
Small Angle X-ray Diffraction as a Tool for Structural Characterization of Muscle Disease



Abstract
:1. Introduction
1.1. X-ray Instrumentation and Data Analysis Software
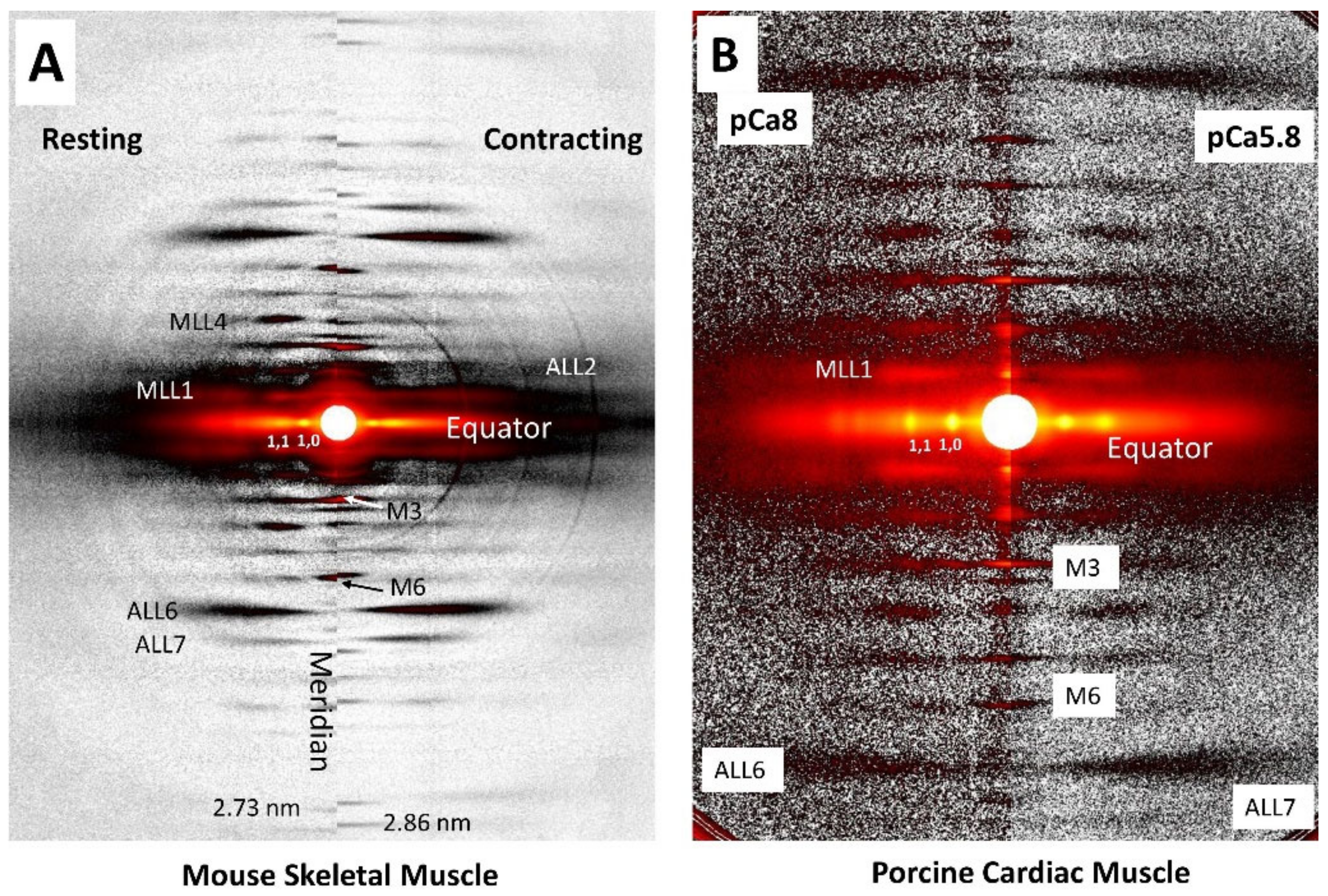
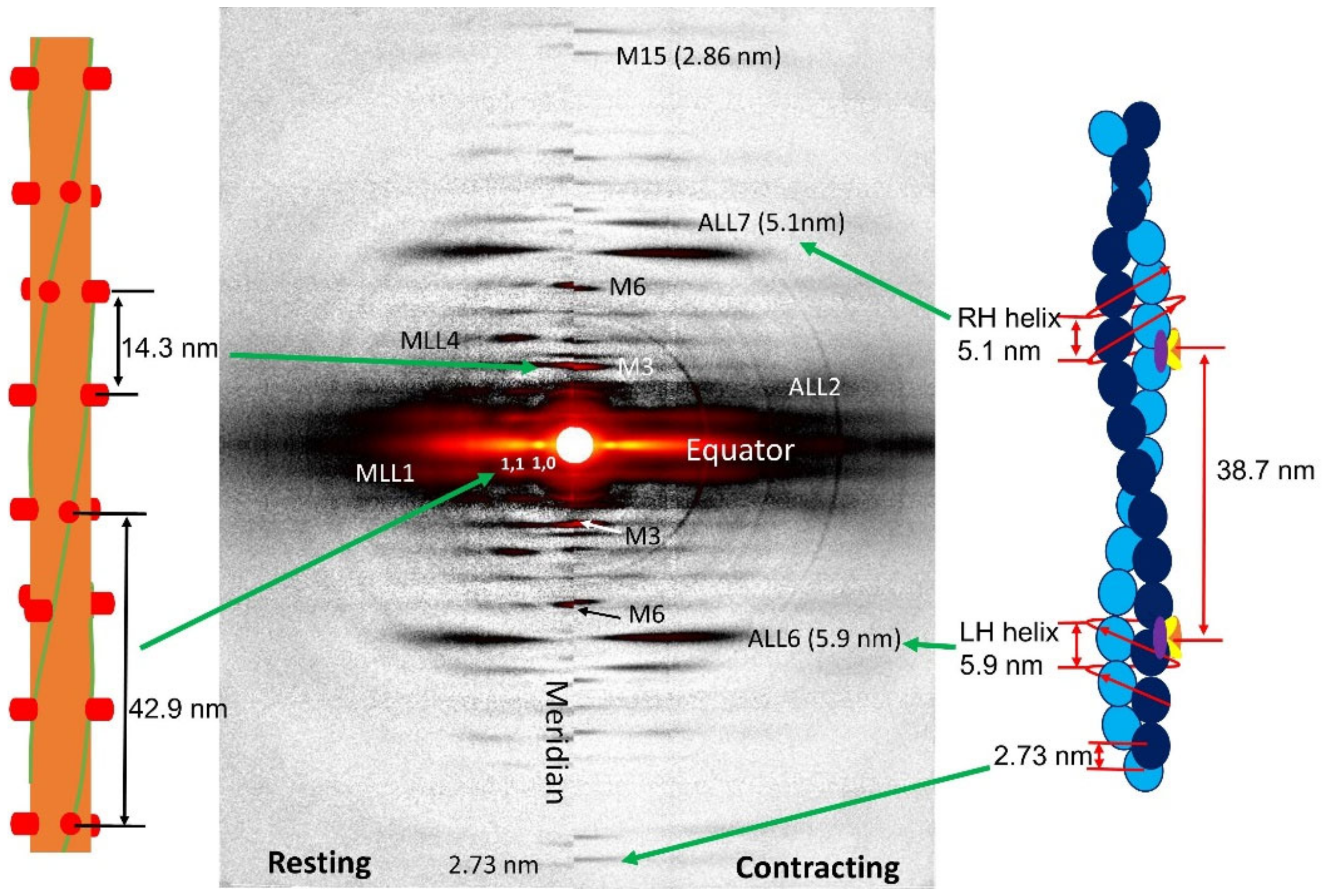
1.2. X-ray Fiber Diffraction Patterns from Vertebrate Muscle
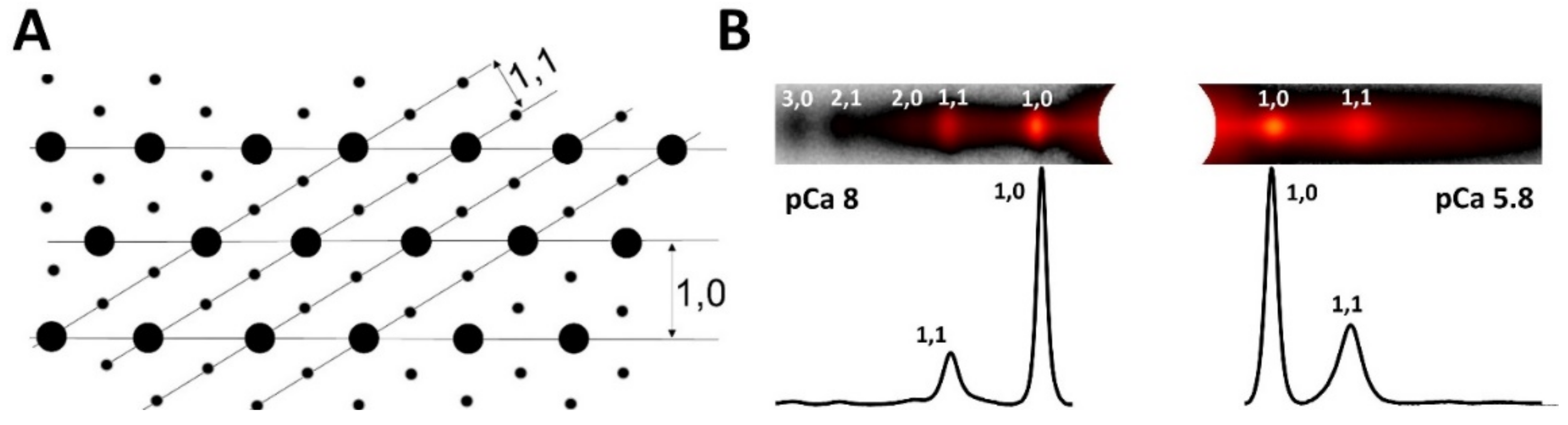
1.3. Equatorial Reflections
1.3.1. Lattice Spacing
1.3.2. The Equatorial Intensity Ratio
1.3.3. Characterizing Lattice Disorder Using the Equatorial Peak Widths
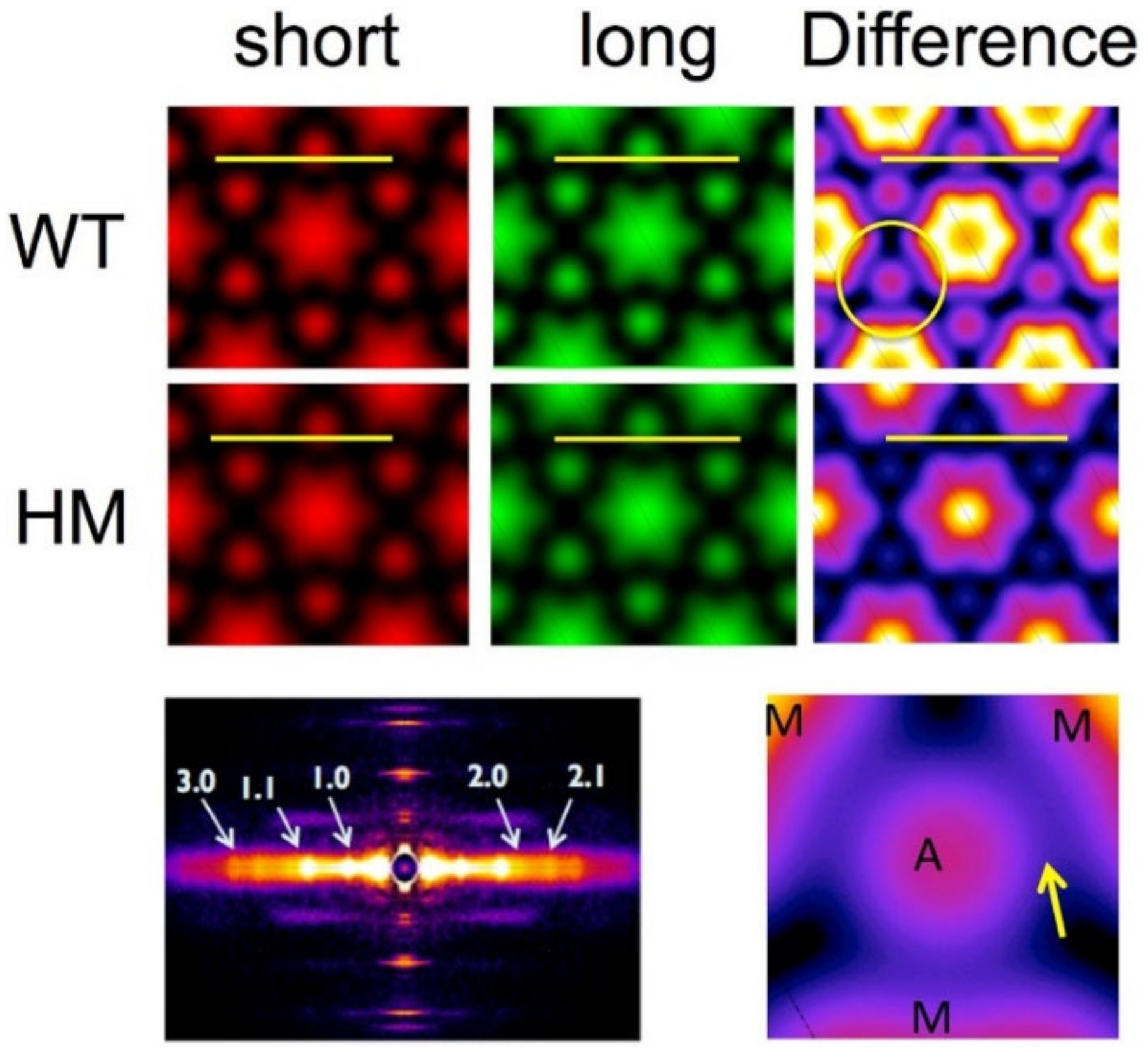
1.3.4. Myofibrillar Orientation from the Angular Spread of Reflections
1.3.5. Electron Density Maps
1.4. Meridional Reflections
1.4.1. Overview
1.4.2. M3 Reflection
1.4.3. M6 Reflection
1.4.4. Thin Filament Meridional Reflections
1.4.5. Reflections from MyBP-C
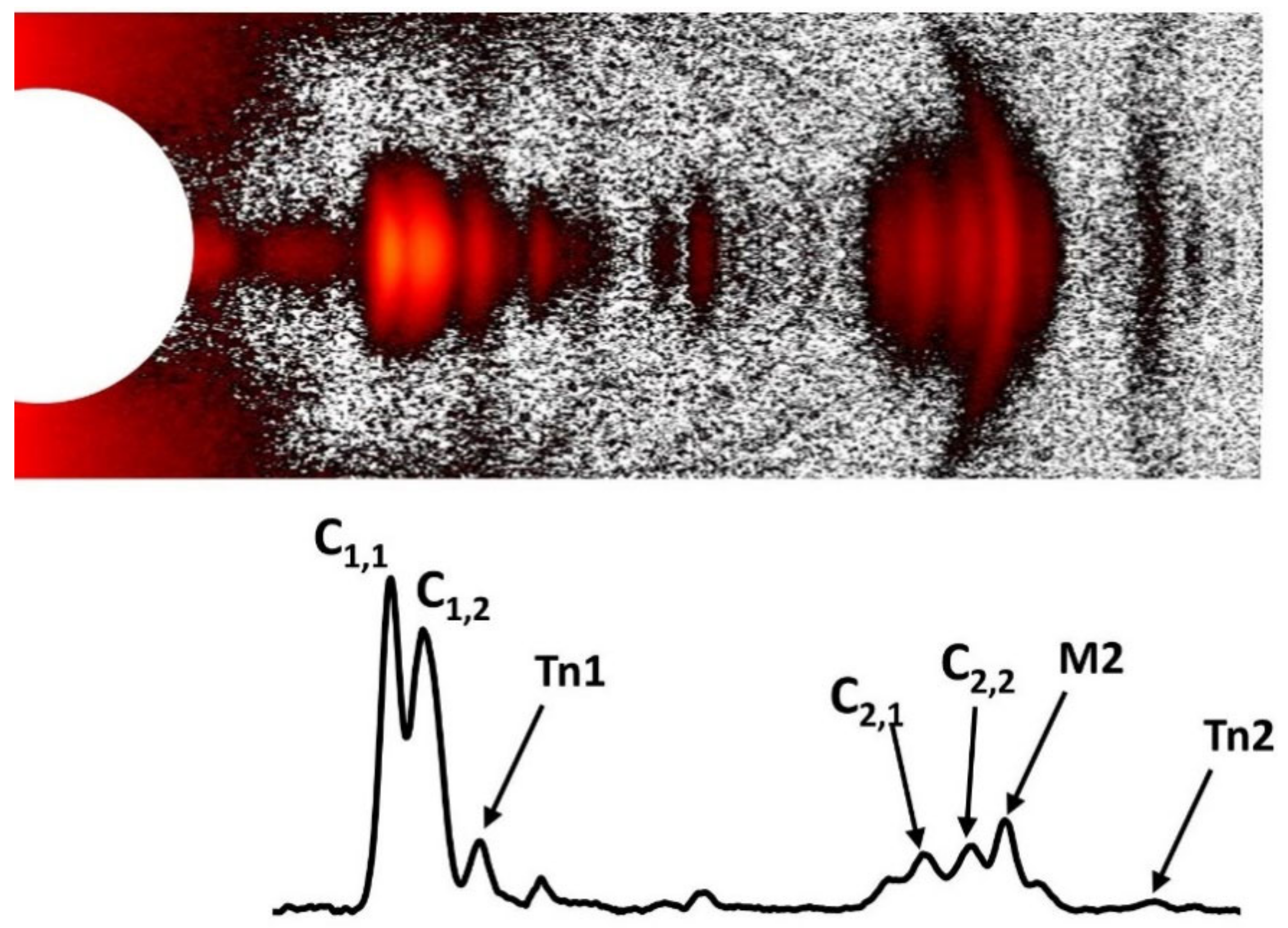
1.5. Actin and Myosin Layer Lines
1.5.1. Actin Layer Lines—Spacings
1.5.2. Actin Layer Lines—Intensity Changes with Contraction
1.5.3. “Tropomyosin” Reflection on the Second Actin Layer Line
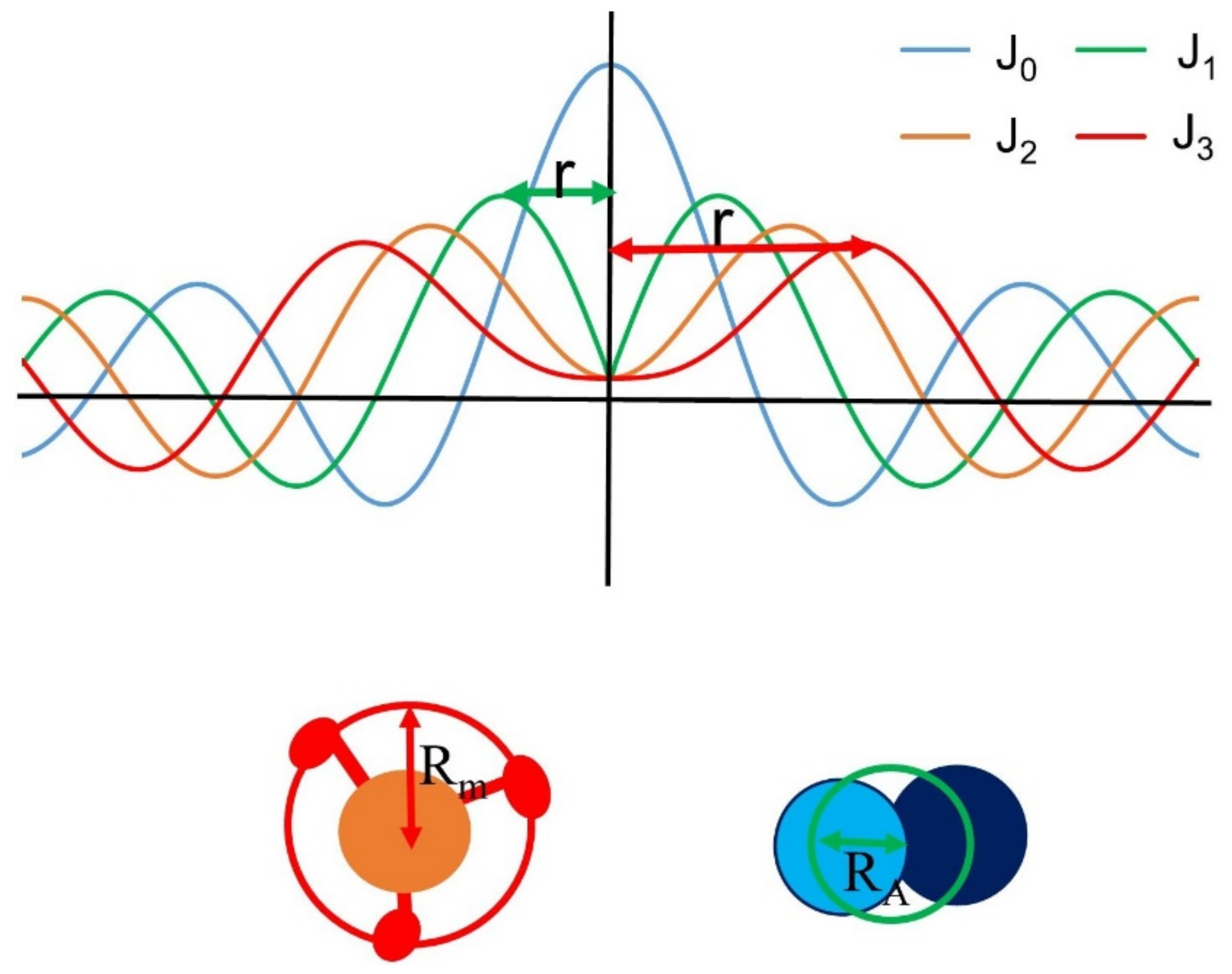
1.5.4. Estimating Myofilament Radius from Layer Lines
2. Examples Where X-ray Diffraction Can Inform Questions Related to Muscle Regulation
2.1. The Super-Relaxed and Disordered Relaxed States of Myosin
2.2. Myosin Binding Protein C
2.3. Myofilament Length Dependent Activation
3. X-ray Diffraction Studies of Muscle Disease
3.1. Introduction
3.2. X-ray Studies of Cardiomyopathies
3.2.1. Overview
3.2.2. HCM
- Myosin Heavy Chain and HCM
- Myosin Light Chain and HCM
- Troponin and HCM
- Tropomyosin and HCM
3.2.3. DCM
- Myosin Mutations and DCM
- Myosin Light Chains and DCM
- Myosin Binding protein C and DCM
3.2.4. Muscular Dystrophy Cardiomyopathy
3.2.5. Ischemia
3.2.6. Diabetic Cardiomyopathy
3.3. Skeletal Muscle Diseases
- Myosin-based Skeletal Myopathies
- Titin-based Skeletal Myopathies
- Nemaline Myopathies
- Tropomyosin Based Myopathies
4. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huxley, H.E. The mechanism of muscular contraction. Science 1969, 164, 1356–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huxley, H.E.; Simmons, R.M.; Faruqi, A.R.; Kress, M.; Bordas, J.; Koch, M.H. Millisecond time-resolved changes in x-ray reflections from contracting muscle during rapid mechanical transients, recorded using synchrotron radiation. Proc. Natl. Acad. Sci. USA 1981, 78, 2297–2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huxley, H.; Faruqi, A.; Kress, M.; Bordas, J.; Koch, M. Time-resolved X-ray diffraction studies of the myosin layer-line reflections during muscle contraction. J. Mol. Biol. 1982, 158, 637–684. [Google Scholar] [CrossRef]
- Huxley, H.; Simmons, R.; Faruqi, A.; Kress, M.; Bordas, J.; Koch, M. Changes in the X-ray reflections from contracting muscle during rapid mechanical transients and their structural implications. J. Mol. Biol. 1983, 169, 469–506. [Google Scholar] [CrossRef]
- Huxley, H.; Reconditi, M.; Stewart, A.; Irving, T. X-ray interference studies of crossbridge action in muscle contraction: Evidence from muscles during steady shortening. J. Mol. Biol. 2006, 363, 762–772. [Google Scholar] [CrossRef]
- Huxley, H.E.; Reconditi, M.; Stewart, A.; Irving, T. X-ray interference evidence concerning the range of crossbridge movement, and backbone contributions to the meridional pattern. Adv. Exp. Med. Biol. 2003, 538, 233–241. [Google Scholar]
- Geeves, A.M.; Holmes, K.C. The molecular mechanism of muscle contraction. Adv. Protein. Chem. 2005, 71, 161–193. [Google Scholar]
- Holmes, C.K.; Geeves, M.A. The structural basis of muscle contraction. Philos. Trans. R Soc. Lond. B Biol. Sci. 2000, 355, 419–431. [Google Scholar] [CrossRef] [Green Version]
- Mckillop, A.D.F.; Geeves, M.A. Regulation of the Interaction between Actin and Myosin Subfragment-1—Evidence for 3 States of the Thin Filament. Biophys. J. 1993, 65, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Lehman, W.; Craig, R.; Vibert, P. Ca2+ induced tropomyosin movement in Limulus thin filaments revealed by three-dimensional reconstruction. Nature 1994, 368, 65–67. [Google Scholar] [CrossRef]
- Gordon, A.M.; Homsher, E.; Regnier, M. Regulation of contraction in striated muscle. Physiol. Rev. 2000, 80, 853–924. [Google Scholar] [CrossRef] [PubMed]
- Perz-Edwards, R.J.; Irving, T.C.; Baumann, B.A.J.; Gore, D.; Hutchinson, D.C.; Kržič, U.; Porter, R.L.; Ward, A.B.; Reedy, M.K. X-ray diffraction evidence for myosin-troponin connections and tropomyosin movement during stretch activation of insect flight muscle. Proc. Natl. Acad. Sci. USA 2010, 108, 120–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekyarova, T.I.; Reedy, M.C.; Baumann, B.A.; Tregear, R.T.; Ward, A.; Krzic, U.; Prince, K.M.; Perz-Edwards, R.J.; Reconditi, M.; Gore, D.; et al. Reverse actin sliding triggers strong myosin binding that moves tropomyosin. Proc. Natl. Acad. Sci. USA 2008, 105, 10372–10377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huxley, H.E. Structural changes during muscle contraction. Biochem. J. 1971, 125, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haselgrove, J.C. X-ray evidence for a conformational change in the actin-containing filaments of vertebrate striated muscle. Cold Spring Harb. Symp. Quant. Biol. 1972, 37, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Parry, A.D.; Squire, J.M. Structural role of tropomyosin in muscle regulation: Analysis of the X-ray diffraction patterns from relaxed and contracting muscles. J. Mol. Biol. 1973, 75, 33–55. [Google Scholar] [CrossRef]
- Huxley, H.E. Structural changes in the actin- and myosin-containing filaments during contraction. Cold Spring Harb. Symp.Quant. Biol. 1972, 37, 361–376. [Google Scholar] [CrossRef]
- Perz-Edwards, J.R.; Reedy, M.K. Electron microscopy and X-ray diffraction evidence for two Z-band structural states. Biophys. J. 2011, 101, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Linari, M.; Brunello, E.; Reconditi, M.; Fusi, L.; Caremani, M.; Narayanan, T.; Piazzesi, G.; Lombardi, V.; Irving, M. Force generation by skeletal muscle is controlled by mechanosensing in myosin filaments. Nature 2015, 528, 276–279. [Google Scholar] [CrossRef] [Green Version]
- Fusi, L.; Brunello, E.; Yan, Z.; Irving, M. Thick filament mechano-sensing is a calcium-independent regulatory mechanism in skeletal muscle. Nat. Commun. 2016, 7, 13281. [Google Scholar] [CrossRef] [Green Version]
