
Epstein-Barr Virus Enhances Cancer-Specific Aberrant Splicing of TSG101 Pre-mRNA



Abstract
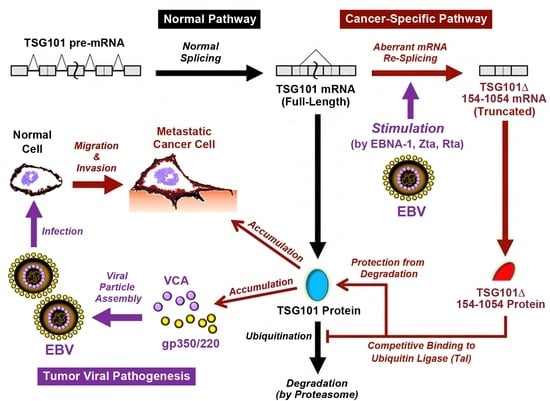
:
1. Introduction
2. Results
2.1. EBV Triggers Cancer-Specific Alternative Splicing of TSG101
2.2. EBV Preferentially Enhances TSG∆154-1054 Production upon Lytic Activation
2.3. TSG∆154-1054 Increases TSG101 Protein Stability
2.4. EBV Exploits TSG101 Stabilization for Viral Late Protein Synthesis
2.5. TSG∆154-1054 Is Correlated with Increasing Anti-VCA Antibodies and EBV Load in NPC Patients
3. Discussion
4. Materials and Methods
4.1. NPC Tissue Collection
4.2. Cell Culture
4.3. Construction and Transfection of Plasmids and siRNAs
4.4. RT-Nested-PCR
4.5. RT-PCR Targeting EBV Viral Gene Products
4.6. Quantification of EBV Copy Number
4.7. Northern Blotting Analysis
4.8. Immunoblotting Analysis
4.9. CHX Chase Assay to Assess TSG101 Stability
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Kieff, E.; Rickinson, A.B. Epstein-Barr virus and its replication. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Griffin, D.E., Lamb, R.A., Martin, M.A., Roizman, B., Straus, S.E., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2007; Volume 2, pp. 2603–2654. [Google Scholar]
- Young, L.S.; Murray, P.G. Epstein-Barr virus and oncogenesis: From latent genes to tumours. Oncogene 2003, 22, 5108–5121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosemarie, Q.; Sugden, B. Epstein-Barr Virus: How Its Lytic Phase Contributes to Oncogenesis. Microorganisms 2020, 8, 1824. [Google Scholar] [CrossRef] [PubMed]
- Adamson, A.L.; Darr, D.; Holley-Guthrie, E.; Johnson, R.A.; Mauser, A.; Swenson, J.; Kenney, S. Epstein-Barr virus immediate-early proteins BZLF1 and BRLF1 activate the ATF2 transcription factor by increasing the levels of phosphorylated p38 and c-Jun N-terminal kinases. J. Virol. 2000, 74, 1224–1233. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.Y.; Hsieh, M.J.; Chen, C.Y.; Chen, Y.J.; Chen, J.Y.; Chen, M.R.; Tsai, C.H.; Lin, S.F.; Hsu, T.Y. Epstein-Barr virus Rta-mediated transactivation of p21 and 14-3-3sigma arrests cells at the G1/S transition by reducing cyclin E/CDK2 activity. J. Gen. Virol. 2012, 93, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Francies, F.Z.; Dlamini, Z. Aberrant Splicing Events and Epigenetics in Viral Oncogenomics: Current Therapeutic Strategies. Cells 2021, 10, 239. [Google Scholar] [CrossRef]
- Ruland, J.; Sirard, C.; Elia, A.; MacPherson, D.; Wakeham, A.; Li, L.; de la Pompa, J.L.; Cohen, S.N.; Mak, T.W. p53 accumulation, defective cell proliferation, and early embryonic lethality in mice lacking tsg101. Proc. Natl. Acad. Sci. USA 2001, 98, 1859–1864. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.U.; Krempler, A.; Qi, Y.; Park, K.; Henry, M.D.; Triplett, A.A.; Riedlinger, G.; Rucker, E.B.; Hennighausen, L. Tsg101 is essential for cell growth, proliferation, and cell survival of embryonic and adult tissues. Mol. Cell Biol. 2003, 23, 150–162. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Gilchrist, R.; Borley, N.; Chng, H.W.; Morgan, M.; Marshall, J.F.; Camplejohn, R.S.; Muir, G.H.; Hart, I.R. Reduction of TSG101 protein has a negative impact on tumor cell growth. Int. J. Cancer 2004, 109, 541–547. [Google Scholar] [CrossRef]
