
Transcriptome Analysis of Populus euphratica under Salt Treatment and PeERF1 Gene Enhances Salt Tolerance in Transgenic Populus alba × Populus glandulosa

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Validation of Salt Stress Treatment in P. euphratica
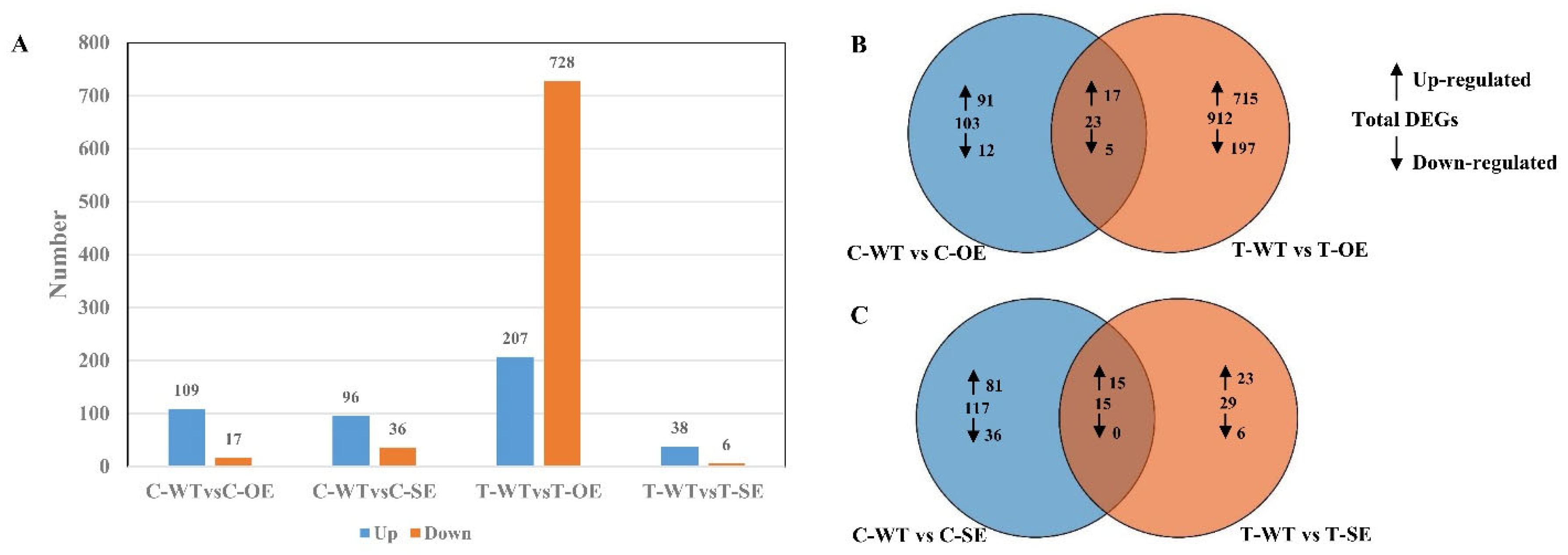
2.2. Analysis of RNA Sequences and Identification of Differentially Expressed Genes in P. euphratica under Salt Stress
2.3. GO and KEGG Enrichment Analysis of DEGs
2.4. Identification of WGCNA Modules Related to Phenotypic Traits
2.5. Cloning of PeERF1 and Protein Sequence Comparisons
2.6. PeERF1 Subcellular Location and Transcriptional Activity Analysis
2.7. Promoter Activity and Relevant Transient Acting Elements Analysis of PeERF1
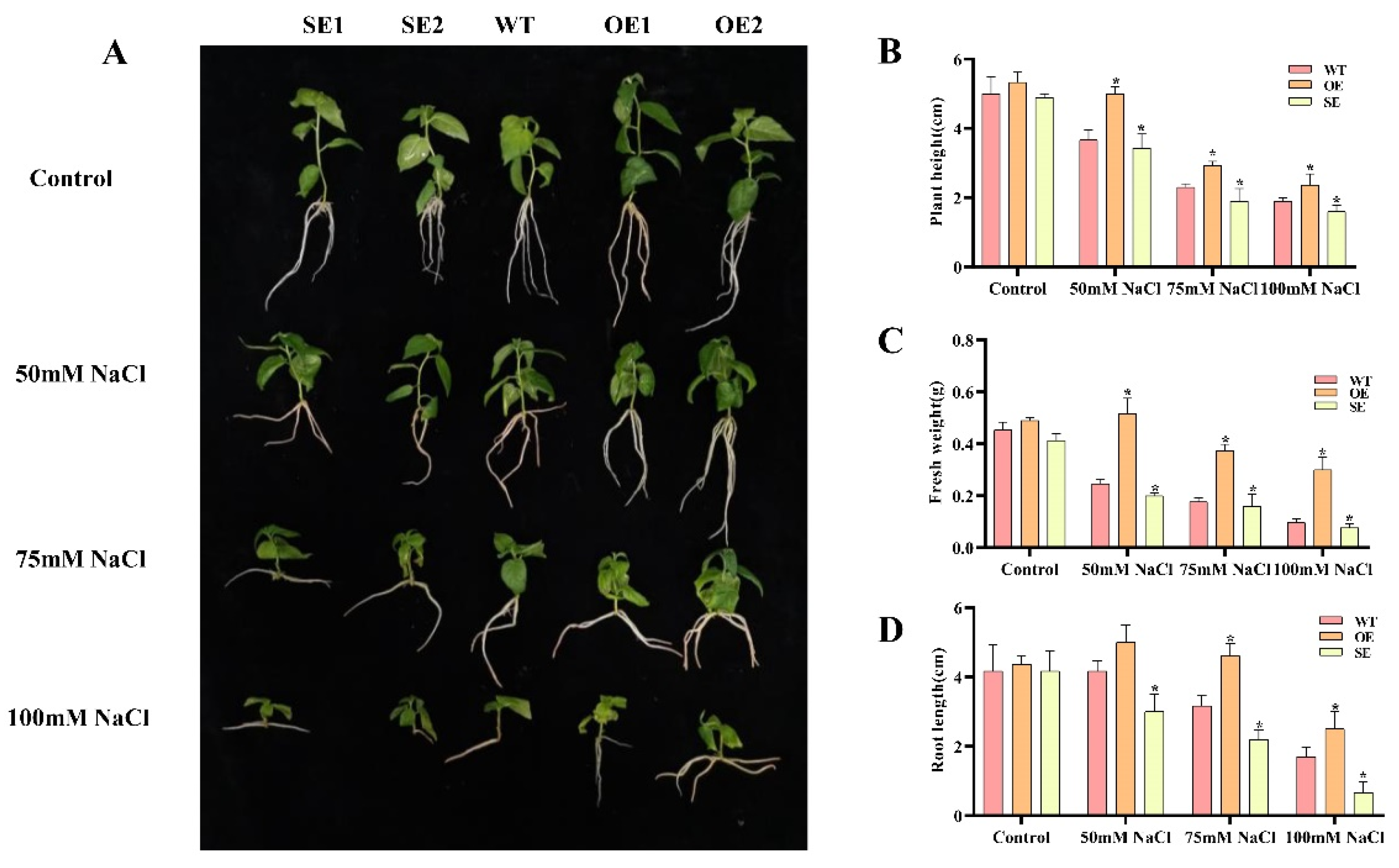
2.8. Morphological Changes of Transgenic P. alba × P. glandulosa under Salt Stress
2.9. Analysis of Physiological Characteristics of PeERF1 Transgenic P. alba × P. glandulosa
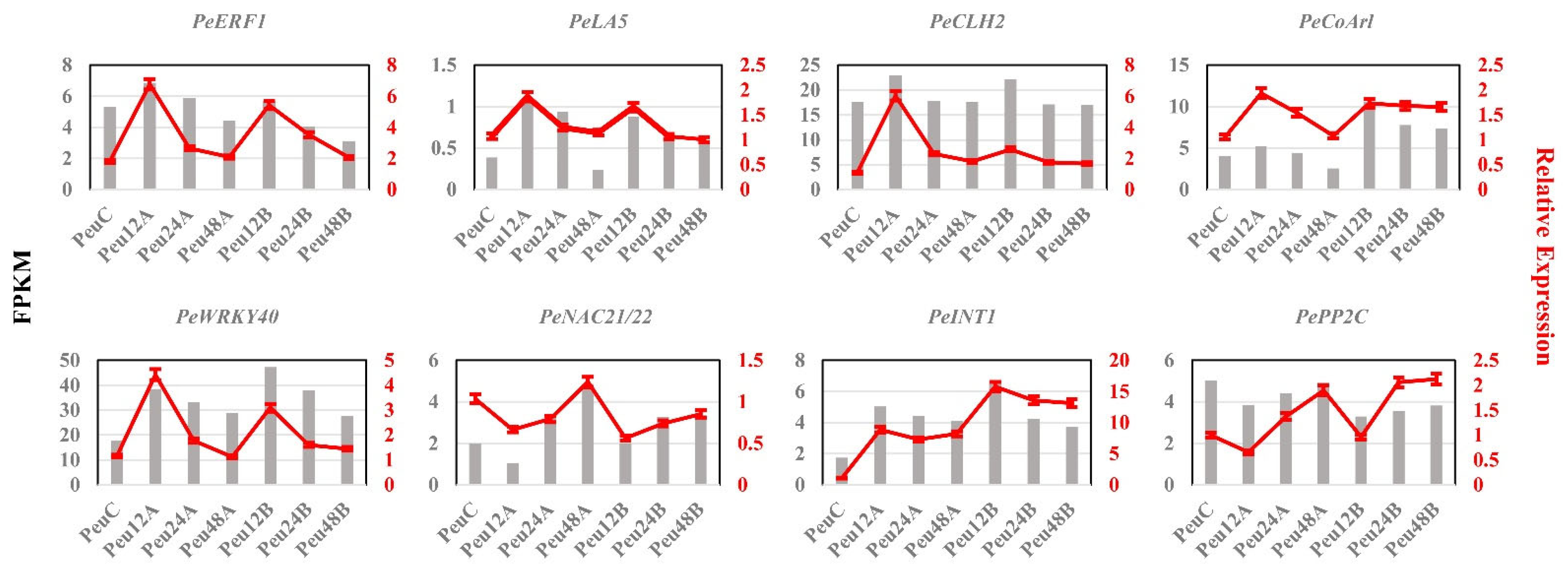
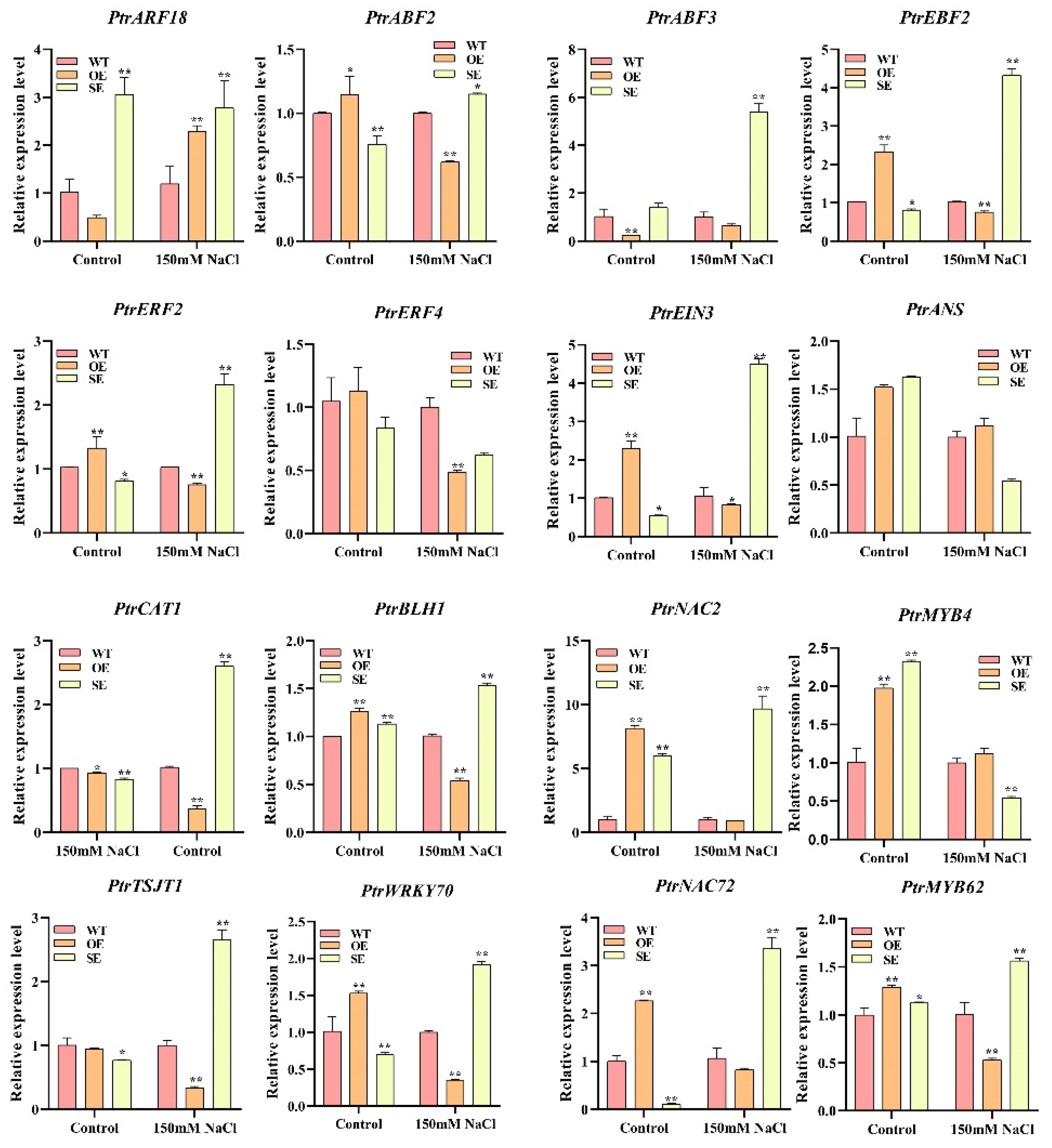
2.10. Expression Analysis of Downstream Genes Regulated of PeERF1
3. Discussion
4. Materials and Methods