- Caremani, M.; Pinzauti, F.; Powers, J.D.; Governali, S.; Narayanan, T.; Stienen, G.J.M.; Reconditi, M.; Linari, M.; Lombardi, V.; Piazzesi, G. Inotropic interventions do not change the resting state of myosin motors during cardiac diastole. J. Gen. Physiol. 2019, 151, 53–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunello, E.; Fusi, L.; Reconditi, M.; Linari, M.; Bianco, P.; Panine, P.; Narayanan, T.; Piazzesi, G.; Lombardi, V.; Irving, M. Structural changes in myosin motors and filaments during relaxation of skeletal muscle. J. Physiol. 2009, 587, 4509–4521. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Childers, M.; Murray, J.; Moussavi-Harami, F.; Gong, H.; Weiss, R.; Daggett, V.; Irving, T.; Regnier, M. Myosin dynamics during relaxation in mouse soleus muscle and modulation by 2’-deoxy-ATP. J. Physiol. 2020, 598, 5165–5182. [Google Scholar] [CrossRef]
- Ma, W.; Gong, H.; Irving, T. Myosin Head Configurations in Resting and Contracting Murine Skeletal Muscle. Int. J. Mol. Sci. 2018, 19, 2643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.; Gong, H.; Kiss, B.; Lee, E.J.; Granzier, H.; Irving, T. Thick-Filament Extensibility in Intact Skeletal Muscle. Biophys. J. 2018, 115, 1580–1588. [Google Scholar] [CrossRef] [PubMed]
- Caremani, M.; Brunello, E.; Linari, M.; Fusi, L.; Irving, T.C.; Gore, D.; Piazzesi, G.; Irving, M.; Lombardi, V.; Reconditi, M. Low temperature traps myosin motors of mammalian muscle in a refractory state that prevents activation. J. Gen. Physiol. 2019, 151, 1272–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.; Irving, T.C. X-ray Diffraction of Intact Murine Skeletal Muscle as a Tool for Studying the Structural Basis of Muscle Disease. J. Vis. Exp. 2019, 149, e59559. [Google Scholar] [CrossRef]
- Joumaa, V.; Smith, I.C.; Fukutani, A.; Leonard, T.R.; Ma, W.; Mijailovich, S.M.; Irving, T.C.; Herzog, W. Effect of Active Lengthening and Shortening on Small-Angle X-ray Reflections in Skinned Skeletal Muscle Fibres. Int. J. Mol. Sci. 2021, 22, 8526. [Google Scholar] [CrossRef]
- Kiss, B.; Lee, E.J.; Ma, W.; Li, F.W.; Tonino, P.; Mijailovich, S.M.; Irving, T.C.; Granzier, H.L. Nebulin stiffens the thin filament and augments cross-bridge interaction in skeletal muscle. Proc. Natl. Acad. Sci. USA 2018, 115, 10369–10374. [Google Scholar] [CrossRef] [Green Version]
- De Winter, J.M.; Molenaar, J.P.; Yuen, M.; van der Pijl, R.; Shen, S.; Conijn, S.; van de Locht, M.; Willigenburg, M.; Bogaards, S.J.; van Kleef, E.S.; et al. KBTBD13 is an actin-binding protein that modulates muscle kinetics. J. Clin. Investig. 2020, 130, 754–767. [Google Scholar] [CrossRef] [Green Version]
- Lindqvist, J.; Ma, W.; Li, F.; Hernandez, Y.; Kolb, J.; Kiss, B.; Tonino, P.; van der Pijl, R.; Karimi, E.; Gong, H.; et al. Triggering typical nemaline myopathy with compound heterozygous nebulin mutations reveals myofilament structural changes as pathomechanism. Nat. Commun. 2020, 11, 2699. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; McNamara, J.W.; Ma, W.; Landim-Vieira, M.; Lee, K.H.; Martin, L.A.; Heiny, J.A.; Lorenz, J.N.; Craig, R.; Pinto, J.R.; et al. Fast skeletal myosin-binding protein-C regulates fast skeletal muscle contraction. Proc. Natl. Acad. Sci. USA 2021, 118, e2003596118. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, J.; Iwamoto, H.; Blanco, G.; Ochala, J. The fraction of strongly bound cross-bridges is increased in mice that carry the myopathy-linked myosin heavy chain mutation MYH4L342Q. Dis. Model. Mech. 2013, 6, 834–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazmierczak, K.; Liang, J.; Yuan, C.-C.; Yadav, S.; Sitbon, Y.H.; Walz, K.; Ma, W.; Irving, T.C.; Cheah, J.X.; Gomes, A.V.; et al. Slow-twitch skeletal muscle defects accompany cardiac dysfunction in transgenic mice with a mutation in the myosin regulatory light chain. FASEB J. 2018, 33, 3152–3166. [Google Scholar] [CrossRef] [Green Version]
- Witayavanitkul, N.; Ait Mou, Y.; Kuster, D.W.; Khairallah, R.J.; Sarkey, J.; Govindan, S.; Chen, X.; Ge, Y.; Rajan, S.; Wieczorek, D.F.; et al. Myocardial infarction-induced N-terminal fragment of cardiac myosin-binding protein C (cMyBP-C) impairs myofilament function in human myocardium. J. Biol. Chem. 2014, 289, 8818–8827. [Google Scholar] [CrossRef] [Green Version]
- Joureau, B.; De Winter, J.M.; Conijn, S.; Bogaards, S.J.P.; Kovacevic, I.; Kalganov, A.; Persson, M.; Lindqvist, J.; Stienen, G.J.M.; Irving, T.C.; et al. Dysfunctional sarcomere contractility contributes to muscle weakness in ACTA1-related nemaline myopathy (NEM3). Ann. Neurol. 2018, 83, 269–282. [Google Scholar] [CrossRef]
- Ochala, J.; Lehtokari, V.L.; Iwamoto, H.; Li, M.; Feng, H.Z.; Jin, J.P.; Yagi, N.; Wallgren-Pettersson, C.; Penisson-Besnier, I.; Larsson, L. Disrupted myosin cross-bridge cycling kinetics triggers muscle weakness in nebulin-related myopathy. FASEB J. 2011, 25, 1903–1913. [Google Scholar] [CrossRef] [Green Version]
- Lassche, S.; Stienen, G.J.; Irving, T.C.; van der Maarel, S.M.; Voermans, N.C.; Padberg, G.W.; Granzier, H.; van Engelen, B.G.; Ottenheijm, C.A. Sarcomeric dysfunction contributes to muscle weakness in facioscapulohumeral muscular dystrophy. Neurology 2013, 80, 733–737. [Google Scholar] [CrossRef] [Green Version]
- Van de Locht, M.; Donkervoort, S.; de Winter, J.M.; Conijn, S.; Begthel, L.; Kusters, B.; Mohassel, P.; Hu, Y.; Medne, L.; Quinn, C.; et al. Pathogenic variants in TNNC2 cause congenital myopathy due to an impaired force response to calcium. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Ma, W.; Gong, H.; Jani, V.; Lee, K.H.; Landim-Vieira, M.; Papadaki, M.; Pinto, J.R.; Aslam, M.I.; Cammarato, A.; Irving, T. Myofibril orientation as a metric for characterizing heart disease. Biophys. J. 2022, 121, 565–574. [Google Scholar] [CrossRef]
- Anderson, R.L.; Trivedi, D.V.; Sarkar, S.S.; Henze, M.; Ma, W.; Gong, H.; Rogers, C.S.; Gorham, J.M.; Wong, F.L.; Morck, M.M. Deciphering the super relaxed state of human beta-cardiac myosin and the mode of action of mavacamten from myosin molecules to muscle fibers. Proc. Natl. Acad. Sci. USA 2018, 115, E8143–E8152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.; Henze, M.; Anderson, R.L.; Gong, H.; Wong, F.L.; del Rio, C.L.; Irving, T. The Super-Relaxed State and Length Dependent Activation in Porcine Myocardium. Circ. Res. 2021, 129, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Squire, J.M.; Knupp, C. The muscle M3 x-ray diffraction peak and sarcomere length: No evidence for disordered myosin heads out of actin overlap. J. Gen. Physiol. 2021, 153, e202012859. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.; Brunello, E.; Fusi, L.; Ovejero, J.G.; Irving, M. Myosin-based regulation of twitch and tetanic contractions in mammalian skeletal muscle. eLife 2021, 10, e68211. [Google Scholar] [CrossRef] [PubMed]
- Javor, J.; Ewoldt, J.K.; Cloonan, P.E.; Chopra, A.; Luu, R.J.; Freychet, G.; Zhernenkov, M.; Ludwig, K.; Seidman, J.G.; Seidman, C.E.; et al. Probing the subcellular nanostructure of engineered human cardiomyocytes in 3D tissue. Microsyst. Nanoeng. 2021, 7, 1–8. [Google Scholar] [CrossRef]