- Shen, H.; Pan, Y.; Han, Z.; Hong, L.; Liu, N.; Han, S.; Yao, L.; Xie, H.; Zhaxi, C.; Shi, Y.; et al. Reversal of multidrug resistance of gastric cancer cells by downregulation of TSG101 with TSG101siRNA. Cancer Biol. Ther. 2004, 3, 561–565. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Yanagi, Y.; Masuhiro, Y.; Yano, T.; Yoshikawa, H.; Yanagisawa, J.; Kato, S. A putative tumor suppressor, TSG101, acts as a transcriptional suppressor through its coiled-coil domain. Biochem. Biophys. Res. Commun. 1998, 245, 900–905. [Google Scholar] [CrossRef]
- Bishop, N.; Woodman, P. TSG101/mammalian VPS23 and mammalian VPS28 interact directly and are recruited to VPS4-induced endosomes. J. Biol. Chem. 2001, 276, 11735–11742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, M.; Lopez-Borges, S.; Lazo, P.A. Expression of a new isoform of the tumor susceptibility TSG101 protein lacking a leucine zipper domain in Burkitt lymphoma cell lines. Oncogene 1999, 18, 2253–2259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Pan, J.; Bubley, G.; Balk, S.P. Frequent abnormalities of TSG101 transcripts in human prostate cancer. Oncogene 1997, 15, 3121–3125. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.P.; Feinberg, A.P. Aberrant splicing but not mutations of TSG101 in human breast cancer. Cancer Res. 1997, 57, 3131–3134. [Google Scholar] [PubMed]
- Lin, P.M.; Liu, T.C.; Chang, J.G.; Chen, T.P.; Lin, S.F. Aberrant TSG101 transcripts in acute myeloid leukaemia. Br. J. Haematol. 1998, 102, 753–758. [Google Scholar] [CrossRef]
- Oh, Y.; Proctor, M.L.; Fan, Y.H.; Su, L.K.; Hong, W.K.; Fong, K.M.; Sekido, Y.S.; Gazdar, A.F.; Minna, J.D.; Mao, L. TSG101 is not mutated in lung cancer but a shortened transcript is frequently expressed in small cell lung cancer. Oncogene 1998, 17, 1141–1148. [Google Scholar] [CrossRef] [Green Version]
- Chua, H.H.; Kameyama, T.; Mayeda, A.; Yeh, T.H. Cancer-Specifically Re-Spliced TSG101 mRNA Promotes Invasion and Metastasis of Nasopharyngeal Carcinoma. Int. J. Mol. Sci. 2019, 20, 773. [Google Scholar] [CrossRef] [Green Version]
- Klaes, R.; Kloor, M.; Willeke, F.; Melsheimer, P.; von Knebel Doeberitz, M.; Ridder, R. Significant increase of a specific variant TSG101 transcript during the progression of cervical neoplasia. Eur. J. Cancer 1999, 35, 733–737. [Google Scholar] [CrossRef]
- Lo, Y.F.; Chen, T.C.; Chen, S.C.; Chao, C.C. Aberrant expression of TSG101 in Taiwan Chinese breast cancer. Breast Cancer Res Treat 2000, 60, 259–266. [Google Scholar] [CrossRef]
- Kameyama, T.; Suzuki, H.; Mayeda, A. Re-splicing of mature mRNA in cancer cells promotes activation of distant weak alternative splice sites. Nucleic Acids Res. 2012, 40, 7896–7906. [Google Scholar] [CrossRef] [Green Version]
