.L.; Zhang, M.Q.; Zhang, Y.; Xu, H.X.; Wang, J.M.; An, G.P.; Wang, Y.Y.; Li, L. Glycyrrhizin protects rat heart against ischemia-reperfusion injury through blockade of HMGB1-dependent phospho-JNK/Bax pathway. Acta Pharmacol. Sin. 2012, 33, 1477–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, T.; Shimada, S.; Fukai, M.; Kimura, T.; Umemoto, K.; Shibata, K.; Fujiyoshi, M.; Fujiyoshi, S.; Hayasaka, T.; Kawamura, N.; et al. Post-reperfusion hydrogen gas treatment ameliorates ischemia reperfusion injury in rat livers from donors after cardiac death: A preliminary study. Surg. Today 2018, 48, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, R.; Tao, G.; Lehwald, N.C.; Liu, B.; Koh, Y.; Sylvester, K.G. Augmented Wnt signaling as a therapeutic tool to prevent ischemia/reperfusion injury in liver: Preclinical studies in a mouse model. Liver Transpl. 2015, 21, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, C.; Ng, K.T.; Liu, J.; Liu, H.; Zhang, W.; Xiao, F.; Li, X.; Lo, C.M.; Lu, L.; et al. IL-17a exacerbates hepatic ischemia-reperfusion injury in fatty liver by promoting neutrophil infiltration and mitochondria-driven apoptosis. J. Leukoc. Biol. 2020, 108, 1603–1613. [Google Scholar] [CrossRef]
- Meyer, J.N.; Leuthner, T.C.; Luz, A.L. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 2017, 391, 42–53. [Google Scholar] [CrossRef]
- Park, K.S.; Wiederkehr, A.; Wollheim, C.B. Defective mitochondrial function and motility due to mitofusin 1 overexpression in insulin secreting cells. Korean J. Physiol. Pharmacol. Off. J. Korean Physiol. Soc. Korean Soc. Pharmacol. 2012, 16, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Szabo, A.; Sumegi, K.; Fekete, K.; Hocsak, E.; Debreceni, B.; Setalo, G., Jr.; Kovacs, K.; Deres, L.; Kengyel, A.; Kovacs, D.; et al. Activation of mitochondrial fusion provides a new treatment for mitochondria-related diseases. Biochem. Pharmacol. 2018, 150, 86–96. [Google Scholar] [CrossRef]
- Yang, M.; Linn, B.S.; Zhang, Y.; Ren, J. Mitophagy and mitochondrial integrity in cardiac ischemia-reperfusion injury. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2293–2302. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Chen, L.; Wu, Y.; Tang, Y.; Tang, L.; Zhong, Y.; Wang, S.; Liu, H.; Wang, X.; Chen, A. Gastrodin pretreatment alleviates myocardial ischemia/reperfusion injury through promoting autophagic flux. Biochem. Biophys. Res. Commun. 2018, 503, 2421–2428. [Google Scholar] [CrossRef] [PubMed]
- Kon, N.; Satoh, A.; Miyoshi, N. A small-molecule DS44170716 inhibits Ca(2+)-induced mitochondrial permeability transition. Sci. Rep. 2017, 7, 3864. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.Q.; Liu, W.; Wang, J.; Huang, Y.Q.; Li, P.Y.; Zhu, Y.; Wang, J.B.; Ma, X.; Li, R.S.; Wei, S.Z.; et al. Paeoniflorin attenuates ANIT-induced cholestasis by inhibiting apoptosis in vivo via mitochondria-dependent pathway. Biomed. Pharm. 2017, 89, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Waseem, M.; Tabassum, H.; Bhardwaj, M.; Parvez, S. Ameliorative efficacy of quercetin against cisplatin-induced mitochondrial dysfunction: Study on isolated rat liver mitochondria. Mol. Med. Rep. 2017, 16, 2939–2945. [Google Scholar] [CrossRef] [PubMed]
- Buko, V.; Kuzmitskaya, I.; Kirko, S.; Belonovskaya, E.; Naruta, E.; Lukivskaya, O.; Shlyahtun, A.; Ilyich, T.; Zakreska, A.; Zavodnik, I. Betulin attenuated liver damage by prevention of hepatic mitochondrial dysfunction in rats with alcoholic steatohepatitis. Physiol. Int. 2019, 106, 323–334. [Google Scholar] [CrossRef]
- Lin, H.C.; Liu, S.Y.; Lai, H.S.; Lai, I.R. Isolated mitochondria infusion mitigates ischemia-reperfusion injury of the liver in rats. Shock 2013, 39, 304–310. [Google Scholar] [CrossRef]
- Zheng, J.; Chen, L.; Lu, T.; Zhang, Y.; Sui, X.; Li, Y.; Huang, X.; He, L.; Cai, J.; Zhou, C.; et al. MSCs ameliorate hepatocellular apoptosis mediated by PINK1-dependent mitophagy in liver ischemia/reperfusion injury through AMPKα activation. Cell Death Dis. 2020, 11, 256. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factors | How | Effects | Results |
|---|---|---|---|
| ROS | excessive ROS produce after reperfusion and CO exposure | promote mitochondrial permeability transition and depolarizes ΔΨm; produce lipid peroxides and other toxic aldehydes | induce MPTP opening |
| Ca2+ | Na+/Ca2+ commutator overburden leads to Ca2+ overload | induce PKC formation activate NFkB activate Ca2+-dependent enzymes; cause mitochondrial integrity impairment | mitochondrial membrane damage |
| ΔΨm | mitochondrial integrity impaired causes the ΔΨm loss | block the synthesis of mitochondrial RNA and protein, uncoupling oxidative phosphorylation, and ATP depletion | release of apoptosis drivers, cyt c |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Rao, S.; Yang, M.; Ma, C.; Hong, F.; Yang, S. Role of Mitochondrial Pathways in Cell Apoptosis during He-Patic Ischemia/Reperfusion Injury. Int. J. Mol. Sci. 2022, 23, 2357. https://doi.org/10.3390/ijms23042357
Zhang S, Rao S, Yang M, Ma C, Hong F, Yang S. Role of Mitochondrial Pathways in Cell Apoptosis during He-Patic Ischemia/Reperfusion Injury. International Journal of Molecular Sciences. 2022; 23(4):2357. https://doi.org/10.3390/ijms23042357
Chicago/Turabian StyleZhang, Sen, Sijing Rao, Meiwen Yang, Chen Ma, Fengfang Hong, and Shulong Yang. 2022. "Role of Mitochondrial Pathways in Cell Apoptosis during He-Patic Ischemia/Reperfusion Injury" International Journal of Molecular Sciences 23, no. 4: 2357. https://doi.org/10.3390/ijms23042357
APA StyleZhang, S., Rao, S., Yang, M., Ma, C., Hong, F., & Yang, S. (2022). Role of Mitochondrial Pathways in Cell Apoptosis during He-Patic Ischemia/Reperfusion Injury. International Journal of Molecular Sciences, 23(4), 2357. https://doi.org/10.3390/ijms23042357