- Jiratrakanvong, J.; Shao, J.; Menendez, M.; Li, X.; Li, J.; Ma, W.; Agam, G.; Irving, T. MuscleX: Software Suite for Diffraction X-ray Imaging. 2018. Available online: https://zenodo.org/record/3360909#.Xin5McZKihc (accessed on 23 January 2020).
- Muscle, X. 2022. Available online: https://musclex.readthedocs.io/en/latest/ (accessed on 11 February 2022).
- Squire, J. The Structural Basis of Muscular Contraction; Plenum Press: New York, NY, USA, 1981; Volume xvii, p. 698. [Google Scholar]
- Ma, W.; Gong, H.; Qi, L.; Nag, S.; Irving, T. Cardiac myosin filaments are directly regulated by calcium. bioRxiv 2022. [Google Scholar] [CrossRef]
- Yuan, C.C.; Kazmierczak, K.; Liang, J.; Ma, W.; Irving, T.C.; Szczesna-Cordary, D. Molecular basis of force-pCa relation in MYL2 cardiomyopathy mice: Role of the super relaxed state of myosin. Proc. Natl. Acad. Sci. USA 2022, 119, e21103281192022. [Google Scholar] [CrossRef]
- Mijailovich, S.M.; Prodanovic, M.; Irving, T.C. Estimation of Forces on Actin Filaments in Living Muscle from X-ray Diffraction Patterns and Mechanical Data. Int. J. Mol. Sci. 2019, 20, 6044. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.D.; Regnier, M.; Daniel, T.L. Axial and Radial Forces of Cross-Bridges Depend on Lattice Spacing. PLOS Comput. Biol. 2010, 6, e1001018. [Google Scholar] [CrossRef] [Green Version]
- Schoenberg, M. Geometrical factors influencing muscle force development. I. The effect of filament spacing upon axial forces. Biophys. J. 1980, 30, 51–67. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.D.; Salcedo, M.K.; Irving, T.C.; Regnier, M.; Daniel, T.L. The length-tension curve in muscle depends on lattice spacing. Proc. Biol. Sci. 2013, 280, 20130697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millman, B.M. The filament lattice of striated muscle. Physiol. Rev. 1998, 78, 359–391. [Google Scholar] [CrossRef] [PubMed]
- Farman, G.P.; Walker, J.S.; De Tombe, P.P.; Irving, T.C. Impact of osmotic compression on sarcomere structure and myofilament calcium sensitivity of isolated rat myocardium. Am. J. Physiol. Circ. Physiol. 2006, 291, H1847–H1855. [Google Scholar] [CrossRef] [PubMed]
- Caremani, M.; Fusi, L.; Linari, M.; Reconditi, M.; Piazzesi, G.; Irving, T.C.; Narayanan, T.; Irving, M.; Lombardi, V.; Brunello, E. Dependence of thick filament structure in relaxed mammalian skeletal muscle on temperature and interfilament spacing. J. Gen. Physiol. 2021, 153, e202012713. [Google Scholar] [CrossRef] [PubMed]
- Ovejero, J.G.; Fusi, L.; Park-Holohan, S.-J.; Ghisleni, A.; Narayanan, T.; Irving, M.; Brunello, E. Cooling intact and demembranated trabeculae from rat heart releases myosin motors from their inhibited conformation. J. Gen. Physiol. 2022, 154, e202113029. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Kawai, M. The effect of the lattice spacing change on cross-bridge kinetics in chemically skinned rabbit psoas muscle fibers. II. Elementary steps affected by the spacing change. Biophys. J. 1993, 64, 197–210. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.C.; Arata, T.; Steven, A.C.; Naylor, G.R.; Gamble, R.C.; Podolsky, R.J. Structural studies of muscle during force development in various states. Adv. Exp. Med. Biol. 1984, 170, 207–220. [Google Scholar]
- Matsubara, I.; Goldman, Y.E.; Simmons, R.M. Changes in the lateral filament spacing of skinned muscle fibres when cross-bridges attach. J. Mol. Biol. 1984, 173, 15–33. [Google Scholar] [CrossRef]
- Konhilas, J.P.; Irving, T.C.; de Tombe, P. Frank-Starling law of the heart and the cellular mechanisms of length-dependent activation. Pflugers Arch. 2002, 445, 305–310. [Google Scholar] [CrossRef]
- McDonald, S.K.; Moss, R.L. Osmotic compression of single cardiac myocytes eliminates the reduction in Ca2+ sensitivity of tension at short sarcomere length. Circ. Res. 1995, 77, 199–205. [Google Scholar] [CrossRef]
- Irving, T.C. X-ray diffraction of Indirect Flight muscle from Drosophila in Vivo. In Nature’s Versatile Engine: Insect Flight Muscle Inside and Out; Springer: Berlin/Heidelberg, Germany, 2006; pp. 197–213. [Google Scholar]
- Matsubara, I. X-ray diffraction studies of the heart. Annu. Rev. Biophys. Bioeng. 1980, 9, 81–105. [Google Scholar] [CrossRef] [PubMed]
- Brunello, E.; Fusi, L.; Ghisleni, A.; Park-Holohan, S.J.; Ovejero, J.G.; Narayanan, T.; Irving, M. Myosin filament-based regulation of the dynamics of contraction in heart muscle. Proc. Natl. Acad. Sci. USA 2020, 117, 8177–8186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powers, J.D.; Yuan, C.C.; McCabe, K.J.; Murray, J.D.; Childers, M.C.; Flint, G.V.; Moussavi-Harami, F.; Mohran, S.; Castillo, R.; Zuzek, C. Cardiac myosin activation with 2-deoxy-ATP via increased electrostatic interactions with actin. Proc. Natl. Acad. Sci. USA 2019, 116, 11502–11507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, B.M.; Sadayappan, S.; Wang, Y.; Weith, A.E.; Previs, M.J.; Bekyarova, T.; Irving, T.C.; Robbins, J.; Maughan, D.W. Roles for cardiac MyBP-C in maintaining myofilament lattice rigidity and prolonging myosin cross-bridge lifetime. Biophys. J. 2011, 101, 1661–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colson, B.A.; Bekyarova, T.; Locher, M.R.; Fitzsimons, D.P.; Irving, T.C.; Moss, R.L. Protein kinase A-mediated phosphorylation of cMyBP-C increases proximity of myosin heads to actin in resting myocardium. Circ. Res. 2008, 103, 244–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, C.C.; Muthu, P.; Kazmierczak, K.; Liang, J.; Huang, W.; Irving, T.C.; Kanashiro-Takeuchi, R.M.; Hare, J.M.; Szczesna-Cordary, D. Constitutive phosphorylation of cardiac myosin regulatory light chain prevents development of hypertrophic cardiomyopathy in mice. Proc. Natl. Acad. Sci. USA 2015, 112, E4138–E4146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malinchik, S.; Xu, S.; Yu, L.C. Temperature-induced structural changes in the myosin thick filament of skinned rabbit psoas muscle. Biophys. J. 1997, 73, 2304–2312. [Google Scholar] [CrossRef] [Green Version]
- Malinchik, S.; Yu, L.C. Analysis of equatorial X-ray diffraction patterns from muscle fibers: Factors that affect the intensities. Biophys. J. 1995, 68, 2023–2031. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.L.; Brenner, B. Structures of actomyosin crossbridges in relaxed and rigor muscle fibers. Biophys. J. 1989, 55, 441–453. [Google Scholar] [CrossRef] [Green Version]
- Irving, T.C.; Millman, B.M. Changes in thick filament structure during compression of the filament lattice in relaxed frog sartorius muscle. J. Muscle Res. Cell Motil. 1989, 10, 385–394. [Google Scholar] [CrossRef]
