
The mRubyFT Protein, Genetically Encoded Blue-to-Red Fluorescent Timer

 and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Developing Blue-to-Red Fluorescent Timer Based on mRuby2 RFP in E. coli
2.2. In Vitro Characterization of Purified mRubyFT Timer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Timer | Form | Abs, Ex/Em (nm) | ε (mM−1 cm−1) a | QY b | Brightness vs. EGFP c(%) | Characteristic times, h d | pKa |
|---|---|---|---|---|---|---|---|
| Fast-FT e | Blue | 403, ND/466 | 49.7 (18.4) * | 0.3 | 44 (16) * | 0.25 | 2.8 |
| Red | 583, ND/606 | 75.0 (19.1) * | 0.09 | 20 (5.1) * | 7.1 | 4.1 | |
| mRubyFT | Blue | 406, 408/457 | 96.8 (26.0) * | 0.63 | 181 (49) * | 5.7 | 3.9 ± 0.5 |
| Red | 577, 582/624 | 60.0 (29.6) * | 0.086 | 15 (7.6) * | 15 | 4.5 ± 0.1 |
2.3. Behavior of the mRubyFT Timer in Cultured Mammalian Cells
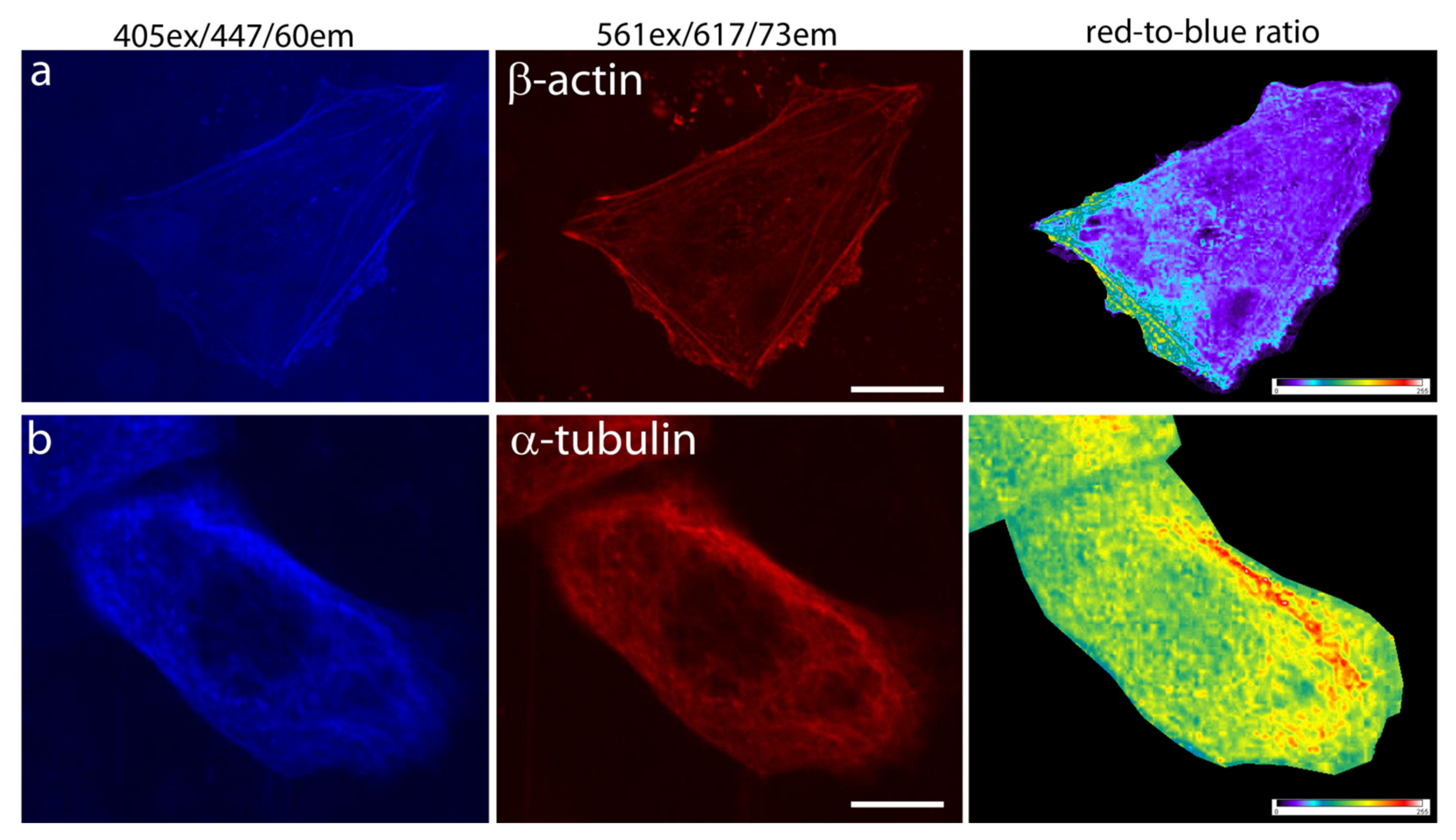
2.4. Behavior of mRubyFT in Fusions with Cytoskeleton Proteins in Mammalian Cells
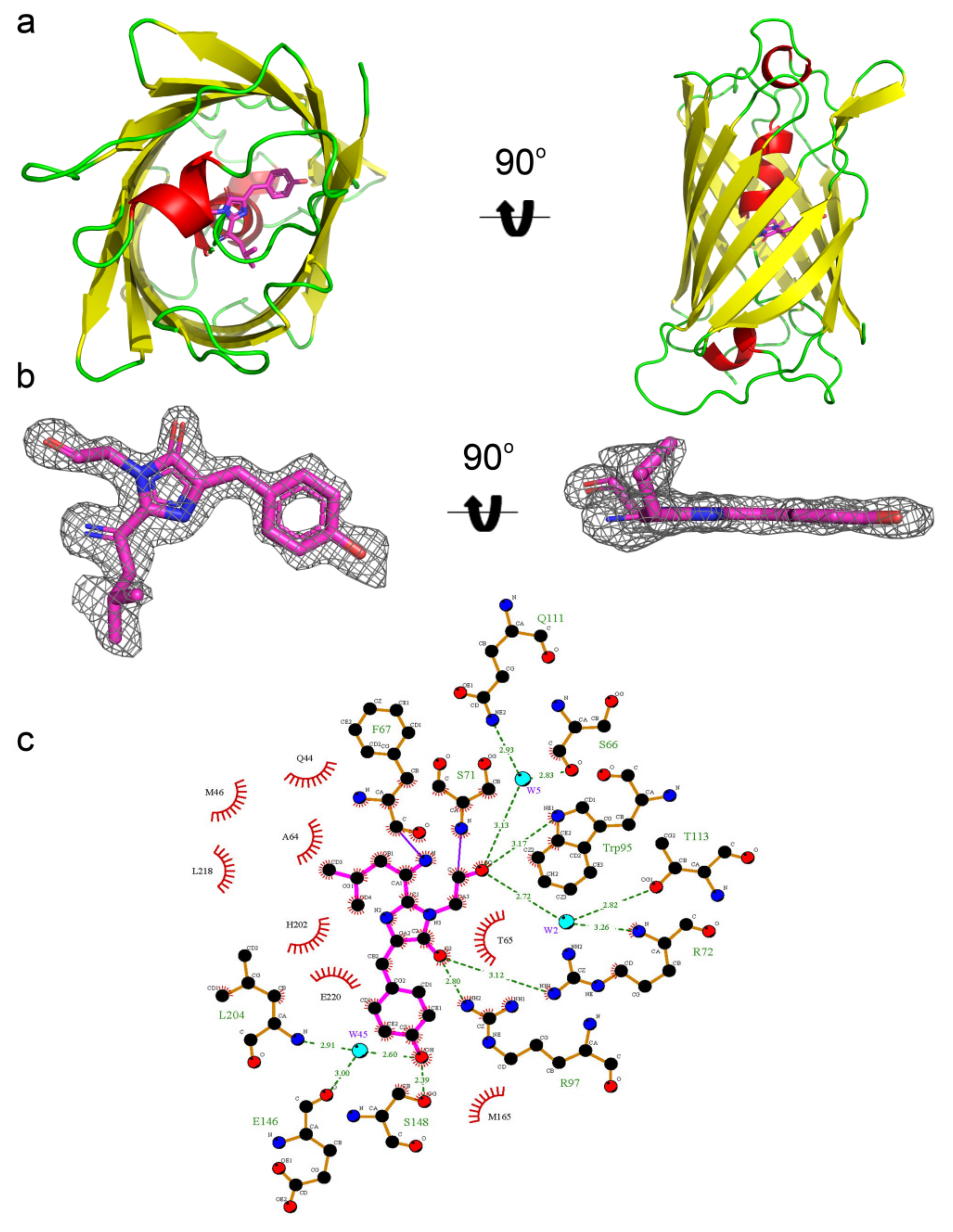
2.5. Structural Characterization of mRubyFT Timer
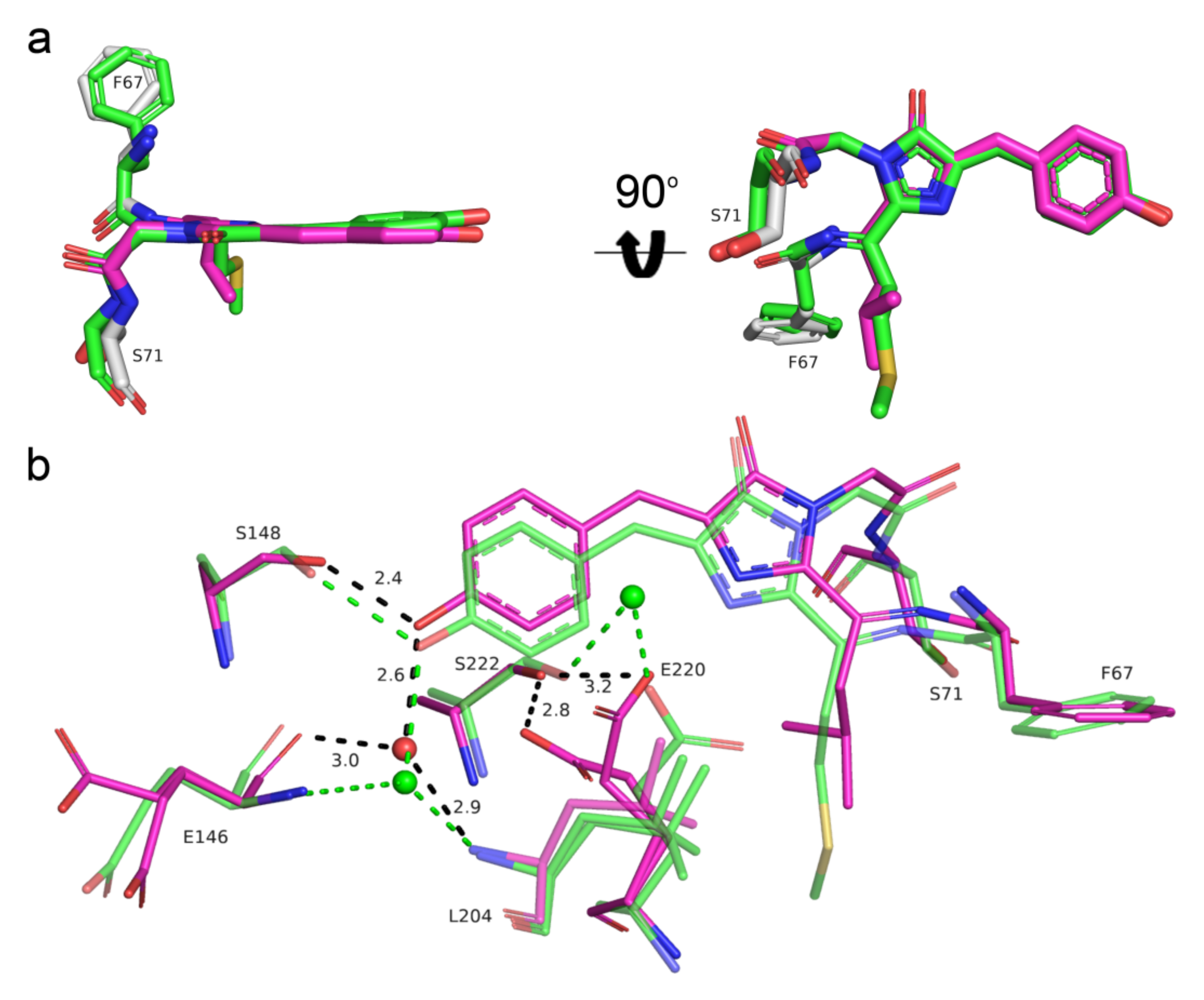
2.6. Directed Mutagenesis of the mRubyFT Blue-to-Red Fluorescent Timer and mRuby2 RFP at Key Positions
3. Materials and Methods
3.1. Cloning of Bacterial Vectors, Mutagenesis and Library Screening
3.2. Proteins’ Purification and Characterization
3.3. Protein Crystallization
3.4. Data Collection, Processing, Structure Solution, and Refinement
3.5. Structure Analysis and Validation
3.6. Mammalian Plasmids Construction
3.7. Mammalian Live Cell Imaging
3.8. Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| RFP | Red fluorescent protein |
| FT | Fluorescent timer |
| FP | Fluorescent protein |
| PBS | Phosphate buffered saline |
| QY | Quantum yield |