- Otani, Y.; Fujita, K.I.; Kameyama, T.; Mayeda, A. The Exon Junction Complex Core Represses Cancer-Specific Mature mRNA Re-splicing: A Potential Key Role in Terminating Splicing. Int. J. Mol. Sci. 2021, 22, 6519. [Google Scholar] [CrossRef] [PubMed]
- Gayther, S.A.; Barski, P.; Batley, S.J.; Li, L.; de Foy, K.A.; Cohen, S.N.; Ponder, B.A.; Caldas, C. Aberrant splicing of the TSG101 and FHIT genes occurs frequently in multiple malignancies and in normal tissues and mimics alterations previously described in tumours. Oncogene 1997, 15, 2119–2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turpin, E.; Dalle, B.; de Roquancourt, A.; Plassa, L.F.; Marty, M.; Janin, A.; Beuzard, Y.; De Thé, H. Stress-induced aberrant splicing of TSG101: Association to high tumor grade and p53 status in breast cancers. Oncogene 1999, 18, 7834–7837. [Google Scholar] [CrossRef] [Green Version]
- Moyret-Lalle, C.; Duriez, C.; Van Kerckhove, J.; Gilbert, C.; Wang, Q.; Puisieux, A. p53 induction prevents accumulation of aberrant transcripts in cancer cells. Cancer Res. 2001, 61, 486–488. [Google Scholar] [PubMed]
- Chua, H.H.; Huang, C.S.; Weng, P.L.; Yeh, T.H. TSGDelta154-1054 splice variant increases TSG101 oncogenicity by inhibiting its E3-ligase-mediated proteasomal degradation. Oncotarget 2016, 7, 8240–8252. [Google Scholar] [CrossRef] [PubMed]
- Boudreault, S.; Armero, V.E.S.; Scott, M.S.; Perreault, J.P.; Bisaillon, M. The Epstein-Barr virus EBNA1 protein modulates the alternative splicing of cellular genes. Virol. J. 2019, 16, 29. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Gradoville, L.; Miller, G. Cellular immediate-early gene expression occurs kinetically upstream of Epstein-Barr virus bzlf1 and brlf1 following cross-linking of the B cell antigen receptor in the Akata Burkitt lymphoma cell line. J. Virol. 2010, 84, 12405–12418. [Google Scholar] [CrossRef] [Green Version]
- Hadar, T.; Rahima, M.; Kahan, E.; Sidi, J.; Rakowsky, E.; Sarov, B.; Sarov, I. Significance of specific Epstein-Barr virus IgA and elevated IgG antibodies to viral capsid antigens in nasopharyngeal carcinoma patients. J. Med. Virol. 1986, 20, 329–339. [Google Scholar] [CrossRef]
- Wong, M.M.; Lye, M.S.; Cheng, H.M.; Sam, C.K. Epstein-Barr virus serology in the diagnosis of nasopharyngeal carcinoma. Asian Pac. J. Allergy Immunol. 2005, 23, 65–67. [Google Scholar]
- White, J.T.; Rives, J.; Tharp, M.E.; Wrabl, J.O.; Thompson, E.B.; Hilser, V.J. Tumor Susceptibility Gene 101 Regulates the Glucocorticoid Receptor through Disorder-Mediated Allostery. Biochemistry 2021, 60, 1647–1657. [Google Scholar] [CrossRef]
