1. Introduction
Most known human gene products execute their molecular function at the protein level. Proteomics, therefore, should theoretically be considered the preferred approach for high-throughput screening of the expression of such genes. This is also true for quantitative assessments of the activities of molecular pathways. However, for various reasons, the current limitations of proteomic techniques do not allow routine screening of most protein-coding genes [
1]. For example, in the US National Cancer Institute’s large-scale project
Clinical Proteomic Tumor Analysis Consortium (CPTAC), devoted to the integrative proteogenomic characterization of human cancers, expression levels for only 15,112 genes have been resolved at the protein level by using tandem mass (MS/MS) spectrometry conducted on Thermo QE series of mass spectrometers (QE, QEplus, QE-HF, and QE-HF-X) [
2]. These limitations are especially severe for the analysis of formalin-fixed paraffin-embedded (FFPE) cancer tissue samples, which is a routine format of biosample storage in clinical oncology [
3]. Formalin fixation produces numerous artifact chemical modifications, including covalent cross-links between unrelated protein molecules [
4].
Thus, it is important to improve current proteomic instruments or to use alternative methods that could be validated at the proteomic level. Transcriptomics offer another way of measuring expression of protein-coding genes [
5]. It has been shown that RNA- and proteomic raw gene expression levels statistically significantly correlate with mean average Speaman Rho 0.16–0.52 [
6,
7,
8]. Furthermore, RNA sequencing (RNAseq) can be used as an alternative to immunohistochemical methods for the assessment of biomarker functional status in clinical cancer samples for proteins such as HER2, ESR1, PGR, and PD-L1 [
9].
The RNAseq results demonstrated high reproducibility and robustness in resolving transcripts of 23,248 protein-coding genes [
10]. This allowed us to incorporate this type of analysis into the pipeline of tumor molecular analysis to estimate the expression of cancer drug target genes and the activation of targeted molecular pathways [
11]. The latter, in turn, made it possible to simulate and predict the activities of cancer drugs with known molecular specificities, and to determine their patient-specific rating [
12]. However, to our knowledge, the question of whether there is a correlation between the quantitative molecular pathway activation data received using RNAseq and proteomic data has never been explored before.
We have shown previously that the aggregation of RNA-level gene expression profiles into quantitative molecular pathway activation metrics results in reduced batch effects and better agreement between different experimental platforms. However, to our knowledge, the question of whether the pathway level of data analysis provides any advantage when comparing transcriptomic and proteomic data has never been explored before.
Here, we examined this question using paired proteomic and transcriptomic gene expression profiles obtained for the same human cancer tissue biosamples and available through The Cancer Genome Atlas (TCGA) and the NCI Clinical Proteomic Tumor Analysis Consortium (CPTAC) portals. We analyzed a total of 755 samples for glioblastoma and breast, liver, lung, ovarian, pancreatic, and uterine cancers. In CPTAC proteomic assay, the expression levels of 15,112 genes were profiled using the tandem mass (MS/MS) spectrometry conducted on Thermo QE series of mass spectrometers (QE, QEplus, QE-HF, and QE-HF-X) [
2]. We used TCGA RNA expression levels of the same genes obtained with Illumina HiSeq 4000 platform for the same biosamples to compare proteomic and transcriptomic data at the individual gene- and pathway activation levels. At the gene level, absolute gene expression values were compared, whereas the pathway-grade comparisons were made between the pathway activation levels (PALs) calculated using average sample-normalized transcriptomic and proteomic profiles. We observed remarkably different average correlations between the primary RNA- and protein expression data for different cancer types: Spearman Rho between 0.017 (
p = 1.7 × 10
−13) and 0.27 (
p < 2.2 × 10
−16). However, at the pathway level, we detected overall statistically significantly higher correlations: averaged Rho between 0.022 (
p < 2.2 × 10
−16) and 0.56 (
p < 2.2 × 10
−16). Thus, we conclude that data analysis at the PAL level enables obtaining results of a greater similarity when comparing high-throughput RNA and protein expression profiles.
2. Results
Intracellular molecular pathways include gene products participating in common molecular processes, i.e., in all major cellular events in health and disease. Traditionally, they are primarily classified as the metabolic, DNA repair, signaling, and cytoskeleton organization pathways [
1]. Quantitative assessments of pathway activation levels (PALs) have given rise to next generation biomarkers in human biology that are, in many contexts, more accurate and robust than individual gene expression levels [
13]. Herein, positive, zero, and negative PAL values mean upregulation, no changes, and downregulation of a molecular pathway, respectively [
14]. Furthermore, the absolute value of PAL corresponds to the extent of a pathway differential regulation [
13]. Thus, a higher PAL value reflects greater pathway activation and vice versa. For several experimental techniques regarding RNA expression analysis, PALs also showed less significant platform bias and batch effects than individual gene expression profiles [
15]. In this study, we compared, for the first time, RNA and protein expression levels for the same biosamples at both the pathway- and individual gene activation levels. The biospecimens were human cancer tissue samples. Primary RNA and protein expression data were extracted from the TCGA and CPTAC project databases, respectively. The platform for RNA expression profiling was Illumina HiSeq 4000, and proteomic profiles were obtained using tandem mass (MS/MS) spectrometry conducted on Thermo QE series of mass spectrometers (QE, QEplus, QE-HF, and QE-HF-X) [
2]. In total, we analyzed 755 paired transcriptomic/proteomic biosamples for seven human cancer types: breast invasive carcinoma (99, 13.11%), glioblastoma (98, 12.98%), hepatocellular carcinoma (87, 11.52%), lung adenocarcinoma (111, 14.70%), ovarian serous cystadenocarcinoma (119, 15.76%), pancreatic ductal adenocarcinoma (140, 18.54%), and uterine corpus endometrial carcinoma (101, 13.38%).
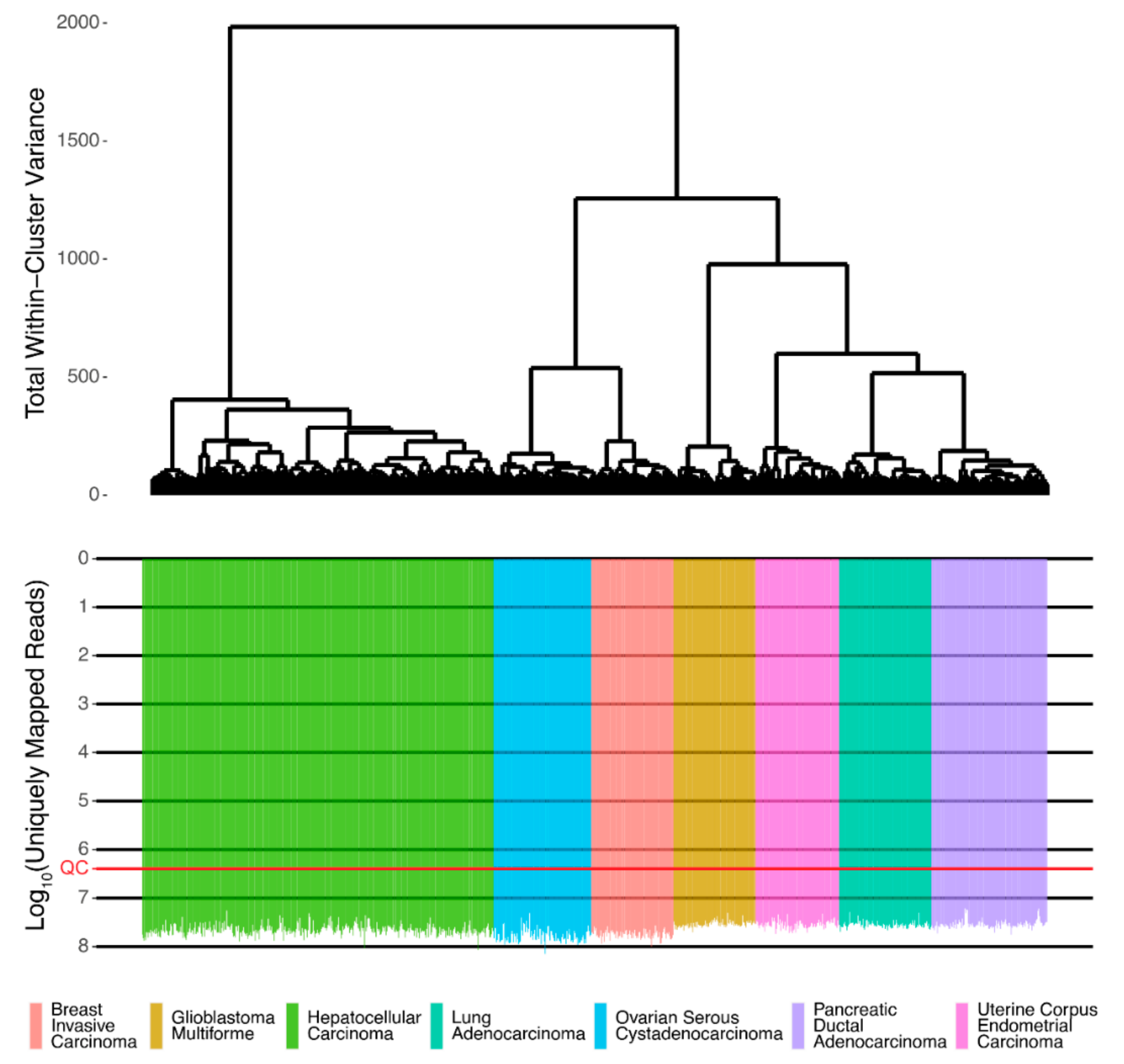
Associations between protein and RNA expressions were assessed using Spearman Rho and Pearson R correlation coefficients for gene expression and pathway activation levels. The initial RNA sequencing profiles were screened and passed technical quality control metrics [
16] (
Figure 1).