- Ait-Mou, Y.A.; Hsu, K.; Farman, G.P.; Kumar, M.; Greaser, M.L.; Irving, T.C.; de Tombe, P.P. Titin strain contributes to the Frank–Starling law of the heart by structural rearrangements of both thin- and thick-filament proteins. Proc. Natl. Acad. Sci. USA 2016, 113, 2306–2311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eakins, F.; Pinali, C.; Gleeson, A.; Knupp, C.; Squire, J.M. X-ray Diffraction Evidence for Low Force Actin-Attached and Rigor-Like Cross-Bridges in the Contractile Cycle. Biology 2016, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- Squire, J.M.; Knupp, C. Analysis methods and quality criteria for investigating muscle physiology using X-ray diffraction. J. Gen. Physiol. 2021, 153, e202012778. [Google Scholar] [CrossRef] [PubMed]
- Reconditi, M.; Brunello, E.; Fusi, L.; Linari, M.; Martínez, M.F.; Lombardi, V.; Irving, M.; Piazzesi, G. Sarcomere-length dependence of myosin filament structure in skeletal muscle fibres of the frog. J. Physiol. 2014, 592, 1119–1137. [Google Scholar] [CrossRef] [Green Version]
- Reconditi, M.; Linari, M.; Lucii, L.; Stewart, A.; Sun, Y.-B.; Boesecke, P.; Narayanan, T.; Fischetti, R.F.; Irving, T.; Piazzesi, G.; et al. The myosin motor in muscle generates a smaller and slower working stroke at higher load. Nature 2004, 428, 578–581. [Google Scholar] [CrossRef]
- Tregear, R.; Edwards, R.; Irving, T.; Poole, K.; Reedy, M.; Schmitz, H.; Towns-Andrews, E. X-Ray Diffraction Indicates That Active Cross-Bridges Bind to Actin Target Zones in Insect Flight Muscle. Biophys. J. 1998, 74, 1439–1451. [Google Scholar] [CrossRef] [Green Version]
- Vainshtein, B.K. Diffraction of X-rays by Chain Molecules; Elsevier: Amsterdam, The Netherlands, 1966. [Google Scholar]
- Yu, L.; Steven, A.; Naylor, G.; Gamble, R.; Podolsky, R. Distribution of mass in relaxed frog skeletal muscle and its redistribution upon activation. Biophys. J. 1985, 47, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Colson, B.A.; Bekyarova, T.; Fitzsimons, D.P.; Irving, T.C.; Moss, R.L. Radial displacement of myosin cross-bridges in mouse myocardium due to ablation of myosin binding protein-C. J. Mol. Biol. 2007, 367, 36–41. [Google Scholar] [CrossRef] [Green Version]
- Green, E.M.; Wakimoto, H.; Anderson, R.L.; Evanchik, M.J.; Gorham, J.M.; Harrison, B.C.; Henze, M.; Kawas, R.; Oslob, J.D.; Rodriguez, H.M.; et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 2016, 351, 617–621. [Google Scholar] [CrossRef] [Green Version]
- Irving, T. Analysis of the Equatorial X-Ray Diffraction Pattern of Vertebrate Striated Muscle. Ph.D. Thesis, University of Guelph, Guelph, ON, Canada, 1989; p. 5425. [Google Scholar]
- Hawkins, J.C.; Bennett, P.M. Evaluation of freeze substitution in rabbit skeletal muscle. Comparison of electron microscopy to X-ray diffraction. J. Muscle Res. Cell Motil. 1995, 16, 303–318. [Google Scholar] [CrossRef]
- Yu, L.C. Analysis of equatorial X-ray diffraction patterns from skeletal muscle. Biophys. J. 1989, 55, 433–440. [Google Scholar] [CrossRef] [Green Version]
- Linari, M.; Piazzesi, G.; Dobbie, I.; Koubassova, N.; Reconditi, M.; Narayanan, T.; Diat, O.; Irving, M.; Lombardi, V. Interference fine structure and sarcomere length dependence of the axial x-ray pattern from active single muscle fibers. Proc. Natl. Acad. Sci. USA 2000, 97, 7226–7231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huxley, H.E. Recent X-ray diffraction studies of muscle contraction and their implications. Philos. Trans. R. Soc. B Biol. Sci. 2004, 359, 1879–1882. [Google Scholar] [CrossRef] [Green Version]
- Reconditi, M. Recent improvements in small angle x-ray diffraction for the study of muscle physiology. Rep. Prog. Phys. 2006, 69, 2709–2759. [Google Scholar] [CrossRef] [PubMed]
- Padrón, R.; Ma, W.; Duno-Miranda, S.; Koubassova, N.; Lee, K.; Pinto, A.; Alamo, L.; Bolaños, P.; Tsaturyan, A.; Irving, T.; et al. The myosin interacting-heads motif present in live tarantula muscle explains tetanic and posttetanic phosphorylation mechanisms. Proc. Natl. Acad. Sci. USA 2020, 117, 11865–11874. [Google Scholar] [CrossRef] [PubMed]
- Huxley, E.H.; Brown, W. The low-angle X-ray diagram of vertebrate striated muscle and its behaviour during contraction and rigor. J. Mol. Biol. 1967, 30, 383–434. [Google Scholar] [CrossRef]
- Haselgrove, J.C. X-ray evidence for conformational changes in the myosin filaments of vertebrate striated muscle. J. Mol. Biol. 1975, 92, 113–143. [Google Scholar] [CrossRef]
- Irving, M.; Piazzesi, G.; Lucii, L.; Sun, Y.-B.; Harford, J.J.; Dobbie, I.M.; Ferenczi, M.A.; Reconditi, M.; Lombardi, V. Conformation of the myosin motor during force generation in skeletal muscle. Nat. Struct. Biol. 2000, 7, 482–485. [Google Scholar] [CrossRef]
- Reconditi, M.; Brunello, E.; Fusi, L.; Linari, M.; Lombardi, V.; Irving, M.; Piazzesi, G. Myosin motors that cannot bind actin leave their folded OFF state on activation of skeletal muscle. J. Gen. Physiol. 2021, 153, e202112896. [Google Scholar] [CrossRef]
- Bordas, J.; Diakun, G.P.; Diaz, F.G.; Harries, J.E.; Lewis, R.A.; Lowy, J.; Mant, G.R.; Martin-Fernandez, M.L.; Towns-Andrews, E. Two-dimensional time-resolved X-ray diffraction studies of live isometrically contracting frog sartorius muscle. J. Muscle Res. Cell Motil. 1993, 14, 311–324. [Google Scholar] [CrossRef]
- Huxley, H.E.; Reconditi, M.; Stewart, A.; Irving, T. Distribution of crossbridge states in contracting muscle. Adv. Exp. Med. Biol. 2005, 565, 37–44. [Google Scholar] [PubMed]
- Huxley, H.; Reconditi, M.; Stewart, A.; Irving, T. X-ray interference studies of crossbridge action in muscle contraction: Evidence from quick releases. J. Mol. Biol. 2006, 363, 743–761. [Google Scholar] [CrossRef] [PubMed]
- Piazzesi, G.; Reconditi, M.; Linari, M.; Lucii, L.; Bianco, P.; Brunello, E.; Decostre, V.; Stewart, A.; Gore, D.B.; Irving, T.C. Skeletal muscle performance determined by modulation of number of myosin motors rather than motor force or stroke size. Cell 2007, 131, 784–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, K.; Sugimoto, Y.; Tanaka, H.; Ueno, Y.; Takezawa, Y.; Amemiya, Y. X-ray diffraction evidence for the extensibility of actin and myosin filaments during muscle contraction. Biophys. J. 1994, 67, 2422–2435. [Google Scholar] [CrossRef] [Green Version]
- Huxley, H.; Stewart, A.; Sosa, H.; Irving, T. X-ray diffraction measurements of the extensibility of actin and myosin filaments in contracting muscle. Biophys. J. 1994, 67, 2411–2421. [Google Scholar] [CrossRef] [Green Version]
- Irving, M. Regulation of Contraction by the Thick Filaments in Skeletal Muscle. Biophys. J. 2017, 113, 2579–2594. [Google Scholar] [CrossRef] [Green Version]
- Huxley, H.E.; Brown, W.; Holmes, K. Constancy of Axial Spacings in Frog Sartorius Muscle during Contraction. Nature 1965, 206, 1358. [Google Scholar] [CrossRef]