- Ismaili, N.; Blind, R.; Garabedian, M.J. Stabilization of the unliganded glucocorticoid receptor by TSG101. J. Biol. Chem. 2005, 280, 11120–11126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felten, A.; Brinckmann, D.; Landsberg, G.; Scheidtmann, K.H. Zipper-interacting protein kinase is involved in regulation of ubiquitination of the androgen receptor, thereby contributing to dynamic transcription complex assembly. Oncogene 2013, 32, 4981–4988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgdorf, S.; Leister, P.; Scheidtmann, K.H. TSG101 interacts with apoptosis-antagonizing transcription factor and enhances androgen receptor-mediated transcription by promoting its monoubiquitination. J. Biol. Chem. 2004, 279, 17524–17534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Pan, J.; Hope, W.X.; Cohen, S.N.; Balk, S.P. Tumor susceptibility gene 101 protein represses androgen receptor transactivation and interacts with p300. Cancer 1999, 86, 689–696. [Google Scholar] [CrossRef]
- Muromoto, R.; Sugiyama, K.; Yamamoto, T.; Oritani, K.; Shimoda, K.; Matsuda, T. Physical and functional interactions between Daxx and TSG101. Biochem. Biophys. Res. Commun. 2004, 316, 827–833. [Google Scholar] [CrossRef] [Green Version]
- Chua, H.H.; Lee, H.H.; Chang, S.S.; Lu, C.C.; Yeh, T.H.; Hsu, T.Y.; Cheng, T.H.; Cheng, J.T.; Chen, M.R.; Tsai, C.H. Role of the TSG101 gene in Epstein-Barr virus late gene transcription. J. Virol. 2007, 81, 2459–2471. [Google Scholar] [CrossRef] [Green Version]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Côté, M.; Rich, R.L.; et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Martin-Serrano, J.; Zang, T.; Bieniasz, P.D. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 2001, 7, 1313–1319. [Google Scholar] [CrossRef]
- Broniarczyk, J.; Bergant, M.; Gozdzicka-Jozefiak, A.; Banks, L. Human papillomavirus infection requires the TSG101 component of the ESCRT machinery. Virology 2014, 460–461, 83–90. [Google Scholar] [CrossRef]
- Kumar, B.; Dutta, D.; Iqbal, J.; Ansari, M.A.; Roy, A.; Chikoti, L.; Pisano, G.; Veettil, M.V.; Chandran, B. ESCRT-I Protein Tsg101 Plays a Role in the Post-macropinocytic Trafficking and Infection of Endothelial Cells by Kaposi’s Sarcoma-Associated Herpesvirus. PLoS Pathog. 2016, 12, e1005960. [Google Scholar] [CrossRef]
- Watanabe, S.M.; Ehrlich, L.S.; Strickland, M.; Li, X.; Soloveva, V.; Goff, A.J.; Stauft, C.B.; Bhaduri-McIntosh, S.; Tjandra, N.; Carter, C. Selective Targeting of Virus Replication by Proton Pump Inhibitors. Sci. Rep. 2020, 10, 4003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannemuddhu, S.S.; Xu, H.; Bleck, C.K.E.; Tjandra, N.; Carter, C.; Bhaduri-McIntosh, S. Prazoles Targeting Tsg101 Inhibit Release of Epstein-Barr Virus following Reactivation from Latency. J. Virol. 2021, 95, e0246620. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, V.; Navarro, L.; Sample, C.E.; David, M.; Sung, S.; Swaminathan, S. The Epstein-Barr virus SM protein induces STAT1 and interferon-stimulated gene expression. J. Virol. 2003, 77, 3690–3701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, D.; Swaminathan, S. Epstein-Barr virus SM protein functions as an alternative splicing factor. J. Virol. 2008, 82, 7180–7188. [Google Scholar] [CrossRef] [Green Version]
- Verma, D.; Bais, S.; Gaillard, M.; Swaminathan, S. Epstein-Barr Virus SM protein utilizes cellular splicing factor SRp20 to mediate alternative splicing. J. Virol. 2010, 84, 11781–11789. [Google Scholar] [CrossRef] [Green Version]