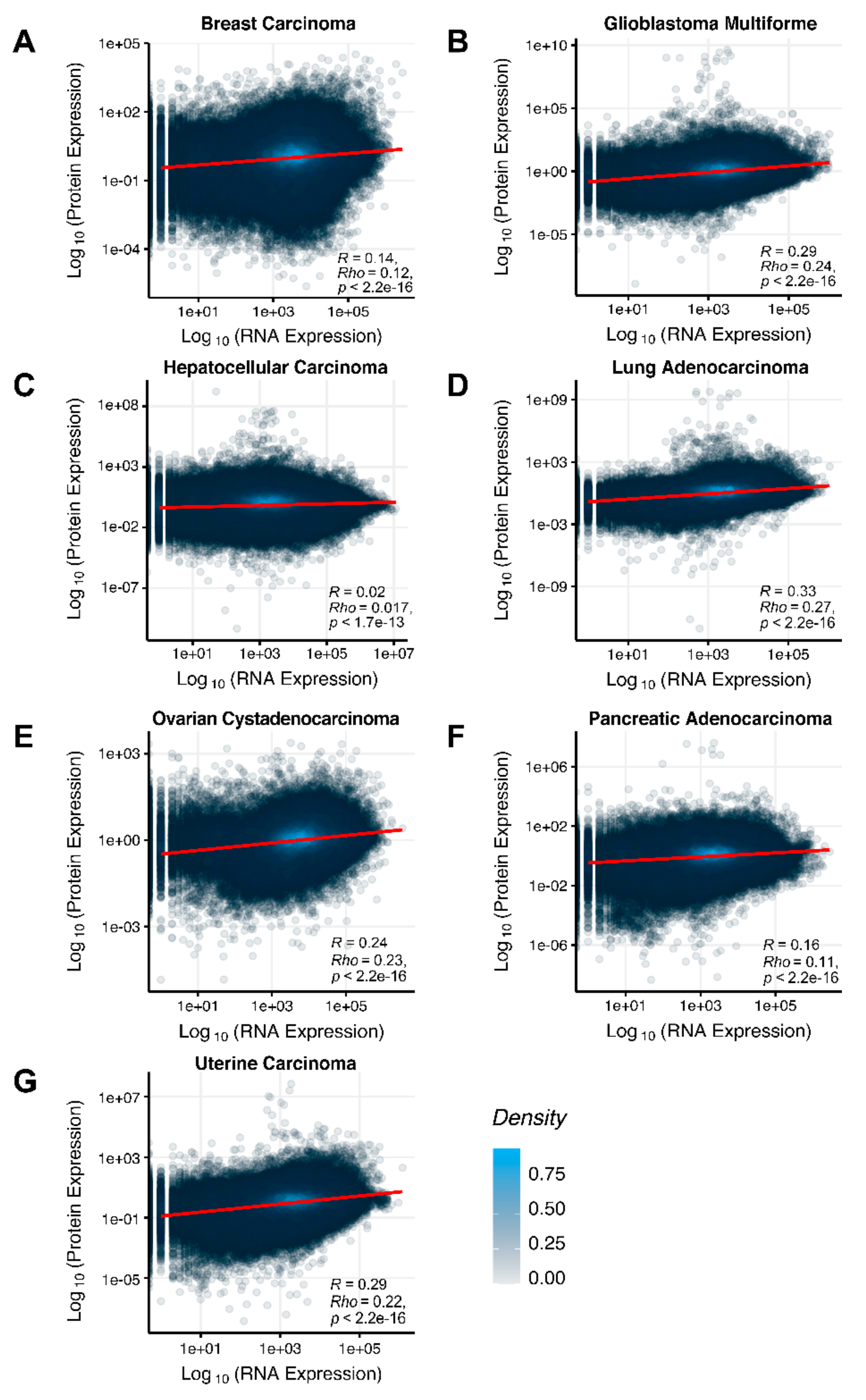
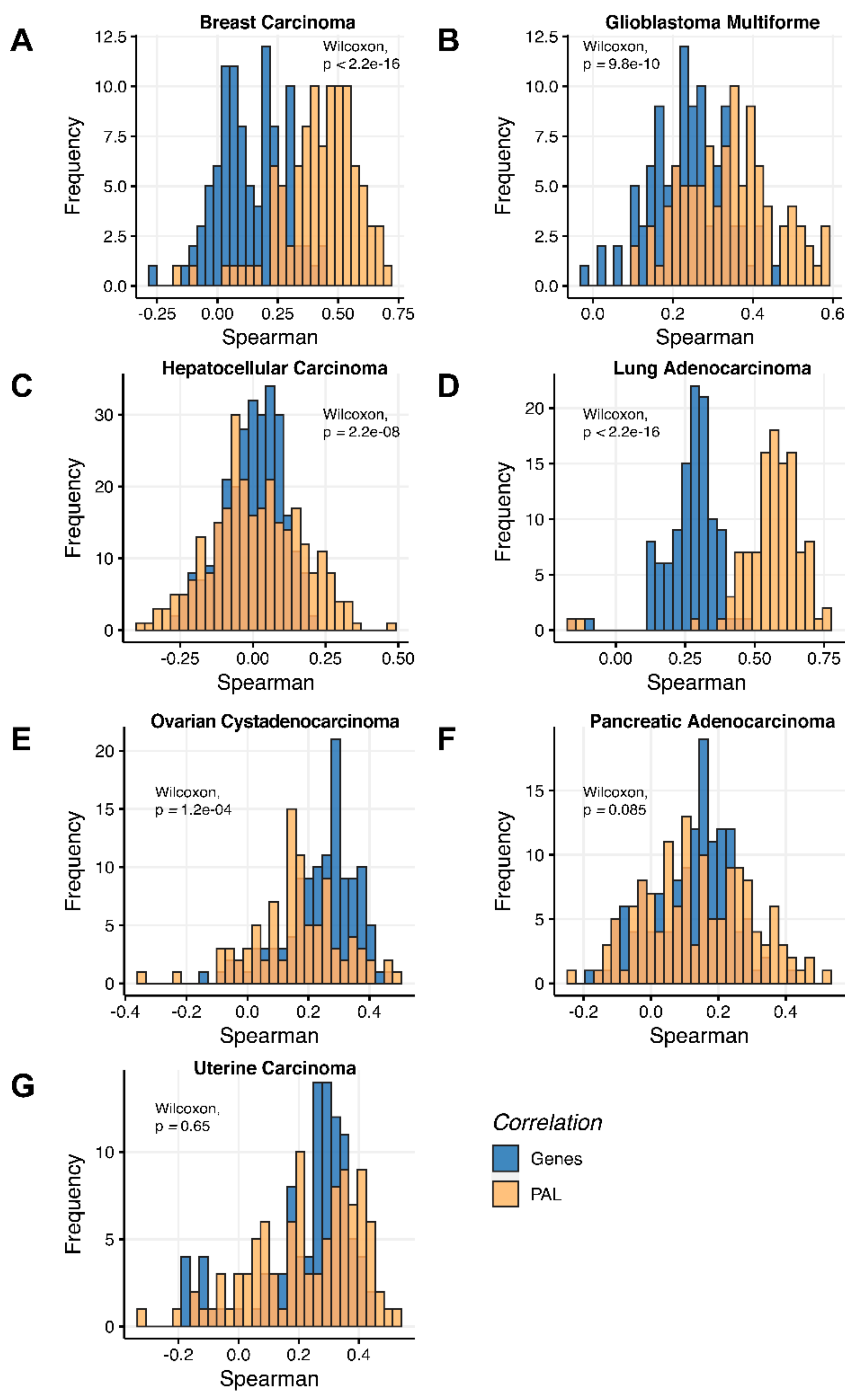
We then assessed at the individual gene level Spearman correlations between RNA and protein expression levels in seven human cancer types. We obtained 0.17 mean Spearman correlation that varied from 0.017 for Hepatocellular Carcinoma till 0.27 for Lung Adenocarcinoma samples (
Figure 2), which is in line with the previous results, i.e., ~0.16–0.35 correlation between the RNA- and proteome-based gene expression levels [
6,
7].