| SD | Standard deviation |
References
- Subach, F.V.; Subach, O.M.; Gundorov, I.S.; Morozova, K.S.; Piatkevich, K.D.; Cuervo, A.M.; Verkhusha, V.V. Monomeric fluorescent timers that change color from blue to red report on cellular trafficking. Nat. Chem. Biol. 2009, 5, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terskikh, A.; Fradkov, A.; Ermakova, G.; Zaraisky, A.; Tan, P.; Kajava, A.V.; Zhao, X.; Lukyanov, S.; Matz, M.; Kim, S.; et al. “Fluorescent timer”: Protein that changes color with time. Science 2000, 290, 1585–1588. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, T.; Kitaguchi, T.; Karasawa, S.; Fukuda, M.; Miyawaki, A. Age-dependent preferential dense-core vesicle exocytosis in neuroendocrine cells revealed by newly developed monomeric fluorescent timer protein. Mol. Biol. Cell 2009, 21, 87–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pletnev, S.; Subach, F.V.; Dauter, Z.; Wlodawer, A.; Verkhusha, V.V. Understanding blue-to-red conversion in monomeric fluorescent timers and hydrolytic degradation of their chromophores. J. Am. Chem. Soc. 2010, 132, 2243–2253. [Google Scholar] [CrossRef] [Green Version]
- Lam, A.J.; St-Pierre, F.; Gong, Y.; Marshall, J.D.; Cranfill, P.J.; Baird, M.A.; McKeown, M.R.; Wiedenmann, J.; Davidson, M.W.; Schnitzer, M.J.; et al. Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods 2012, 9, 1005–1012. [Google Scholar] [CrossRef] [Green Version]
- Subach, O.M.; Gundorov, I.S.; Yoshimura, M.; Subach, F.V.; Zhang, J.; Gruenwald, D.; Souslova, E.A.; Chudakov, D.M.; Verkhusha, V.V. Conversion of red fluorescent protein into a bright blue probe. Chem. Biol. 2008, 15, 1116–1124. [Google Scholar] [CrossRef] [Green Version]