- Meissl, K.; Simonovic, N.; Amenitsch, L.; Witalisz-Siepracka, A.; Klein, K.; Lassnig, C.; Puga, A.; Vogl, C.; Andrea Poelzl, A.; Bosmann, M.; et al. STAT1 Isoforms Differentially Regulate NK Cell Maturation and Anti-tumor Activity. Front. Immunol. 2020, 11, 2189. [Google Scholar] [CrossRef]
- Lu, C.C.; Jeng, Y.Y.; Tsai, C.H.; Liu, M.Y.; Yeh, S.W.; Hsu, T.Y.; Chen, M.R. Genome-wide transcription program and expression of the Rta responsive gene of Epstein-Barr virus. Virology 2006, 345, 358–372. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, N.; Yoshiyama, H.; Takada, K. Clonal propagation of Epstein-Barr virus (EBV) recombinants in EBV-negative Akata cells. J. Virol. 1996, 70, 7260–7263. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.; Tung, C.H.; Huang, Y.T.; Lu, J.; Chen, J.Y.; Tsai, C.H. Requirement for cell-to-cell contact in Epstein-Barr virus infection of nasopharyngeal carcinoma cells and keratinocytes. J. Virol. 1999, 73, 8857–8866. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.; Lee, H.H.; Chen, Y.T.; Lu, J.; Wu, S.Y.; Chen, C.W.; Takada, K.; Tsai, C.H. Induction of the early growth response 1 gene by Epstein-Barr virus lytic transactivator Zta. J. Virol. 2006, 80, 7748–7755. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Chen, S.Y.; Chua, H.H.; Liu, Y.S.; Huang, Y.T.; Chang, Y.; Chen, J.Y.; Sheen, T.S.; Tsai, C.H. Upregulation of tyrosine kinase TKT by the Epstein-Barr virus transactivator Zta. J. Virol. 2000, 74, 7391–7399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.J.; Chen, J.Y.; Hsu, T.Y.; Yu, W.C.; Su, I.J.; Yang, C.S. Induction of apoptosis in epithelial cells by Epstein-Barr virus latent membrane protein 1. J. Gen. Virol. 1996, 77 Pt 8, 1883–1892. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, N.; Tanabe-Tochikura, A.; Kuroiwa, Y.; Takada, K. Isolation of Epstein-Barr virus (EBV)-negative cell clones from the EBV-positive Burkitt’s lymphoma (BL) line Akata: Malignant phenotypes of BL cells are dependent on EBV. J. Virol. 1994, 68, 6069–6073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flemington, E.K.; Borras, A.M.; Lytle, J.P.; Speck, S.H. Characterization of the Epstein-Barr virus BZLF1 protein transactivation domain. J. Virol. 1992, 66, 922–929. [Google Scholar] [CrossRef] [Green Version]
- Lo, Y.M.; Chan, L.Y.; Lo, K.W.; Leung, S.F.; Zhang, J.; Chan, A.T.; Lee, J.C.; Hjelm, N.M.; Johnson, P.J.; Huang, D.P. Quantitative analysis of cell-free Epstein-Barr virus DNA in plasma of patients with nasopharyngeal carcinoma. Cancer Res. 1999, 59, 1188–1191. [Google Scholar]
- Chang, Y.; Cheng, S.D.; Tsai, C.H. Chromosomal integration of Epstein-Barr virus genomes in nasopharyngeal carcinoma cells. Head Neck 2002, 24, 143–150. [Google Scholar] [CrossRef]
- Hsu, T.Y.; Chang, Y.; Wang, P.W.; Liu, M.Y.; Chen, M.R.; Chen, J.Y.; Tsai, C.H. Reactivation of Epstein-Barr virus can be triggered by an Rta protein mutated at the nuclear localization signal. J. Gen. Virol. 2005, 86, 317–322. [Google Scholar] [CrossRef]