4.1. Plant Growth and NaCl Treatment
4.2. Measurement of the Enzyme Activity of POD, MDA, and SOD
4.3. RNA Extraction and Illumina Deep Sequencing
4.4. Sequencing Data Processing and Analysis
4.5. qRT-PCR Analysis of Illumina Sequencing Results
4.6. Sequence Comparisons and Phylogenetic Analysis
4.7. Plasmid Construction and P. alba × P. glandulosa Transformation
4.8. Subcellular Localization and Self-Activation Experiment of PeERF1
4.9. Promoter Assay of PeERF1
4.10. One-Hybrid Yeast Experiment
4.11. Analysis of Physiological Characteristics of Transgenic P. alba × P. glandulosa Plants
4.12. Gene Expression Characterization Using RNA-Seq
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Godfray, H.C.; Beddington, J.R.; Crute, I.R.; Haddad, L.; Lawrence, D.; Muir, J.F.; Pretty, J.; Robinson, S.; Thomas, S.M.; Toulmin, C. Food security: The challenge of feeding 9 billion people. Science 2010, 327, 812–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Luo, W.; Li, Y.; Zhang, X.; Bai, X.; Niu, Z.; Zhang, X.; Li, Z.; Wan, D. Transcriptomic analysis of seed germination under salt stress in two desert sister species (Populus euphratica and P. pruinosa). Front. Genet. 2019, 10, 231. [Google Scholar] [CrossRef] [PubMed]