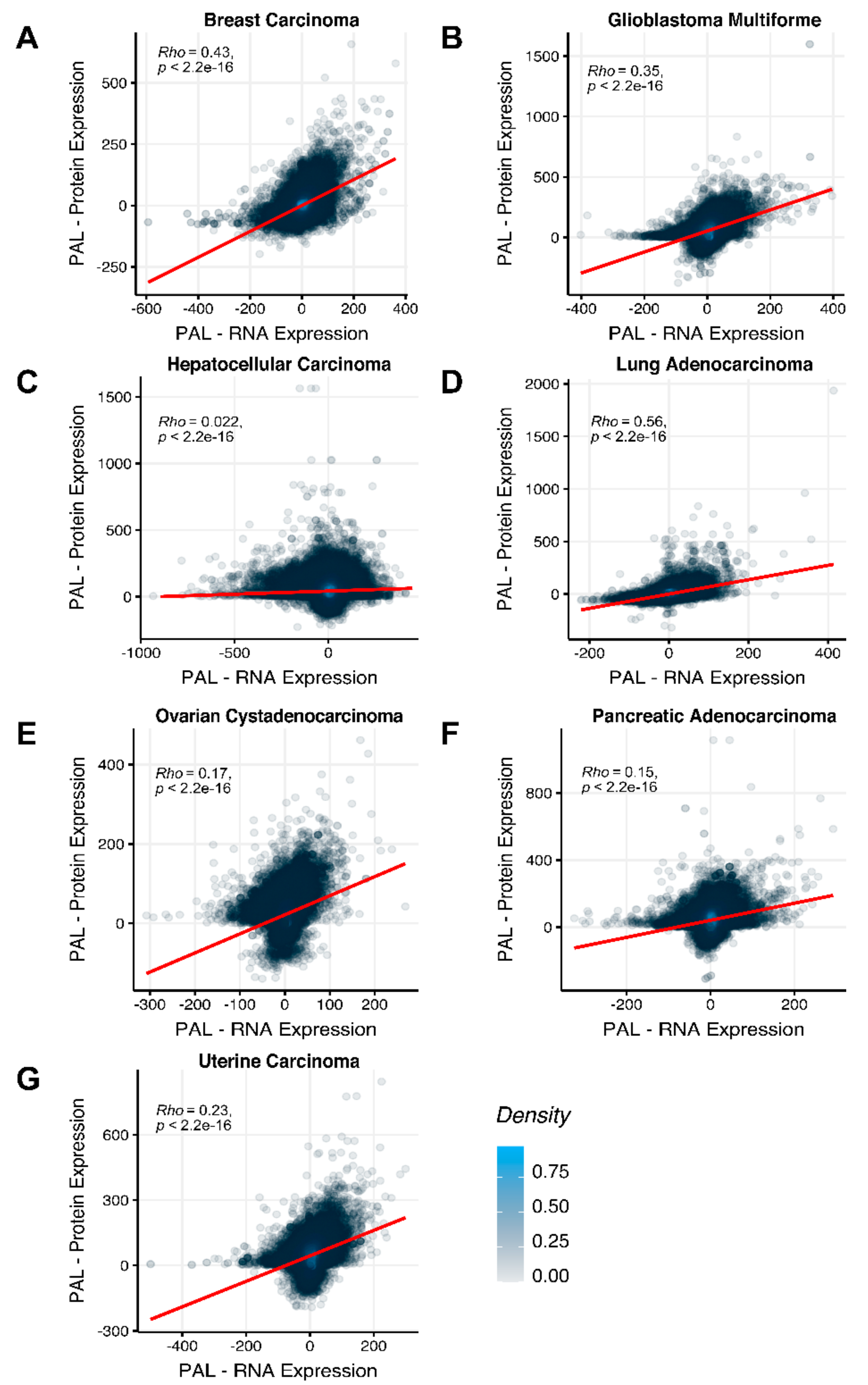
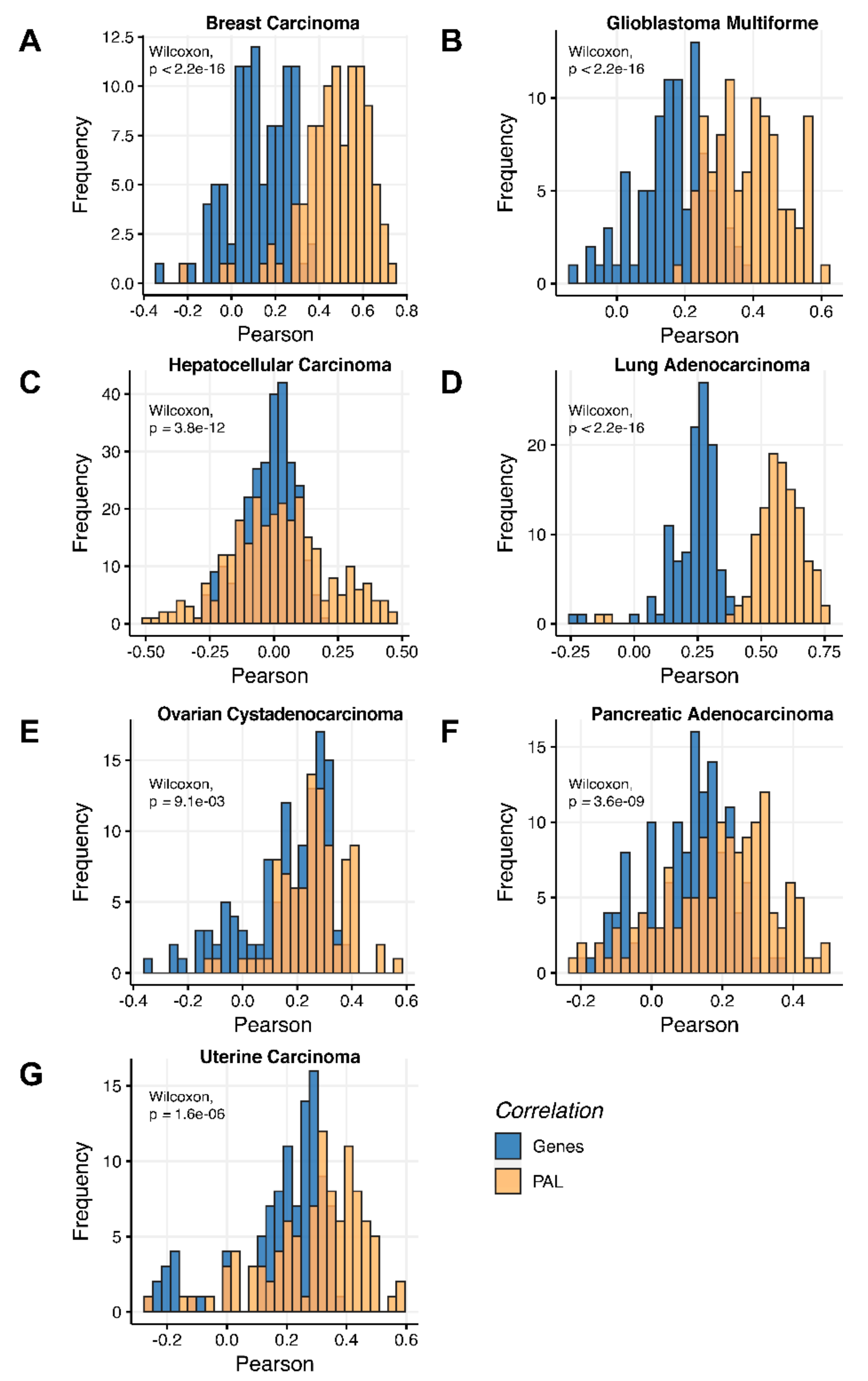
To compare RNA and protein expression data at the pathway level, we used the PAL approach for the same biosamples. We calculated Spearman correlations between PAL values for all tumor types under investigation (
Figure 3). We observed 0.27 mean Spearman correlation between PAL values, which varied from 0.022 for Hepatocellular Carcinoma up to 0.56 for Lung Adenocarcinoma samples. Thus, we obtained 1.58-fold-change between the averaged gene-to-gene and PAL-to-PAL correlations.
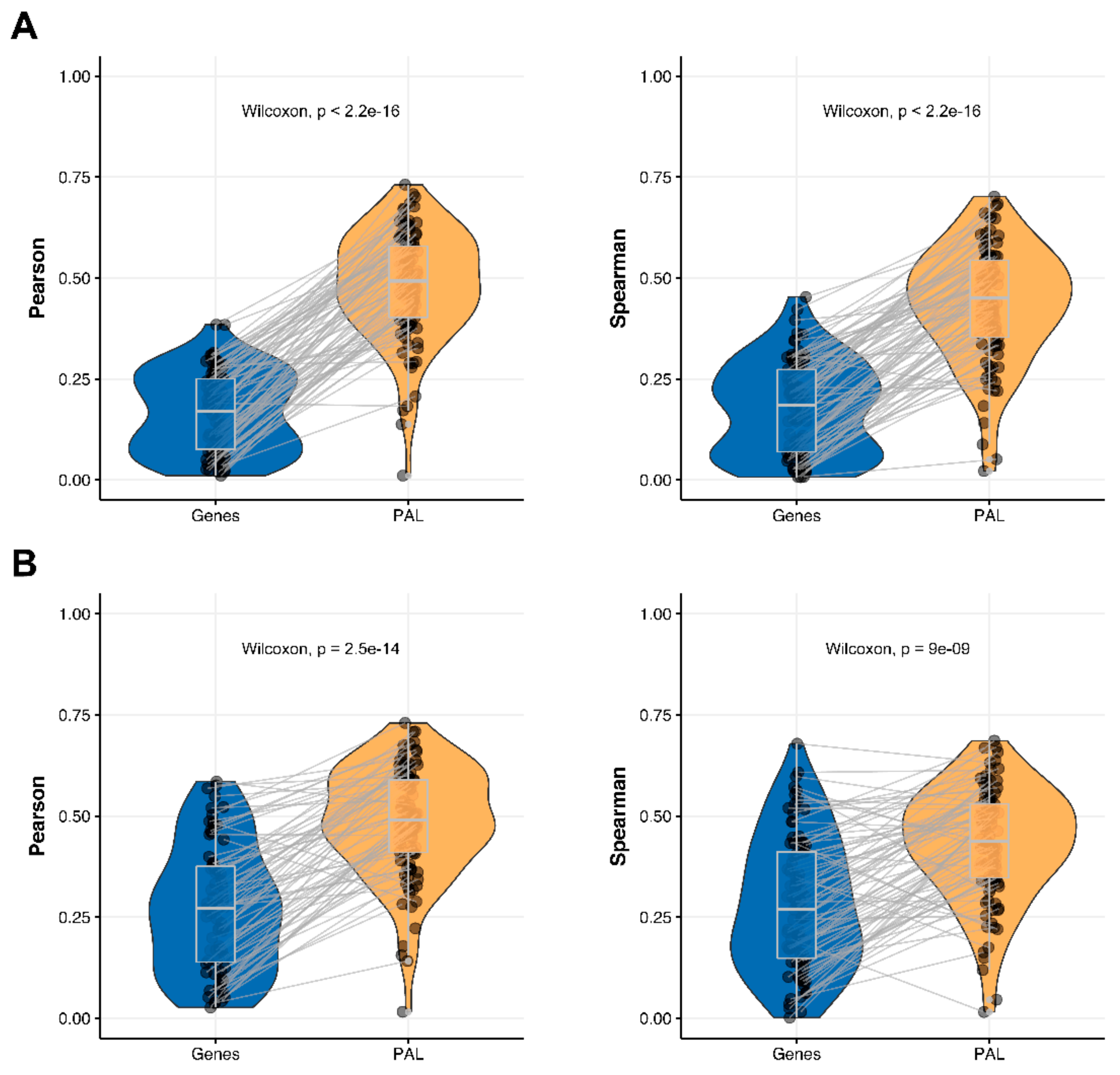
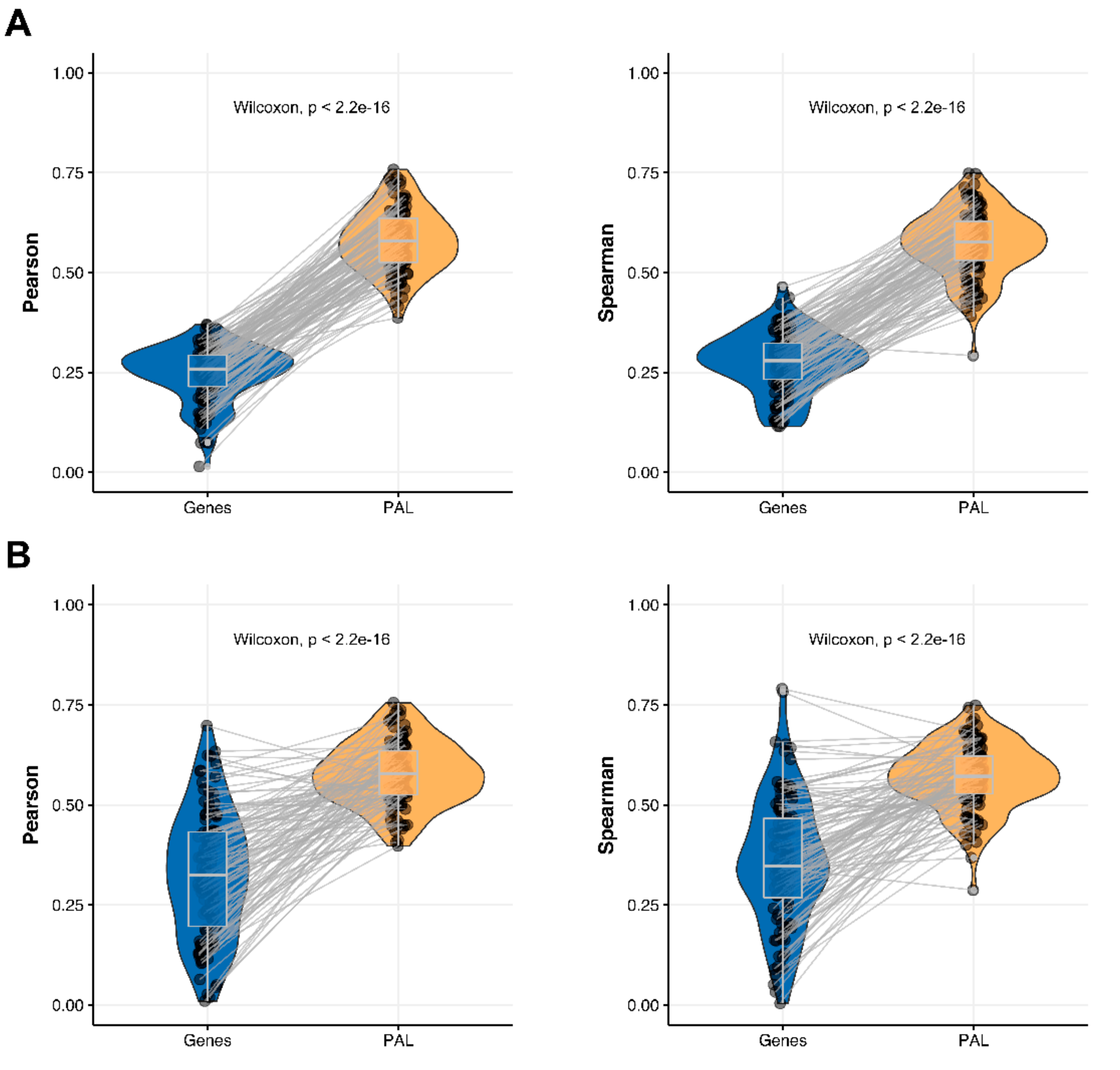
We then measured statistical significance of the differences between gene (transcriptome)-to-gene(proteome) vs. PAL (transcriptome)-to-PAL (proteome) correlations. Using Wilcoxon statistical test, we observed (
Figure 4,
Figure 5,
Figure 6,
Figure 7,
Figure 8,
Figure 9 and
Figure 10) that both Pearson and Spearman correlations calculated for pathway activation levels were statistically significantly higher for the PAL level compared to the gene level in five out of seven