- Subach, F.V.; Malashkevich, V.N.; Zencheck, W.D.; Xiao, H.; Filonov, G.S.; Almo, S.C.; Verkhusha, V.V. Photoactivation mechanism of PAmCherry based on crystal structures of the protein in the dark and fluorescent states. Proc. Natl. Acad. Sci. USA 2009, 106, 21097–21102. [Google Scholar] [CrossRef] [Green Version]
- Subach, O.M.; Malashkevich, V.N.; Zencheck, W.D.; Morozova, K.S.; Piatkevich, K.D.; Almo, S.C.; Verkhusha, V.V. Structural characterization of acylimine-containing blue and red chromophores in mTagBFP and TagRFP fluorescent proteins. Chem. Biol. 2010, 17, 333–341. [Google Scholar] [CrossRef] [Green Version]
- Akerboom, J.; Carreras Calderon, N.; Tian, L.; Wabnig, S.; Prigge, M.; Tolo, J.; Gordus, A.; Orger, M.B.; Severi, K.E.; Macklin, J.J.; et al. Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front. Mol. Neurosci. 2013, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Okuyama, T.; Kitamura, T.; Roy, D.S.; Itohara, S.; Tonegawa, S. Ventral CA1 neurons store social memory. Science 2016, 353, 1536–1541. [Google Scholar] [CrossRef] [Green Version]
- Subach, O.M.; Patterson, G.H.; Ting, L.M.; Wang, Y.; Condeelis, J.S.; Verkhusha, V.V. A photoswitchable orange-to-far-red fluorescent protein, PSmOrange. Nat. Methods 2011, 8, 771–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdanov, A.M.; Mishin, A.S.; Yampolsky, I.V.; Belousov, V.V.; Chudakov, D.M.; Subach, F.V.; Verkhusha, V.V.; Lukyanov, S.; Lukyanov, K.A. Green fluorescent proteins are light-induced electron donors. Nat. Chem. Biol. 2009, 5, 459–461. [Google Scholar] [CrossRef] [PubMed]
- Subach, O.M.; Cranfill, P.J.; Davidson, M.W.; Verkhusha, V.V. An enhanced monomeric blue fluorescent protein with the high chemical stability of the chromophore. PLoS ONE 2011, 6, e28674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [Green Version]