- Kazuo, S.; Kazuko, Y.S. Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 2007, 58, 221–227. [Google Scholar]
- Zeng, F.; Shabala, S.; Maksimović, J.D.; Maksimović, V.; Bonales-Alatorre, E.; Shabala, L.; Yu, M.; Zhang, G.; Živanović, B.D. Revealing mechanisms of salinity tissue tolerance in succulent halophytes: A case study for Carpobrotus rossi. Plant Cell Environ. 2018, 41, 2654–2667. [Google Scholar] [CrossRef] [PubMed]
- Kurt-Kızıldoğan, A.; Abanoz, B.; Okay, S. Global transcriptome analysis of Halolamina sp. to decipher the salt tolerance in extremely halophilic archaea. Gene 2017, 601, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, K.; Bishi, S.K.; Goswami, N.; Singh, A.L.; Zala, P.V. Differential fine-regulation of enzyme driven ROS detoxification network imparts salt tolerance in contrasting peanut genotypes. Environ. Exp. Bot. 2016, 128, 79–90. [Google Scholar] [CrossRef]
- Shen, X.; Guo, X.; Guo, X.; Zhao, D.; Zhao, W.; Chen, J.; Li, T. PacMYBA, a sweet cherry R2R3-MYB transcription factor, is a positive regulator of salt stress tolerance and pathogen resistance. Plant Physiol. Biochem. 2017, 112, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q.; Ma, T.; Hu, Q.; Liu, B.; Wu, Y.; Zhou, H.; Wang, Q.; Wang, J.; Liu, J. Genome-scale transcriptome analysis of the desert poplar, Populus euphratica. Tree Physiol. 2011, 31, 452–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Li, L.; Ye, H.; Tu, W. Weighted gene co-expression network analysis in biomedicine research. Chin. J. Biotechnol. 2017, 33, 1791. [Google Scholar]
- Yang, X.H.; Yan, J.B.; Zheng, Y.P.; Yu, J.M.; Li, J.S. Reviews of Association Analysis for Quantitative Traits in Plants. Acta Agron. Sin. 2007, 33, 8. [Google Scholar]
- Jia, H.; Liu, G.; Li, J.; Zhang, J.; Sun, P.; Zhao, S.; Zhou, X.; Lu, M.; Hu, J. Genome resequencing reveals demographic history and genetic architecture of seed salinity tolerance in Populus euphratica. J. Exp. Bot. 2020, 71, 4308–4320. [Google Scholar] [CrossRef] [PubMed]
- Koenig, D.; Jiménez-Gómez, J.M.; Kimura, S.F.D.; Chitwood, D.H.; Headland, L.R.; Kumar, R.; Covington, M.F.; Devisetty, U.K.; Tat, A.V.; Tohge, T.; et al. Comparative transcriptomics