cancer types investigated. In the remaining two cancer types, the results for Pearson and Spearman correlations were inconsistent or statistically not significant, as for ovarian serous cystadenocarcinoma (
Figure 8) and uterine corpus endometrial carcinoma (
Figure 10).
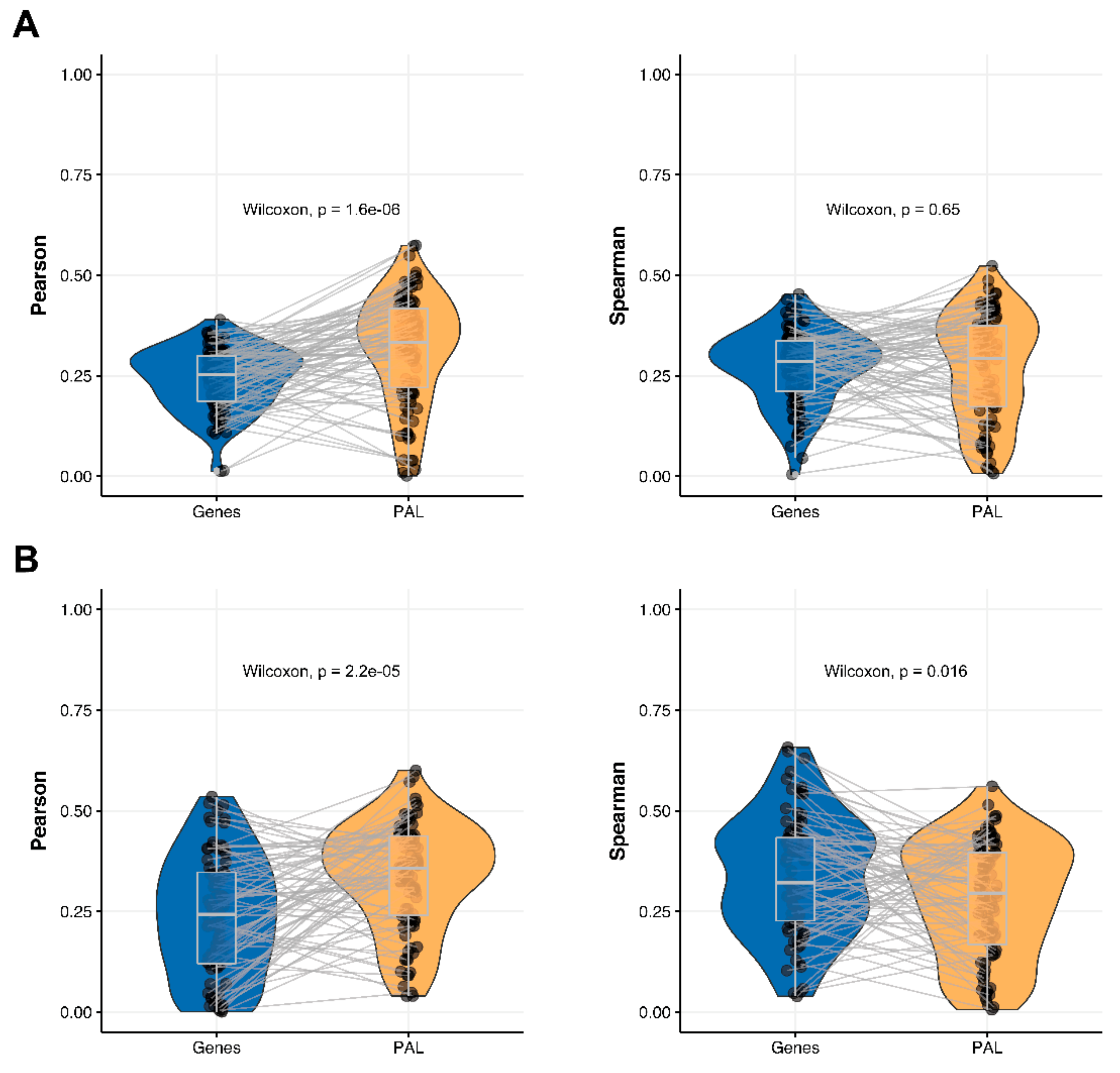
We then performed this analysis for only 78 genes encoding molecular targets for the NCCN-recommended drugs in the seven cancer types considered in this study [
17]. We compared 78-gene expression profiles and the profiles of molecular pathways including these genes at the RNA and protein levels. We observed that Pearson and Spearman correlations for pathway activation levels were statistically significantly higher compared to the single gene expression levels in the same four out of seven cancer types (
Figure 4,
Figure 5,
Figure 9 and
Figure 11). For the remaining three cancer types, i.e., hepatocellular carcinoma, ovarian serous cystadenocarcinoma, and uterine corpus endometrial carcinoma, Pearson and Spearman correlations showed poor statistical significance (
Figure 6,
Figure 8 and
Figure 10).