- Elliott, G.F.; Lowy, J.; Millman, B.M. X-ray diffraction from living striated muscle during contraction. Nature 1965, 206, 1357–1358. [Google Scholar] [CrossRef]
- Dominguez, R.; Holmes, K.C. Actin structure and function. Annu. Rev. Biophys. 2011, 40, 169–186. [Google Scholar] [CrossRef] [Green Version]
- Reedy, M.K.; Reedy, M.C. Rigor crossbridge structure in tilted single filament layers and flared-X formations from insect flight muscle. J. Mol. Biol. 1985, 185, 145–176. [Google Scholar] [CrossRef]
- Al-Khayat, H.A.; Hudson, L.; Reedy, M.K.; Irving, T.C.; Squire, J.M. Myosin Head Configuration in Relaxed Insect Flight Muscle: X-ray Modeled Resting Cross-Bridges in a Pre-Powerstroke State Are Poised for Actin Binding. Biophys. J. 2003, 85, 1063–1079. [Google Scholar] [CrossRef] [Green Version]
- Maeda, Y.; Popp, D.; Stewart, A.A. Time-resolved X-ray diffraction study of the troponin-associated reflexions from the frog muscle. Biophys. J. 1992, 63, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, Y.; Takezawa, Y.; Matsuo, T.; Ueno, Y.; Minakata, S.; Tanaka, H.; Wakabayashi, K. Structural changes of the regulatory proteins bound to the thin filaments in skeletal muscle contraction by X-ray fiber diffraction. Biochem. Biophys. Res. Commun. 2008, 369, 100–108. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Kimura, M.; Ohno, T.; Nakahara, N.; Akiyama, N.; Takemori, S.; Yagi, N. Crossbridge Recruitment Capacity of Wild-Type and Hypertrophic Cardiomyopathy-Related Mutant Troponin-T Evaluated by X-ray Diffraction and Mechanical Study of Cardiac Skinned Fibers. Int. J. Mol. Sci. 2020, 21, 3520. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Ueno, Y.; Takezawa, Y.; Sugimoto, Y.; Oda, T.; Wakabayashi, K. X-ray fiber diffraction modeling of structural changes of the thin filament upon activation of live vertebrate skeletal muscles. Biophysics 2010, 6, 13–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rome, E.; Offer, G.; Pepe, F.A. X-ray diffraction of muscle labelled with antibody to C-protein. Nat. New Biol. 1973, 244, 152–154. [Google Scholar] [CrossRef] [PubMed]
- Squire, J.M.; Luther, P.K.; Knupp, C. Structural evidence for the interaction of C-protein (MyBP-C) with actin and sequence identification of a possible actin-binding domain. J. Mol. Biol. 2003, 331, 713–724. [Google Scholar] [CrossRef]
- Luther, P.K.; Winkler, H.; Taylor, K.; Zoghbi, M.; Craig, R.; Padron, R.; Squire, J.; Liu, J. Direct visualization of myosin-binding protein C bridging myosin and actin filaments in intact muscle. Proc. Natl. Acad. Sci. USA 2011, 108, 11423–11428. [Google Scholar] [CrossRef] [Green Version]
- Rahmanseresht, S.; Lee, K.H.; O’Leary, T.S.; McNamara, J.W.; Sadayappan, S.; Robbins, J.; Warshaw, D.M.; Craig, R.; Previs, M.J. The N terminus of myosin-binding protein C extends toward actin filaments in intact cardiac muscle. J. Gen. Physiol. 2021, 153, e202012726. [Google Scholar] [CrossRef]
- Kulikovskaya, I.; McClellan, G.; Flavigny, J.; Carrier, L.; Winegrad, S. Effect of MyBP-C binding to actin on contractility in heart muscle. J. Gen. Physiol. 2003, 122, 761–774. [Google Scholar] [CrossRef] [Green Version]
- Cochran, W.; Crick, F.H.; Vand, V. The structure of synthetic polypeptides. I. The transform of atoms on a helix. Acta Cryst. 1952, 5, 581–585. [Google Scholar] [CrossRef] [Green Version]
- Haselgrove, J.C.; Huxley, H.E. X-ray evidence for radial cross-bridge movement and for the sliding filament model in actively contracting skeletal muscle. J. Mol. Biol. 1973, 77, 549–568. [Google Scholar] [CrossRef]
- Bordas, J.; Svensson, A.; Rothery, M.; Lowy, J.; Diakun, G.P.; Boesecke, P. Extensibility and symmetry of actin filaments in contracting muscles. Biophys. J. 1999, 77, 3197–3207. [Google Scholar] [CrossRef] [Green Version]
- Tamura, T.; Wakayama, J.; Inoue, K.; Yagi, N.; Iwamoto, H. Dynamics of thin-filament activation in rabbit skeletal muscle fibers examined by time-resolved X-ray diffraction. Biophys. J. 2009, 96, 1045–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagi, N. An X-ray diffraction study on early structural changes in skeletal muscle contraction. Biophys. J. 2003, 84, 1093–1102. [Google Scholar] [CrossRef] [Green Version]
- Huxley, H.E. Structural difference between resting and rigor muscle; evidence from intensity changes in the lowangle equatorial X-ray diagram. J. Mol. Biol. 1968, 37, 507–520. [Google Scholar] [CrossRef]
- Koubassova, N.A.; Bershitsky, S.Y.; Ferenczi, M.A.; Tsaturyan, A.K. Direct modeling of X-ray diffraction pattern from contracting skeletal muscle. Biophys. J. 2008, 95, 2880–2894. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Xu, S.; Leepo, C.Y. A model of cross-bridge attachment to actin in the A*M*ATP state based on X-ray diffraction from permeabilized rabbit psoas muscle. Biophys. J. 2002, 82, 2123–2133. [Google Scholar] [CrossRef] [Green Version]
- Yagi, N.; Iwamoto, H.; Wakayama, J.; Inoue, K. Structural changes of actin-bound myosin heads after a quick length change in frog skeletal muscle. Biophys. J. 2005, 89, 1150–1164. [Google Scholar] [CrossRef] [Green Version]
- Yagi, N.; Iwamoto, H.; Inoue, K. Structural changes of cross-bridges on transition from isometric to shortening state in frog skeletal muscle. Biophys. J. 2006, 91, 4110–4120. [Google Scholar] [CrossRef] [Green Version]
- Al-Khayat, H.A.; Yagi, N.; Squire, J.M. Structural changes in actin-tropomyosin during muscle regulation: Computer modelling of low-angle X-ray diffraction data. J. Mol. Biol. 1995, 252, 611–632. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.H.; Cohen, C. Regulation of muscle contraction by tropomyosin and troponin: How structure illuminates function. Adv. Protein Chem. 2005, 71, 21–59. [Google Scholar]
- Lehrer, S.S.; Geeves, M.A. The muscle thin filament as a classical cooperative/allosteric regulatory system. J. Mol. Biol. 1998, 277, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Galinska-Rakoczy, A.; Engel, P.; Xu, C.; Jung, H.; Craig, R.; Tobacman, L.S.; Lehman, W. Structural basis for the regulation of muscle contraction by troponin and tropomyosin. J. Mol. Biol. 2008, 379, 929–935. [Google Scholar] [CrossRef] [Green Version]
- Boussouf, S.E.; Geeves, M.A. Tropomyosin and troponin cooperativity on the thin filament. Adv. Exp. Med. Biol. 2007, 592, 99–109. [Google Scholar]
- Koubassova, N.A.; Bershitsky, S.Y.; Ferenczi, M.A.; Narayanan, T.; Tsaturyan, A.K. Tropomyosin movement is described by a quantitative high-resolution model of X-ray diffraction of contracting muscle. Eur. Biophys. J. Biophys. Lett. 2017, 46, 335–342. [Google Scholar] [CrossRef]