- Tsai, C.H.; Liu, M.T.; Chen, M.R.; Lu, J.; Yang, H.L.; Chen, J.Y.; Yang, C.S. Characterization of Monoclonal Antibodies to the Zta and DNase Proteins of Epstein-Barr Virus. J. Biomed. Sci. 1997, 4, 69–77. [Google Scholar] [CrossRef]
- Tsai, C.H.; Glaser, R. A comparison of Epstein-Barr virus specific proteins expressed by three Epstein-Barr virus isolates using specific monoclonal antibodies. Intervirology 1991, 32, 376–382. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes (Amplicon Sizes) | Primer Sequences | PCR Condition |
|---|---|---|
| Nested-PCR TSG101 (Full-length: 1148 bp, TSG∆154-1054: 247 bp) | First round PCR Forward primer (P1): 5′-CGGTGTCGGAGAGCCAGCTCAAGAAA-3′ Reverse primer (P2): 5′-CCTCCAGCTGGTATCAGAGAAGTCAGT-3′ Second round PCR Forward primer (P3): 5′-AGCCAGCTCAAGAAAATGGTGTCCAAG-3′ Reverse primer (P4): 5′-TCACTGAGACCGGCAGTCTTTCTTGCTT-3′ | 95 °C, 50 s 65 °C, 30 s 72 °C, 1 min (25 cycles) 95 °C, 50 s 67 °C, 30 s 72 °C, 1 min (30 cycles) |
| PCR | ||
| VCA (1268 bp) | Forward primer (LMRC 181): 5′-CGGGATCCGGTCGTGTACTTGGGATTG-3′ Reverse primer (LMRC 182): 5′-CGGGATCCCCCCATCTCCCTCTTACC-3′ | 95 °C, 1 min 57 °C, 1 min 72 °C, 1 min 30 s (30 cycles) |
| gp350/220 (1008 bp) | Forward primer (LMRC 256): 5′-CGGGATCCGGCACTGAATAGGTAGCA-3′ Reverse primer (LMRC 257): 5′-CGGGATCCGAGACAATGGAGGCAG-3′ | 95 °C, 1 min 52 °C, 1 min 72 °C, 1 min 30 s (30 cycles) |
| Zta (182 bp) | Forward primer (Z1): 5′- TTCCACACAGCCTGCACCAGTG-3′ Reverse primer (Z2): 5′-GGCAGCAGCCACCTCACGGT-3′ | 95 °C, 1 min 55 °C, 1 min 72 °C, 1 min (30 cycles) |
| DAD-1 (240 bp) | Forward primer: 5′-GCAGTTATGTCGGCGTCGGTAG-3′ Reverse primer: 5′-GTTCTGTGGGTTGATCTGTATTC-3′ | 94 °C, 15 s 60 °C, 15 s 72 °C, 30 s (25 cycles) |
| qPCR | ||
| EBNA-1 | Forward primer (EBNA-1162F): 5′-TCATCATCATCCGGGTCTCC-3′ Reverse primer (EBNA-1229R): 5′-CCTACAGGGTGGAAAAATGGC-3′ Probe (EBNA-1186T): 5′-(FAM)*CGCAGGCCCCCTCCAGGTAGAA(TAMRA)*-3′ | 95 °C, 15 s 60 °C, 1 min (40 cycles) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chua, H.-H.; Kameyama, T.; Mayeda, A.; Yeh, T.-H. Epstein-Barr Virus Enhances Cancer-Specific Aberrant Splicing of TSG101 Pre-mRNA. Int. J. Mol. Sci. 2022, 23, 2516. https://doi.org/10.3390/ijms23052516
Chua H-H, Kameyama T, Mayeda A, Yeh T-H. Epstein-Barr Virus Enhances Cancer-Specific Aberrant Splicing of TSG101 Pre-mRNA. International Journal of Molecular Sciences. 2022; 23(5):2516. https://doi.org/10.3390/ijms23052516
Chicago/Turabian StyleChua, Huey-Huey, Toshiki Kameyama, Akila Mayeda, and Te-Huei Yeh. 2022. "Epstein-Barr Virus Enhances Cancer-Specific Aberrant Splicing of TSG101 Pre-mRNA" International Journal of Molecular Sciences 23, no. 5: 2516. https://doi.org/10.3390/ijms23052516
APA StyleChua, H. -H., Kameyama, T., Mayeda, A., & Yeh, T. -H. (2022). Epstein-Barr Virus Enhances Cancer-Specific Aberrant Splicing of TSG101 Pre-mRNA. International Journal of Molecular Sciences, 23(5), 2516. https://doi.org/10.3390/ijms23052516