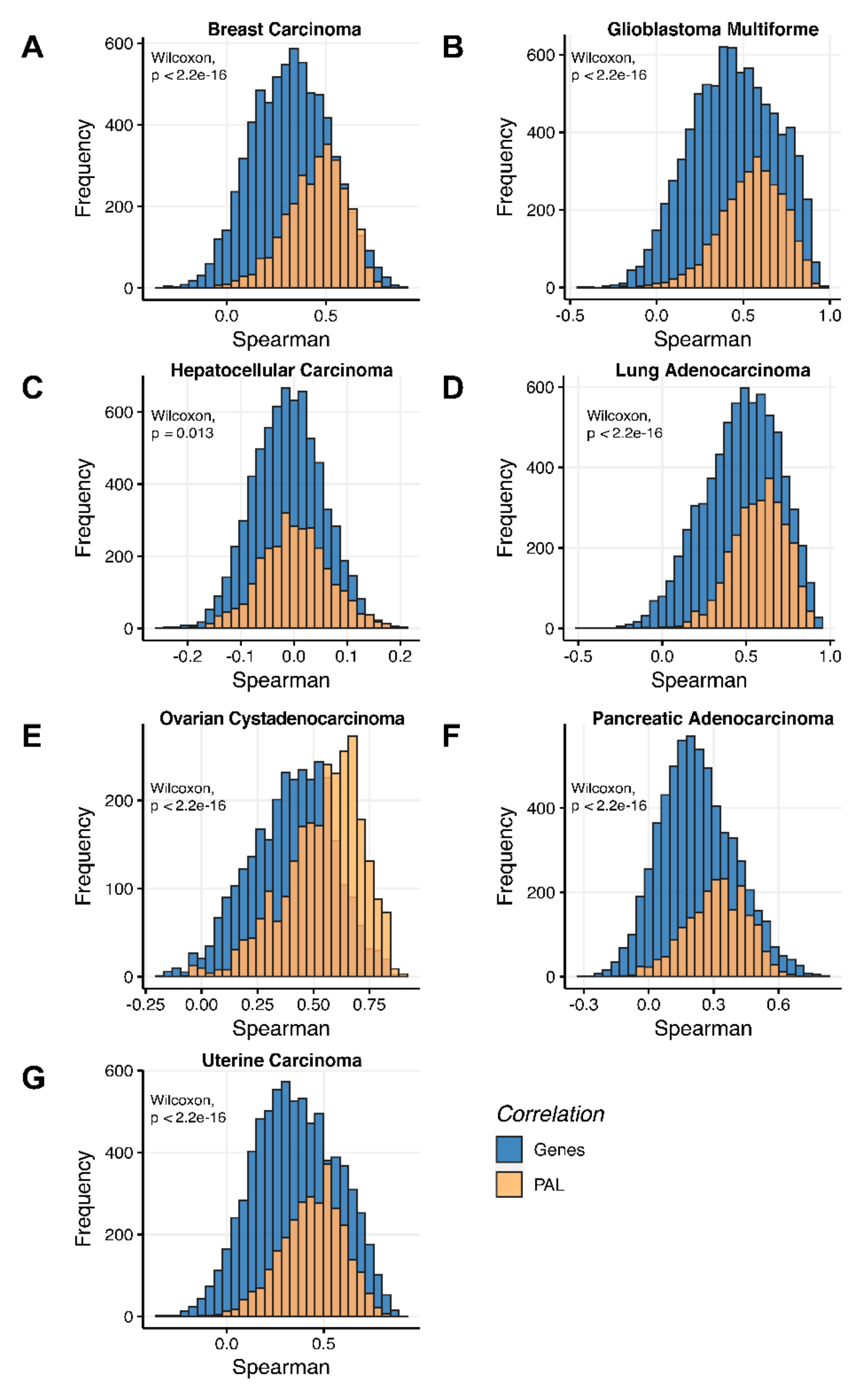
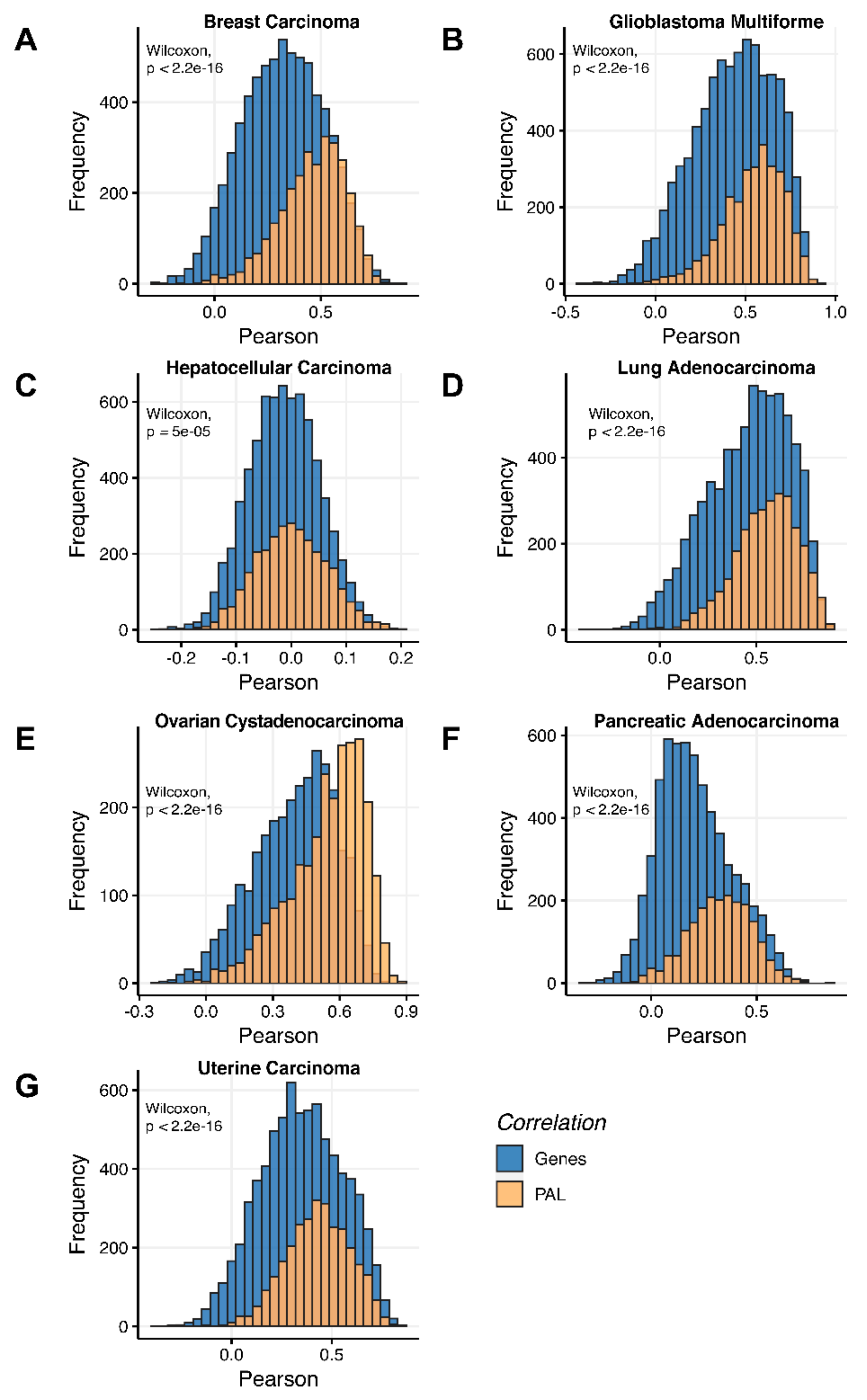
Additionally, we investigated how activation patterns of individual molecular pathways correlated between the different biosamples and compared this with the gene-specific patterns. We calculated Pearson and Spearman correlations for every gene and every molecular pathway among all biosamples of a given tumor type (
Figure 12 and
Figure 13). We observed that these correlations were statistically significantly higher for pathway activation levels, with the lowest statistical significance (highest
p = 0.013) being observed for hepatocellular carcinoma.
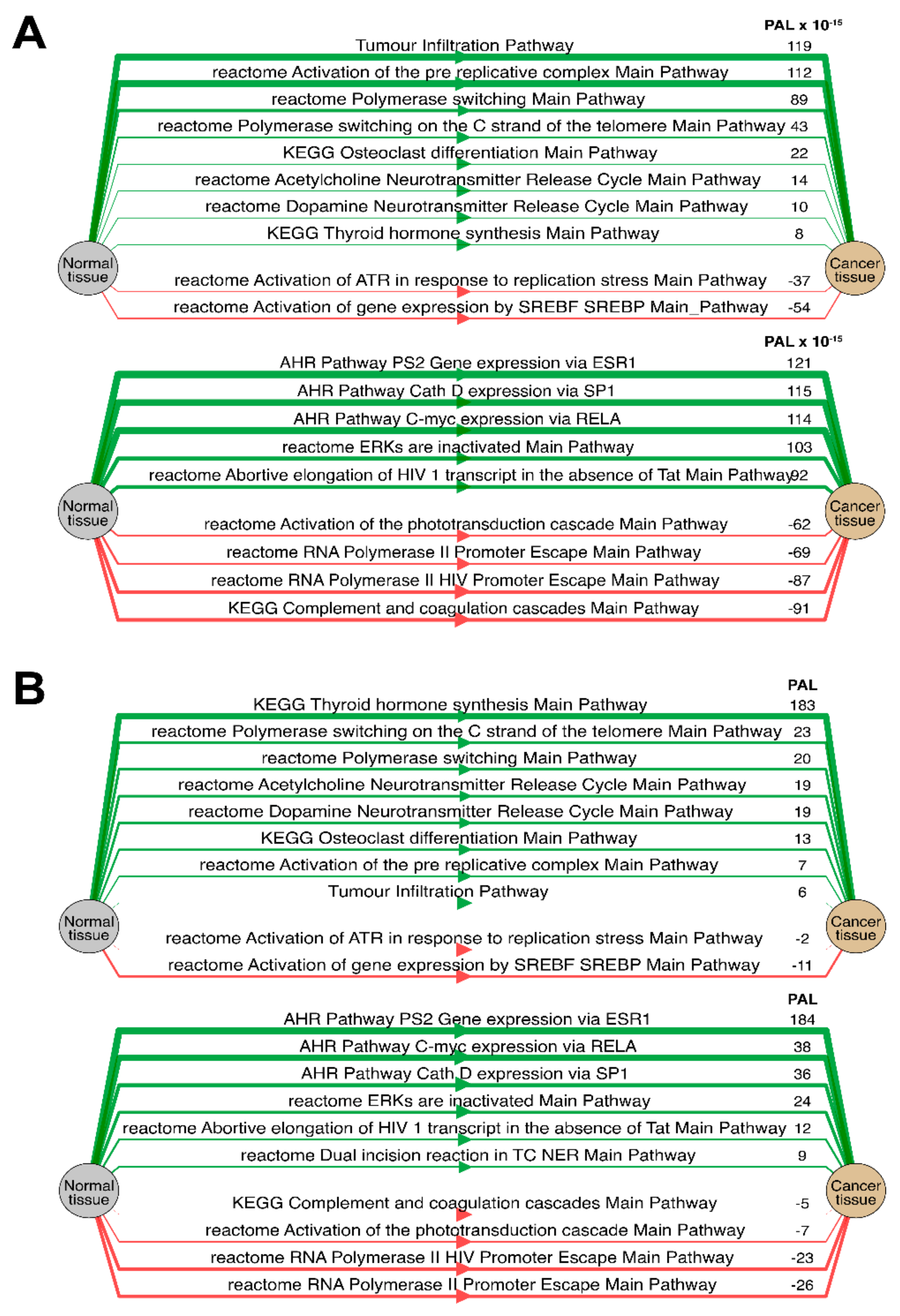
Separately for each cancer type, we then analyzed the “top-10” of the most and the least correlated molecular pathways between RNA and protein expression data (see
Table 1,
Table 2,
Table 3,
Table 4,
Table 5,
Table 6 and
Table 7 and
Supplementary Table S1). Notably, such highly correlated pathways with regard to tumor infiltration, immune response, and regulation of DNA polymerase alpha, delta and epsilon activity were consistent among most tumor types (Spearman correlation 0.61–0.91).