- Kress, M.; Huxley, H.E.; Faruqi, A.R.; Hendrix, J. Structural changes during activation of frog muscle studied by time-resolved X-ray diffraction. J. Mol. Biol. 1986, 188, 325–342. [Google Scholar] [CrossRef]
- Squire, J.; Al-Khayat, H.A.; Yagi, N. Muscle thin-filament structure and regulation. Actin sub-domain movements and the tropomyosin shift modelled from low-angle X-ray diffraction. J. Chem. Soc. Faraday Trans. 1993, 89, 2717–2726. [Google Scholar] [CrossRef]
- Poole, K.J.V.; Lorenz, M.; Evans, G.; Rosenbaum, G.; Pirani, A.; Craig, R.; Tobacman, L.S.; Lehman, W.; Holmes, K.C. A comparison of muscle thin filament models obtained from electron microscopy reconstructions and low-angle X-ray fibre diagrams from non-overlap muscle. J. Struct. Biol. 2006, 155, 273–284. [Google Scholar] [CrossRef]
- Phillips, G.N., Jr.; Fillers, J.; Cohen, C. Tropomyosin crystal structure and muscle regulation. J. Mol. Biol. 1986, 192, 111–131. [Google Scholar] [CrossRef]
- Vinogradova, M.V.; Stone, D.B.; Malanina, G.G.; Karatzaferi, C.; Cooke, R.; Mendelson, R.A.; Fletterick, R.J. Ca2+-regulated structural changes in troponin. Proc. Natl. Acad. Sci. USA 2005, 102, 5038–5043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, K.C.; Popp, D.; Gebhard, W.; Kabsch, W. Atomic model of the actin filament. Nature 1990, 347, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Namba, K.; Fujii, T. Cardiac muscle thin filament structures reveal calcium regulatory mechanism. Nat. Commun. 2020, 11, 153. [Google Scholar] [CrossRef] [PubMed]
- Risi, C.M.; Pepper, I.; Belknap, B.; Landim-Vieira, M.; White, H.D.; Dryden, K.; Pinto, J.R.; Chase, P.B.; Galkin, V.E. The structure of the native cardiac thin filament at systolic Ca2+ levels. Proc. Natl. Acad. Sci. USA 2021, 118, e2024288118. [Google Scholar] [CrossRef]
- Landim-Vieira, M.; Ma, W.; Johnston, J.; Galkin, V.E.; Chase, P.B.; Irving, T.C.; Pinto, J.R.D. Modulation of Cardiac Thin Filament Azimuthal Rigidity by Calcium and Cross-Bridges. Biophys. J. 2021, 120, 342a–343a. [Google Scholar] [CrossRef]
- Eakins, F.; Harford, J.J.; Knupp, C.; Roessle, M.; Squire, J.M. Different Myosin Head Conformations in Bony Fish Muscles Put into Rigor at Different Sarcomere Lengths. Int. J. Mol. Sci. 2018, 19, 2091. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Lee, K.H.; Yang, S.; Irving, T.C.; Craig, R. Lattice arrangement of myosin filaments correlates with fiber type in rat skeletal muscle. J. Gen. Physiol. 2019, 151, 1404–1412. [Google Scholar] [CrossRef]
- Squire, J.M.; Knupp, C.; Roessle, M.; Al-Khayat, H.A.; Irving, T.C.; Eakins, F.; Mok, N.S.; Harford, J.J.; Reedy, M.K. X-ray diffraction studies of striated muscles. Adv. Exp. Med. Biol. 2005, 565, 45–60. [Google Scholar]
- Cooke, R. The role of the myosin ATPase activity in adaptive thermogenesis by skeletal muscle. Biophys. Rev. 2011, 3, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Hooijman, P.; Stewart, M.A.; Cooke, R. A New State of Cardiac Myosin with Very Slow ATP Turnover: A Potential Cardioprotective Mechanism in the Heart. Biophys. J. 2011, 100, 1969–1976. [Google Scholar] [CrossRef] [Green Version]
- McNamara, J.W.; Li, A.; Dos Remedios, C.G.; Cooke, R. The role of super-relaxed myosin in skeletal and cardiac muscle. Biophys. Rev. 2015, 7, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alamo, L.; Qi, D.; Wriggers, W.; Pinto, A.; Zhu, J.; Bilbao, A.; Gillilan, R.E.; Hu, S.; Padron, R. Conserved Intramolecular Interactions Maintain Myosin Interacting-Heads Motifs Explaining Tarantula Muscle Super-Relaxed State Structural Basis. J. Mol. Biol. 2016, 428, 1142–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, R.; Padron, R. Structural basis of the super- and hyper-relaxed states of myosin II. J. Gen. Physiol. 2022, 154, e202113012. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Duno-Miranda, S.; Irving, T.; Craig, R.; Padron, R. Relaxed tarantula skeletal muscle has two ATP energy-saving mechanisms. J. Gen. Physiol. 2021, 153, e202012780. [Google Scholar] [CrossRef]
- Chu, S.; Muretta, J.M.; Thomas, D.D. Direct detection of the myosin super-relaxed state and interacting-heads motif in solution. J. Biol. Chem. 2021, 297, 101157. [Google Scholar] [CrossRef]
- Spudich, J.A. Three perspectives on the molecular basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Pflugers Arch. 2019, 471, 701–717. [Google Scholar] [CrossRef] [Green Version]
- Sadayappan, S.; de Tombe, P. Cardiac myosin binding protein-C: Redefining its structure and function. Biophys. Rev. 2012, 4, 93–106. [Google Scholar] [CrossRef]
- McNamara, J.W.; Sadayappan, S. Skeletal myosin binding protein-C: An increasingly important regulator of striated muscle physiology. Arch. Biochem. Biophys. 2018, 660, 121–128. [Google Scholar] [CrossRef]
- Barefield, D.; Sadayappan, S. Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J. Mol. Cell Cardiol. 2010, 48, 866–875. [Google Scholar] [CrossRef]
- Harris, S.P.; Lyons, E.G.; Bezold, K.L. In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ. Res. 2011, 108, 751–764. [Google Scholar] [CrossRef] [Green Version]
- Kensler, R.W.; Craig, R.; Moss, R.L. Phosphorylation of cardiac myosin binding protein C releases myosin heads from the surface of cardiac thick filaments. Proc. Natl. Acad. Sci. USA 2017, 114, E1355–E1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razumova, M.V.; Shaffer, J.F.; Tu, A.Y.; Flint, G.V.; Regnier, M.; Harris, S.P. Effects of the N-terminal domains of myosin binding protein-C in an in vitro motility assay: Evidence for long-lived cross-bridges. J. Biol. Chem. 2006, 281, 35846–35854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaffer, J.F.; Kensler, R.W.; Harris, S. The myosin-binding protein C motif binds to F-actin in a phosphorylation-sensitive manner. J. Biol. Chem. 2009, 284, 12318–12327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colson, B.A.; Patel, J.R.; Chen, P.P.; Bekyarova, T.; Abdalla, M.I.; Tong, C.W.; Fitzsimons, D.P.; Irving, T.C.; Moss, R.L. Myosin binding protein-C phosphorylation is the principal mediator of protein kinase A effects on thick filament structure in myocardium. J. Mol. Cell Cardiol. 2012, 53, 609–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alamo, L.; Ware, J.S.; Pinto, A.; Gillilan, R.E.; Seidman, J.G.; Seidman, C.E.; Padron, R. Effects of myosin variants on interacting-heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. Elife 2017, 6, e24634. [Google Scholar] [CrossRef] [PubMed]
- McNamara, J.W.; Li, A.; Lal, S.; Bos, J.M.; Harris, S.P.; van der Velden, J.; Ackerman, M.J.; Cooke, R.; Dos Remedios, C.G. MYBPC3 mutations are associated with a reduced super-relaxed state in patients with hypertrophic cardiomyopathy. PLoS ONE 2017, 12, e01800642017. [Google Scholar] [CrossRef] [PubMed]