- Nienhaus, K.; Nar, H.; Heilker, R.; Wiedenmann, J.; Nienhaus, G.U. Trans-cis isomerization is responsible for the red-shifted fluorescence in variants of the red fluorescent protein eqFP611. J. Am. Chem. Soc. 2008, 130, 12578–12579. [Google Scholar] [CrossRef]
- Ho, S.N.; Hunt, H.D.; Horton, R.M.; Pullen, J.K.; Pease, L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 1989, 77, 51–59. [Google Scholar] [CrossRef]
- Shaner, N.C.; Campbell, R.E.; Steinbach, P.A.; Giepmans, B.N.; Palmer, A.E.; Tsien, R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004, 22, 1567–1572. [Google Scholar] [CrossRef]
- Svetogorov, R.; Dorovatovskii, P.; Lazarenko, V. Belok/XSA Diffraction Beamline for Studying Crystalline Samples at Kurchatov Synchrotron Radiation Source. Cryst. Res. Technol. 2020, 55, 1900184. [Google Scholar] [CrossRef]
- Winter, G.; Waterman, D.G.; Parkhurst, J.M.; Brewster, A.S.; Gildea, R.J.; Gerstel, M.; Fuentes-Montero, L.; Vollmar, M.; Michels-Clark, T.; Young, I.D.; et al. DIALS: Implementation and evaluation of a new integration package. Acta Crystallogr. D Struct. Biol. 2018, 74 Pt 2, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62 Pt 1, 72–82. [Google Scholar] [CrossRef]
- Vagin, A.; Teplyakov, A. MOLREP: An automated program for molecular replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994, 50 Pt 5, 760–763. [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60 Pt 12, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Krissinel, E.; Henrick, K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 2004, 60 Pt 12, 2256–2268. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Vriend, G. WHAT IF: A molecular modeling and drug design program. J. Mol. Graph. 1990, 8, 52–56. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Subach, O.M.; Barykina, N.V.; Chefanova, E.S.; Vlaskina, A.V.; Sotskov, V.P.; Ivashkina, O.I.; Anokhin, K.V.; Subach, F.V. FRCaMP, a Red Fluorescent Genetically Encoded Calcium Indicator Based on Calmodulin from Schizosaccharomyces Pombe Fungus. Int. J. Mol. Sci. 2020, 22, 111. [Google Scholar] [CrossRef] [PubMed]








| Timer | Form | Brightness vs. EGFP (%) | Brightness vs. Fast-FT (%) |
|---|---|---|---|
| mRubyFT | Blue | 10.7 ± 0.9 | 127 |
| Red | 10 ± 2 | 128 | |
| Fast-FT | Blue | 8.4 ± 0.7 | 100 |
| Red | 8 ± 2 | 100 |
| Protein | Characteristic Times (h) | |
|---|---|---|
| Blue | Red | |
| mRubyFT | 5.7 | 15 |
| mRubyFT/T62S | 6.3 | 0.57 |
| mRubyFT/R69K | Non-fluorescent | Non-fluorescent |
| mRubyFT/R69K/H203Y | 0.17 | 0.7 |
| mRubyFT/S148F | 1.1 | Non-fluorescent |
| mRubyFT/S148I | (5.8) a | Non-fluorescent |
| mRubyFT/T165N | 3.4 | Non-fluorescent |
| mRubyFT/167Q | 7.2 | Non-fluorescent |
| mRubyFT/H203Y | (0.72) a | 0.65 |
| mRubyFT/S224C | Non-fluorescent | Non-fluorescent |
| mRubyFT/S224A | 1.6 | 0.35 |
| Data Collection | |
| Diffraction source | BL41XU, SPring8 |
| Wavelength (Å) | 1.0 |
| Temperature (K) | 100 |
| Detector | EIGER |
| Crystal-to-detector distance (mm) | 200.00 |
| Rotation range per image (°) | 1.0 |
| Total rotation range (°) | 280 |
| Space group | P212121 |
| a, b, c (Å) | 31.34; 66.25; 96.50 |
| α, β, γ (°) | 90.0; 90.0; 90.0 |
| Unique reflections | 33,034 (1590) |
| Resolution range (Å) | 96.5–1.50 (1.53–1.50) |
| Completeness (%) | 99.8 (100.0) |
| Average redundancy | 7.9 (7.2) |
| 〈I/σ(I)〉 | 40.2 (4.3) |
| Rmeas (%) | 2.7 (22.5) |
| CC1/2 | 100.0 (97.3) |
| Refinement | |
| Rfact (%) | 17.4 |
| Rfree. (%) | 19.6 |
| Bonds (Å) | 0.01 |
| Angles (°) | 2.06 |