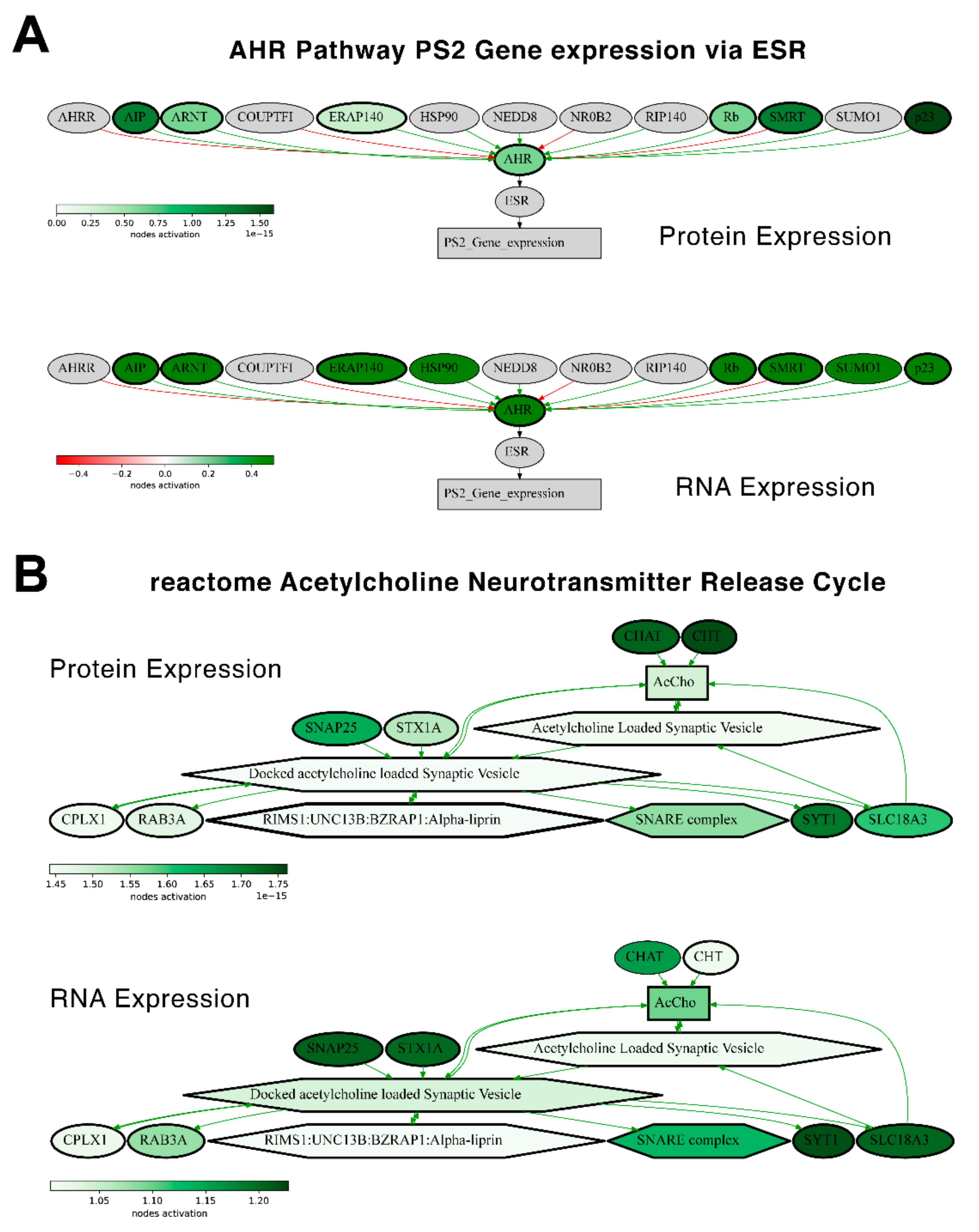
We then checked the consistencies of pathway activation schemes built using transcriptomic or proteomic data for the best and the least correlated molecular pathways. We calculated PAL levels for the “top-10” such pathways for Lung Adenocarcinoma biosamples (
Figure 14) and compared activation charts for the chosen “AHR Pathway PS2 Gene expression via ESR” and “reactome Acetylcholine Neurotransmitter Release Cycle Main Pathway” pathways, which have the highest and the lowest correlations, respectively, for Lung Adenocarcinoma (
Figure 15). Notably, all the pathways from both the best and the least correlated “top-10” group showed congruent activation patterns for RNAseq and proteomic data. Similarly, most of the components of the two molecular pathways that were considered more in-depth also showed congruent activation trends (
Figure 15).
The number of gene products in a pathway theoretically may have an impact on the extent of the data aggregation effect [
15] and, consequently, influence the gain of correlation in the gene-pathway comparisons. Therefore, we separately estimated gene-pathway correlations for the groups of bigger and smaller pathways including, respectively, at least 10, 20, and 40 gene products (
Supplementary Figures S1–S7). We noticed, however, that the pathway size did not have any detectable impact on the pathway-level correlation gain.
Specifically, we obtained the following results for the individual cancer types under investigation.
2.1. Breast Invasive Carcinoma
The Pearson and Spearman gene-to-gene correlations between RNA and protein expression for an averaged biosample (
Figure 2A) were 0.14 and 0.12, respectively, while on the PAL-to-PAL level (
Figure 3A), they were 0.47 and 0.43, respectively. Thus, data analysis at the pathway activation levels resulted in ~3.5 times higher transcriptome-proteome correlation compared to the gene level, which was statistically significant (Wilcoxon
p < 2.2 × 10
−16); see
Figure 4A,
Figure 16A and
Figure 17A. This difference also remained significant (
p < 2.2 × 10
−16) for drug target genes and molecular pathways (
Figure 4B).
Pearson and Spearman correlations for individual genes or molecular pathways were statistically significantly higher for pathway activation levels (Wilcoxon, both
p < 2.2 × 10
−16,
Figure 12A and
Figure 13A).
The “top-10” most strongly and weakly correlated molecular pathways are shown on
Table 1.
2.2. Glioblastoma Multiforme
The Pearson and Spearman gene-to-gene correlations for an average biosample (
Figure 2B) were 0.29 and 0.24, respectively, and PAL-to-PAL correlations (
Figure 3B) were 0.39 and 0.35, respectively. Thus, in this case, we detected ~1.5 times pathway-level gain of correlation, which was statistically significant (
p < 2.2 × 10
−16 and
p < 10
−9 for Pearson and Spearman correlations, respectively); see
Figure 5A,
Figure 16B and
Figure 17B. This difference also remained statistically significant for the cancer drug-targeted genes and molecular pathways (
Figure 5B).
Pearson and Spearman correlations for individual genes or pathways were statistically significantly higher for pathway activation levels (Wilcoxon, both
p < 2.2 × 10
−16,
Figure 12B and
Figure 13B).
The “top-10” of the most and the least correlated pathways are shown on
Table 2.