- McNamara, J.W.; Li, A.; Smith, N.J.; Lal, S.; Graham, R.M.; Kooiker, K.B.; van Dijk, S.J.; Remedios, C.G.D.; Harris, S.P.; Cooke, R. Ablation of cardiac myosin binding protein-C disrupts the super-relaxed state of myosin in murine cardiomyocytes. J. Mol. Cell Cardiol. 2016, 94, 65–71. [Google Scholar] [CrossRef] [Green Version]
- De Tombe, P.P.; Mateja, R.D.; Tachampa, K.; Ait Mou, Y.; Farman, G.P.; Irving, T.C. Myofilament length dependent activation. J. Mol. Cell Cardiol. 2010, 48, 851–858. [Google Scholar] [CrossRef] [Green Version]
- Ter Keurs, H.E.; Rijnsburger, W.H.; van Heuningen, R.; Nagelsmit, M.J. Tension development and sarcomere length in rat cardiac trabeculae. Evidence of length-dependent activation. Circ. Res. 1980, 46, 703–714. [Google Scholar] [CrossRef] [Green Version]
- Allen, D.G.; Kentish, J.C. The cellular basis of the length-tension relation in cardiac muscle. J. Mol. Cell Cardiol. 1985, 17, 821–840. [Google Scholar] [CrossRef]
- McDonald, K.S.; Hanft, L.M.; Robinett, J.C.; Guglin, M.; Campbell, K.S. Regulation of Myofilament Contractile Function in Human Donor and Failing Hearts. Front. Physiol. 2020, 11, 468. [Google Scholar] [CrossRef] [PubMed]
- Schwinger, R.H.; Bohm, M.; Koch, A.; Schmidt, U.; Morano, I.; Eissner, H.J.; Uberfuhr, P.; Reichart, B.; Erdmann, E. The failing human heart is unable to use the Frank-Starling mechanism. Circ. Res. 1994, 74, 959–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequeira, V.; Wijnker, P.J.; Nijenkamp, L.L.; Kuster, D.W.; Najafi, A.; Witjas-Paalberends, E.R.; Regan, J.A.; Boontje, N.; Ten Cate, F.J.; Germans, T.; et al. Perturbed length-dependent activation in human hypertrophic cardiomyopathy with missense sarcomeric gene mutations. Circ. Res. 2013, 112, 1491–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reconditi, M.; Caremani, M.; Pinzauti, F.; Powers, J.D.; Narayanan, T.; Stienen, G.J.; Linari, M.; Lombardi, V.; Piazzesi, G. Myosin filament activation in the heart is tuned to the mechanical task. Proc. Natl. Acad. Sci. USA 2017, 114, 3240–3245. [Google Scholar] [CrossRef] [Green Version]
- Piazzesi, G.; Caremani, M.; Linari, M.; Reconditi, M.; Lombardi, V. Thick Filament Mechano-Sensing in Skeletal and Cardiac Muscles: A Common Mechanism Able to Adapt the Energetic Cost of the Contraction to the Task. Front. Physiol. 2018, 9, 736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stachowski-Doll, M.J.; Papadaki, M.; Martin, T.G.; Ma, W.; Gong, H.M.; Shao, S.; Shen, S.; Muntu, N.A.; Kumar, M.; Perez, E.; et al. GSK-3beta Localizes to the Cardiac Z-Disc to Maintain Length Dependent Activation. Circ. Res. 2022, CIRCRESAHA121319491. [Google Scholar] [CrossRef]
- Irving, T.; Craig, R. Getting into the thick (and thin) of it. J. Gen. Physiol. 2019, 151, 610–613. [Google Scholar] [CrossRef] [Green Version]
- Muthu, P.; Wang, L.; Yuan, C.C.; Kazmierczak, K.; Huang, W.; Hernandez, O.M.; Kawai, M.; Irving, T.C.; Szczesna-Cordary, D. Structural and functional aspects of the myosin essential light chain in cardiac muscle contraction. FASEB J. 2011, 25, 4394–4405. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Martinez, D.; Johnston, J.R.; Landim-Vieira, M.; Ma, W.; Antipova, O.; Awan, O.; Irving, T.C.; Bryant Chase, P.; Pinto, J.R. Structural and functional impact of troponin C-mediated Ca2+ sensitization on myofilament lattice spacing and cross-bridge mechanics in mouse cardiac muscle. J. Mol. Cell Cardiol. 2018, 123, 26–37. [Google Scholar] [CrossRef]
- Trujillo, A.S.; Hsu, K.H.; Puthawala, J.; Viswanathan, M.C.; Loya, A.; Irving, T.C.; Cammarato, A.; Swank, D.M.; Bernstein, S.I. Myosin dilated cardiomyopathy mutation S532P disrupts actomyosin interactions, leading to altered muscle kinetics, reduced locomotion, and cardiac dilation in Drosophila. Mol. Biol. Cell 2021, 32, 1690–1706. [Google Scholar] [CrossRef]
- Yuan, C.C.; Kazmierczak, K.; Liang, J.; Zhou, Z.; Yadav, S.; Gomes, A.V.; Irving, T.C.; Szczesna-Cordary, D. Sarcomeric perturbations of myosin motors lead to dilated cardiomyopathy in genetically modified MYL2 mice. Proc. Natl. Acad. Sci. USA 2018, 115, E2338–E2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, T.L.T.; Kumar, M.; McNamara, J.W.; Kuster, D.W.D.; Sivaguru, M.; Singh, R.R.; Previs, M.J.; Lee, K.H.; Kuffel, G.; Zilliox, M.J.; et al. Amino terminus of cardiac myosin binding protein-C regulates cardiac contractility. J. Mol. Cell Cardiol. 2021, 156, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Ait Mou, Y.; Lacampagne, A.; Irving, T.; Scheuermann, V.; Blot, S.; Ghaleh, B.; de Tombe, P.P.; Cazorla, O. Altered myofilament structure and function in dogs with Duchenne muscular dystrophy cardiomyopathy. J. Mol. Cell Cardiol. 2018, 114, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.T.; Shirai, M.; Ito, H.; Tokunaga, N.; Tsuchimochi, H.; Nishiura, N.; Schwenke, D.O.; Ishibashi-Ueda, H.; Akiyama, R.; Mori, H.; et al. In situ measurements of crossbridge dynamics and lattice spacing in rat hearts by X-ray diffraction: Sensitivity to regional ischemia. Circulation 2004, 109, 2976–2979. [Google Scholar] [CrossRef] [Green Version]
- Waddingham, M.T.; Edgley, A.J.; Tsuchimochi, H.; Kelly, D.J.; Shirai, M.; Pearson, J.T. Contractile apparatus dysfunction early in the pathophysiology of diabetic cardiomyopathy. World J. Diabetes 2015, 6, 943–960. [Google Scholar] [CrossRef]
- Jenkins, M.J.; Pearson, J.T.; Schwenke, D.O.; Edgley, A.J.; Sonobe, T.; Fujii, Y.; Ishibashi-Ueda, H.; Kelly, D.J.; Yagi, N.; Shirai, M. Myosin heads are displaced from actin filaments in the in situ beating rat heart in early diabetes. Biophys. J. 2013, 104, 1065–1072. [Google Scholar] [CrossRef] [Green Version]
- Waddingham, M.T.; Sonobe, T.; Tsuchimochi, H.; Edgley, A.J.; Sukumaran, V.; Chen, Y.C.; Hansra, S.S.; Schwenke, D.O.; Umetani, K.; Aoyama, K.; et al. Diastolic dysfunction is initiated by cardiomyocyte impairment ahead of endothelial dysfunction due to increased oxidative stress and inflammation in an experimental prediabetes model. J. Mol. Cell Cardiol. 2019, 137, 119–131. [Google Scholar] [CrossRef] [Green Version]
- Ottenheijm, C.A.; Voermans, N.C.; Hudson, B.D.; Irving, T.; Stienen, G.J.; van Engelen, B.G.; Granzier, H. Titin-based stiffening of muscle fibers in Ehlers-Danlos Syndrome. J. Appl. Physiol. 2012, 112, 1157–1165. [Google Scholar] [CrossRef]
- Ochala, J.; Ravenscroft, G.; McNamara, E.; Nowak, K.J.; Iwamoto, H. X-ray recordings reveal how a human disease-linked skeletal muscle alpha-actin mutation leads to contractile dysfunction. J. Struct. Biol. 2015, 192, 331–335. [Google Scholar] [CrossRef]
- Ochala, J.; Iwamoto, H.; Larsson, L.; Yagi, N. A myopathy-linked tropomyosin mutation severely alters thin filament conformational changes during activation. Proc. Natl. Acad. Sci. USA 2010, 107, 9807–9812. [Google Scholar] [CrossRef] [Green Version]