| Ramachandran plot | |
| Most favored (%) | 98.1 |
| Allowed (%) | 1.9 |
| No. atoms | |
| Protein | 1808 |
| Water | 158 |
| Chromophore | 23 |
| Magnesium ions | 3 |
| Other ligands | 0 |
| B-factors (Å2) | |
| Protein | 17.70 |
| Water | 31.0 |
| Chromophore | 24.6 |
| Magnesium ions | 24.60 |
| Other ligands | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subach, O.M.; Tashkeev, A.; Vlaskina, A.V.; Petrenko, D.E.; Gaivoronskii, F.A.; Nikolaeva, A.Y.; Ivashkina, O.I.; Anokhin, K.V.; Popov, V.O.; Boyko, K.M.; et al. The mRubyFT Protein, Genetically Encoded Blue-to-Red Fluorescent Timer. Int. J. Mol. Sci. 2022, 23, 3208. https://doi.org/10.3390/ijms23063208
Subach OM, Tashkeev A, Vlaskina AV, Petrenko DE, Gaivoronskii FA, Nikolaeva AY, Ivashkina OI, Anokhin KV, Popov VO, Boyko KM, et al. The mRubyFT Protein, Genetically Encoded Blue-to-Red Fluorescent Timer. International Journal of Molecular Sciences. 2022; 23(6):3208. https://doi.org/10.3390/ijms23063208
Chicago/Turabian StyleSubach, Oksana M., Aleksandr Tashkeev, Anna V. Vlaskina, Dmitry E. Petrenko, Filipp A. Gaivoronskii, Alena Y. Nikolaeva, Olga I. Ivashkina, Konstantin V. Anokhin, Vladimir O. Popov, Konstantin M. Boyko, and et al. 2022. "The mRubyFT Protein, Genetically Encoded Blue-to-Red Fluorescent Timer" International Journal of Molecular Sciences 23, no. 6: 3208. https://doi.org/10.3390/ijms23063208
APA StyleSubach, O. M., Tashkeev, A., Vlaskina, A. V., Petrenko, D. E., Gaivoronskii, F. A., Nikolaeva, A. Y., Ivashkina, O. I., Anokhin, K. V., Popov, V. O., Boyko, K. M., & Subach, F. V. (2022). The mRubyFT Protein, Genetically Encoded Blue-to-Red Fluorescent Timer. International Journal of Molecular Sciences, 23(6), 3208. https://doi.org/10.3390/ijms23063208