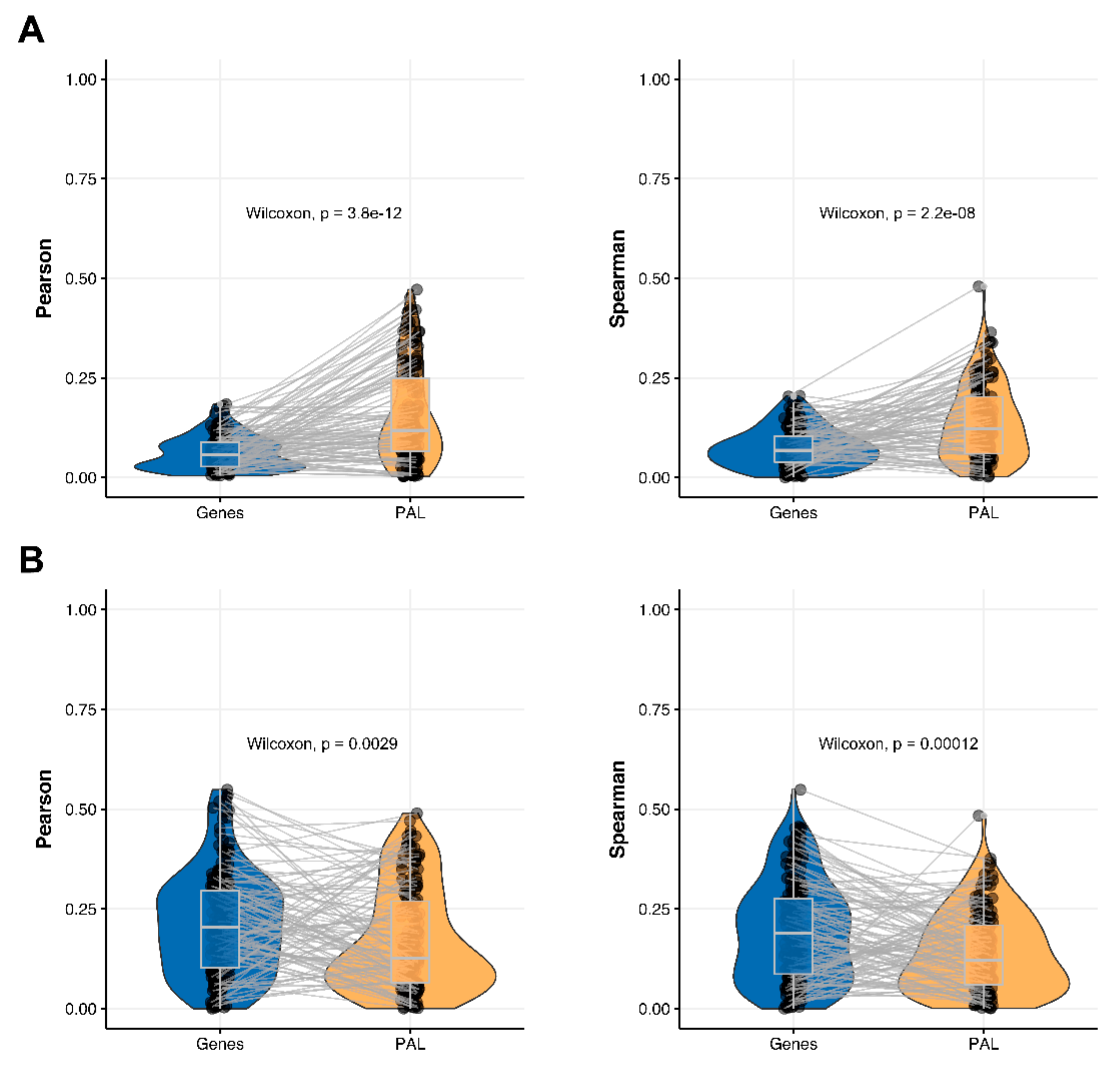
2.3. Hepatocellular Carcinoma
The Pearson and Spearman gene-to-gene correlations for an average biosample (
Figure 2C) were 0.02 and 0.017, respectively, and PAL-to-PAL correlations (
Figure 3C) were 0.061 and 0.022, respectively. This corresponded to ~3 times pathway-level gain for the Pearson correlation (
p < 2.2 × 10
−16), whereas the fold-change for the Spearman correlation was only ~1.2, which was also statistically significant (
p = 2.2 × 10
−8); see
Figure 6A,
Figure 16C and
Figure 17C. This difference, however, had the opposite effect on cancer drug targeted genes and molecular pathways (
Figure 6B).
The Pearson and Spearman correlations for individual genes or molecular pathways were statistically significantly higher for pathway activation levels (Wilcoxon,
p = 5 × 10
−5 and
p = 0.013, respectively); see
Figure 12C and
Figure 13C.
The “top-10” of the most and the least correlated molecular pathways are shown on
Table 3.
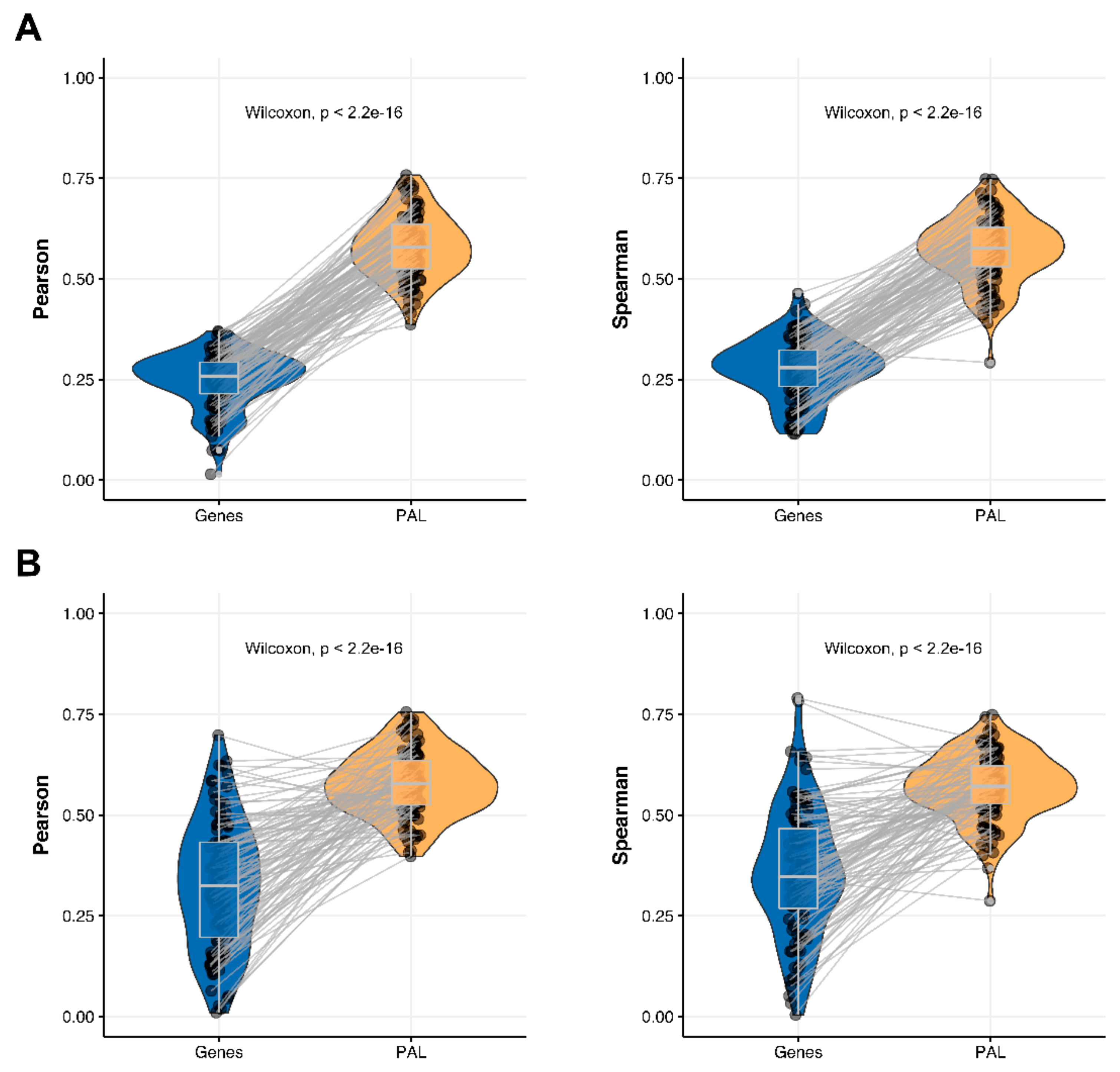
2.4. Lung Adenocarcinoma
The Pearson and Spearman gene-to-gene correlations for an average biosample (
Figure 2D) were 0.33 and 0.27, respectively, and PAL-to-PAL correlations (
Figure 3D) were 0.58 and 0.56, respectively. Thus, the detected pathway-level gain of correlation was ~2 times (
p < 2.2 × 10
−16); see
Figure 7A,
Figure 16D and
Figure 17D. The correlation gain also remained statistically significant for cancer drug-targeted genes and molecular pathways (
Figure 7B).
Pearson and Spearman correlations for individual genes or molecular pathways were statistically significantly higher for pathway activation levels (Wilcoxon, both
p < 2.2 × 10
−16,
Figure 12D and
Figure 13D).
The “top-10” of the most and the least correlated molecular pathways are shown on
Table 4.
We also compared PAL levels for “top-10” lists of the most and the least correlated molecular pathways in Lung Adenocarcinoma biosamples (
Figure 14) for the transcriptomic and proteomic data. All of the most and the least correlated pathways showed common activation or inhibition trends for the RNA and protein expression data.
Similarly, the activation charts for “AHR Pathway PS2 Gene expression via ESR” and “reactome Acetylcholine Neurotransmitter Release Cycle Main Pathway” pathways were compared (
Figure 15). Similarly, most of the components in the two considered molecular pathways were also congruently activated.