- Garfinkel, A.C.; Seidman, J.G.; Seidman, C.E. Genetic Pathogenesis of Hypertrophic and Dilated Cardiomyopathy. Heart Fail. Clin. 2018, 14, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Gardin, J.M.; Flack, J.M.; Gidding, S.S.; Kurosaki, T.T.; Bild, D.E. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation 1995, 92, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.A.; Cook, S.A.; Seidman, J.G.; Seidman, C.E. Clinical and Mechanistic Insights Into the Genetics of Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 68, 2871–2886. [Google Scholar] [CrossRef] [PubMed]
- Alfares, A.A.; Kelly, M.A.; McDermott, G.; Funke, B.H.; Lebo, M.S.; Baxter, S.B.; Shen, J.; McLaughlin, H.M.; Clark, E.H.; Babb, L.J.; et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: Expanded panels offer limited additional sensitivity. Genet. Med. 2015, 17, 880–888. [Google Scholar] [CrossRef] [Green Version]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of Mendelian gene pathogenicity using 7855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 2017, 19, 192–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farman, G.P.; Miller, M.S.; Reedy, M.C.; Soto-Adames, F.N.; Vigoreaux, J.O.; Maughan, D.W.; Irving, T.C. Phosphorylation and the N-terminal extension of the regulatory light chain help orient and align the myosin heads in Drosophila flight muscle. J. Struct. Biol. 2009, 168, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.S.; Farman, G.P.; Braddock, J.M.; Soto-Adames, F.N.; Irving, T.C.; Vigoreaux, J.O.; Maughan, D.W. Regulatory light chain phosphorylation and N-terminal extension increase cross-bridge binding and power output in Drosophila at in vivo myofilament lattice spacing. Biophys. J. 2011, 100, 1737–1746. [Google Scholar] [CrossRef] [Green Version]
- Pieples, K.; Arteaga, G.; Solaro, R.J.; Grupp, I.; Lorenz, J.N.; Boivin, G.P.; Jagatheesan, G.; Labitzke, E.; DeTombe, P.P.; Konhilas, J.P.; et al. Tropomyosin 3 expression leads to hypercontractility and attenuates myofilament length-dependent Ca(2+) activation. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1344–H1353. [Google Scholar] [CrossRef] [Green Version]
- Reichart, D.; Magnussen, C.; Zeller, T.; Blankenberg, S. Dilated cardiomyopathy: From epidemiologic to genetic phenotypes: A translational review of current literature. J. Intern. Med. 2019, 286, 362–372. [Google Scholar] [CrossRef] [Green Version]
- Vikhorev, P.G.; Smoktunowicz, N.; Munster, A.B.; Copeland, O.N.; Kostin, S.; Montgiraud, C.; Messer, A.E.; Toliat, M.R.; Li, A.; Dos Remedios, C.G.; et al. Author Correction: Abnormal contractility in human heart myofibrils from patients with dilated cardiomyopathy due to mutations in TTN and contractile protein genes. Sci. Rep. 2018, 8, 14485. [Google Scholar] [CrossRef]
- Oldfors, A. Hereditary myosin myopathies. Neuromuscul. Disord. 2007, 17, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Kurapati, R.; McKenna, C.; Lindqvist, J.; Williams, D.; Simon, M.; LeProust, E.; Baker, J.; Cheeseman, M.; Carroll, N.; Denny, P.; et al. Myofibrillar myopathy caused by a mutation in the motor domain of mouse MyHC IIb. Hum. Mol. Genet. 2012, 21, 1706–1724. [Google Scholar] [CrossRef] [PubMed]
- Ochala, J. Thin filament proteins mutations associated with skeletal myopathies: Defective regulation of muscle contraction. J. Mol. Med. 2008, 86, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Schnell, C.; Kan, A.; North, K.N. ‘An artefact gone awry’: Identification of the first case of nemaline myopathy by Dr R.D.K. Reye. Neuromuscul. Disord. 2000, 10, 307–312. [Google Scholar] [CrossRef]
- Malfatti, E.; Romero, N.B. Nemaline myopathies: State of the art. Rev. Neurol. 2016, 172, 614–619. [Google Scholar] [CrossRef]
- Li, F.; Buck, D.; De Winter, J.; Kolb, J.; Meng, H.; Birch, C.; Slater, R.; Escobar, Y.N.; Smith, J.E., 3rd; Yang, L.; et al. Nebulin deficiency in adult muscle causes sarcomere defects and muscle-type-dependent changes in trophicity: Novel insights in nemaline myopathy. Hum. Mol. Genet. 2015, 24, 5219–5233. [Google Scholar] [CrossRef] [Green Version]
- Romero, N.B.; Sandaradura, S.A.; Clarke, N.F. Recent advances in nemaline myopathy. Curr. Opin. Neurol. 2013, 26, 519–526. [Google Scholar] [CrossRef]
- Mijailovich, S.M.; Kayser-Herold, O.; Stojanovic, B.; Nedic, D.; Irving, T.C.; Geeves, M.A. Three-dimensional stochastic model of actin-myosin binding in the sarcomere lattice. J. Gen. Physiol. 2016, 148, 459–488. [Google Scholar] [CrossRef] [Green Version]
- Mijailovich, S.M.; Stojanovic, B.; Nedic, D.; Svicevic, M.; Geeves, M.A.; Irving, T.C.; Granzier, H.L. Nebulin and titin modulate cross-bridge cycling and length-dependent calcium sensitivity. J. Gen. Physiol. 2019, 151, 680–704. [Google Scholar] [CrossRef] [Green Version]
- Prodanovic, M.; Irving, T.C.; Mijailovich, S.M. X-ray diffraction from nonuniformly stretched helical molecules. J. Appl. Crystallogr. 2016, 49, 784–797. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Muscle Type | Disease | References |
|---|---|---|
| Cardiac | Heavy Chain HCM | [40,41] |
| Cardiac | Essential Light Chain HCM | [174] |
| Cardiac | Regulatory Light Chain HCM | [50,70] |
| Cardiac | Troponin HCM | [109,175] |
| Cardiac | Tropomyosin HCM | |
| Cardiac 1 | Heavy Chain DCM | [176] |
| Cardiac | Regulatory Light Chain DCM | [50,177] |
| Cardiac | cMyBP-C DCM | [178] |
| Cardiac | Muscular Dystrophy Cardiomyopathy | [179] |
| Cardiac | Ischemia | [180] |
| Cardiac | Diabetic Cardiomyopathy | [181,182,183] |
| Skeletal | Myosin-based myopathies | [33] |
| Skeletal | Titin-based myopathies | [38,184] |
| Skeletal | Nemaline myopathies | [29,30,31,36,37,185] |
| Skeletal | Tropomyosin-based myopathies | [186] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, W.; Irving, T.C. Small Angle X-ray Diffraction as a Tool for Structural Characterization of Muscle Disease. Int. J. Mol. Sci. 2022, 23, 3052. https://doi.org/10.3390/ijms23063052
Ma W, Irving TC. Small Angle X-ray Diffraction as a Tool for Structural Characterization of Muscle Disease. International Journal of Molecular Sciences. 2022; 23(6):3052. https://doi.org/10.3390/ijms23063052
Chicago/Turabian StyleMa, Weikang, and Thomas C. Irving. 2022. "Small Angle X-ray Diffraction as a Tool for Structural Characterization of Muscle Disease" International Journal of Molecular Sciences 23, no. 6: 3052. https://doi.org/10.3390/ijms23063052
APA StyleMa, W., & Irving, T. C. (2022). Small Angle X-ray Diffraction as a Tool for Structural Characterization of Muscle Disease. International Journal of Molecular Sciences, 23(6), 3052. https://doi.org/10.3390/ijms23063052