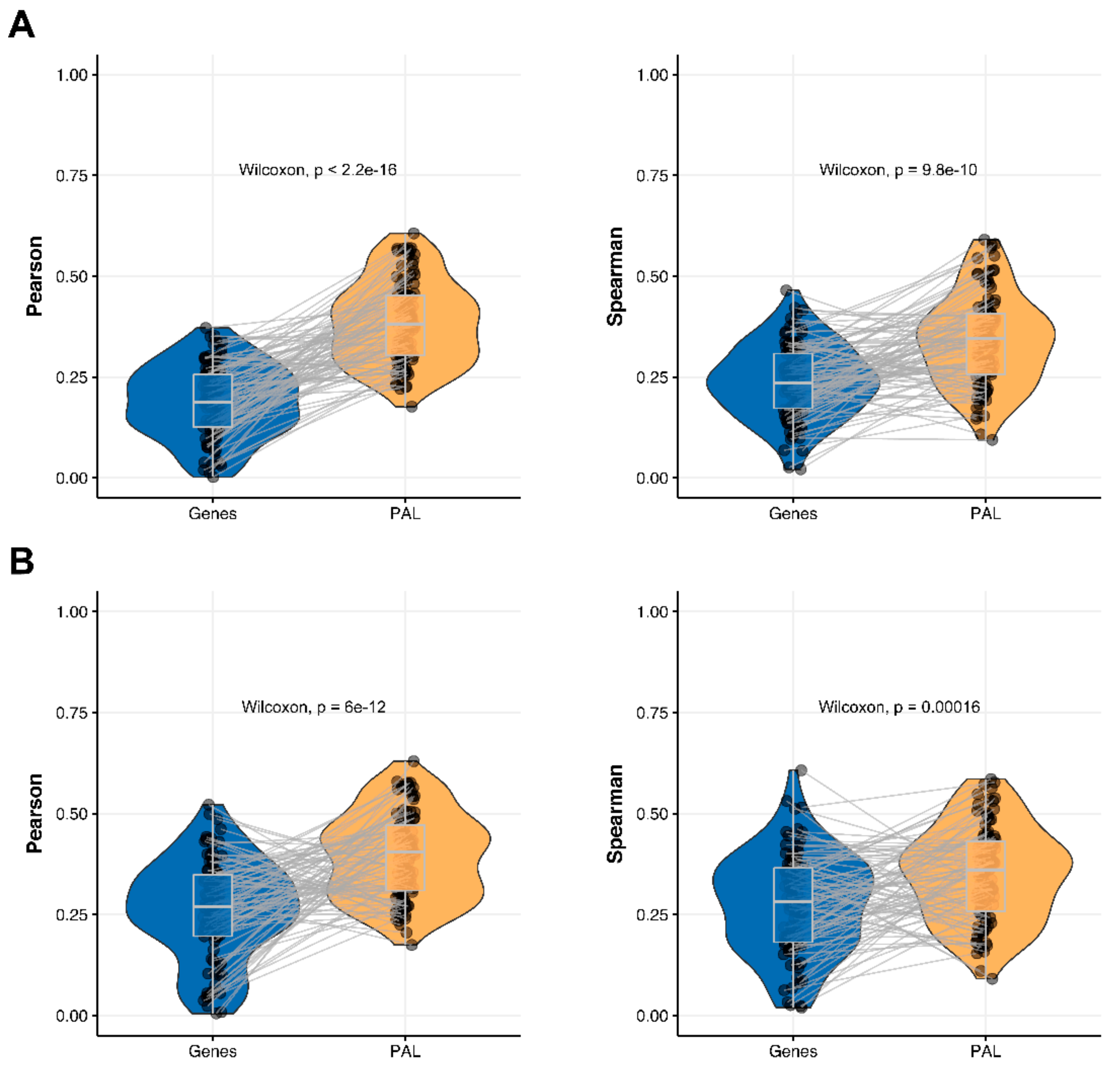
2.5. Ovarian Serous Cystadenocarcinoma
The Pearson and Spearman gene-to-gene correlations for an average biosample (
Figure 2E) were 0.24 and 0.23, respectively, and PAL-to-PAL correlations (
Figure 3E) were 0.25 and 0.17, respectively. In this case, no pathway-level correlation gain was detected (Pearson ~1; Spearman ~0.74 times) either for drug target genes or molecular pathways (
Figure 8B).
The Pearson and Spearman correlations for individual genes or molecular pathways were statistically significantly higher for pathway activation levels (Wilcoxon, both
p < 2.2 × 10
−16); see
Figure 12E and
Figure 13E.
The “top-10” of the most and the least correlated molecular pathways between RNA and protein expression levels are shown on
Table 5.
2.6. Pancreatic Ductal Adenocarcinoma
Pearson and Spearman gene-to-gene correlations for an average biosample (
Figure 2F) were 0.16 and 0.11, respectively, and PAL-to-PAL correlations (
Figure 3F) were 0.19 and 0.15, respectively. This suggests ~1.4 times pathway-level gain of correlation which was statistically significant only for Spearman (
p = 3.6 × 10
−9) correlation; see
Figure 5F,
Figure 15A and
Figure 16F. Differences between Spearman correlations were also statistically significant for drug target genes and molecular pathways (
Figure 9B).
The Pearson and Spearman correlations for individual genes or molecular pathways were statistically significantly higher for pathway activation levels (Wilcoxon, both
p < 2.2 × 10
−16); see
Figure 12F and
Figure 13F.
The “top-10” of the most and the least correlated molecular pathways are shown in
Table 6.
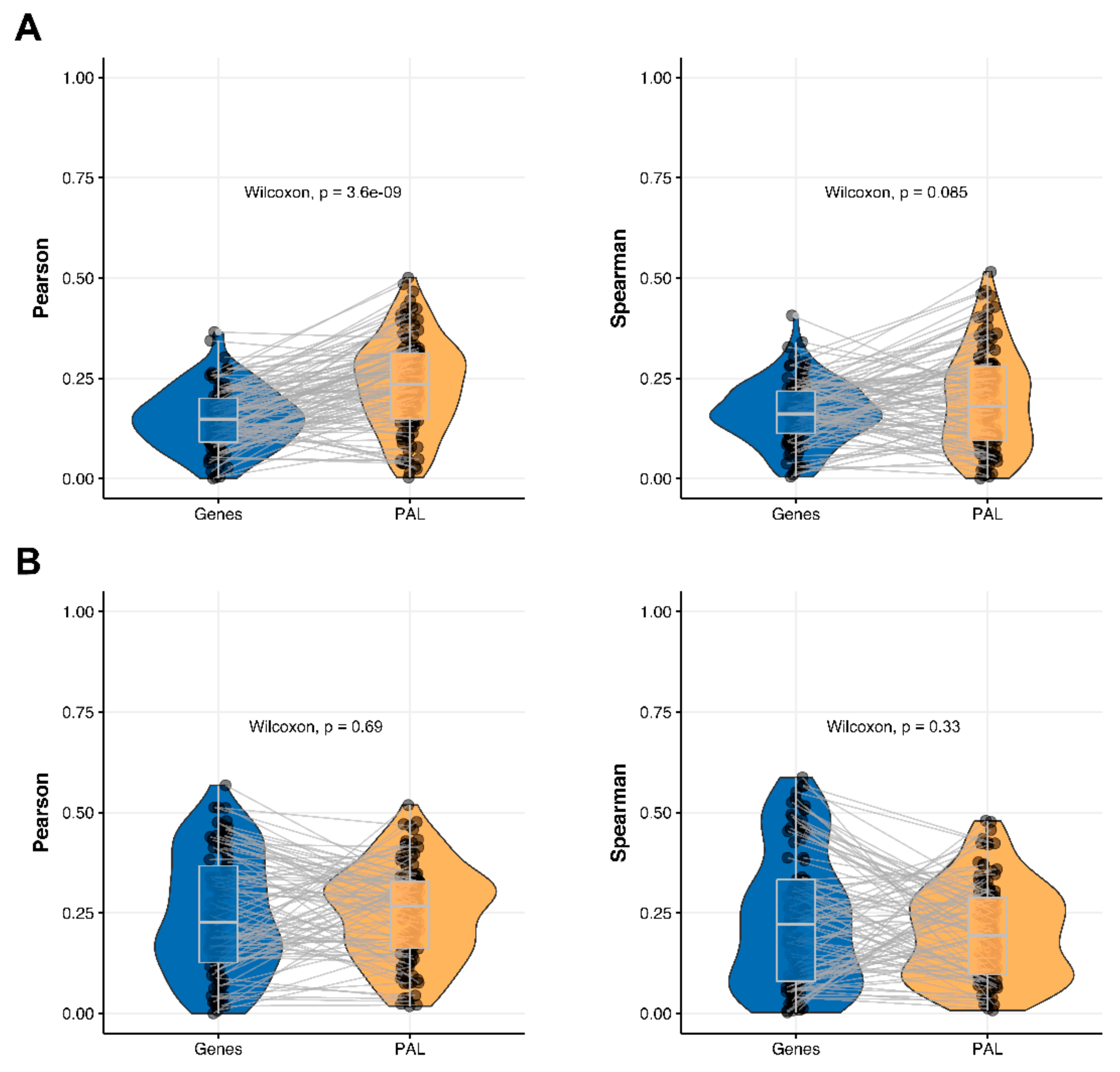
2.7. Uterine Corpus Endometrial Carcinoma
The Pearson and Spearman gene-to-gene correlations for an average biosample (
Figure 2G) were 0.29 and 0.22, respectively, and PAL-to-PAL correlations (
Figure 3G) were 0.29 and 0.23, respectively. In this case, correlations remained essentially the same for both gene- and pathway levels; see
Figure 10A,
Figure 16G and
Figure 17G. The same trend was also seen for drug target genes and pathways (
Figure 10B).
Pearson and Spearman correlations for individual genes or molecular pathways were statistically significantly higher for pathway activation levels (Wilcoxon, both
p < 2.2 × 10
−16,
Figure 12G and
Figure 13G).
The “top-10” of the most and the least correlated molecular pathways between RNA and protein expression levels are shown on
Table 7.
3. Discussion
Quantitative gene expression profiles at the mRNA and protein levels are fundamentally different because of differential mRNA and protein stability patterns, epigenetic and protein translation regulatory mechanisms [
18] and technical differences in the screening methods [
19]. However, there is an overall correlation between the quantitative gene expression profiles that was detected for various organisms and cell types [
18].
In this study we aimed to compare correlations of RNA- and protein-level gene expression data at the single gene and molecular pathway levels.
At the gene level, the correlations observed were congruent with the literature data (e.g., we observed ~0.23–0.24 correlations for ovarian cancer at the gene level, compared to ~0.3 in the previous report [
7], or ~0.24–0.29 for glioblastoma in this study compared to previously observed ~0.15 [
6]).
On the other hand, we detected remarkably different correlations between RNA expression and proteomic data on both gene- and pathway levels between the different tumor types investigated simultaneously in TCGA and CPTAC projects. For example, the lowest correlation could be observed for hepatocellular carcinoma (0.017–0.06), whereas the biggest was detected for lung adenocarcinoma (0.33–0.58).
Such dramatic difference from the first view could be explained by vulnerabilities of the experimental expression platforms to the nature of the tested biospecimens. However, some biological mechanisms could also be involved, e.g., tumor type-specific pH alteration, which can lead to dramatic differences in the repertoire of translated proteins while not strongly affecting RNA transcription [
20].
In any case, we noticed that in five out of seven cancer types tested (glioblastoma, breast, liver, lung, pancreatic cancers), the expression data analysis at the pathway level was beneficial in terms of improving the correlation between the quantitative mRNA and protein data for the same biospecimens. In the two remaining tumor types (endometrial and ovarian cancers), we observed no pathway-level gain of correlation between mRNA and proteomic data. These results did not depend on the pathway size, as they were reproduced here with pathways of any size, and with pathways with 10-, 20-, and 40-participant size limits.
Similarly, we observed a gain of correlation between mRNA and protein expression in four out of seven cancer types in a correlation analysis of cancer drug-targeted genes and molecular pathways.



 ,
, 



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}