reveals patterns of selection in domesticated and wild tomato. Proc. Natl. Acad. Sci. USA 2013, 110, 2655–2662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, J.L.; Zhou, H.W.; Peng, Q.; Zhong, P.A.; Zhang, H.Y.; He, C.; Huang, Y.J. Transcriptome changes in rice (Oryza sativa L.) in response to high night temperature stress at the early milky stage. BMC Genom. 2015, 16, 18. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.Y.; Yang, H.; Wei, C.L.; Yu, O.; Zhang, Z.Z.; Jiang, C.J.; Sun, J.; Li, Y.Y.; Chen, Q.; Xia, T.; et al. Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC Genom. 2011, 12, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, L.; Wang, L.; Guo, Y.; Li, Y.; Ümüt, H.; Wang, Y. An ERF transcription factor from Tamarix hispida, ThCRF1, can adjust osmotic potential and reactive oxygen species scavenging capability to improve salt tolerance. Plant Sci. 2017, 265, 154–166. [Google Scholar] [CrossRef]
- Mao, J.; Miao, Z.; Wang, Z.; Yu, L.; Cai, X. Arabidopsis ERF1 mediates cross-talk between ethylene and auxin biosynthesis during primary root elongation by regulating ASA1 expression. PLoS Genet. 2016, 12, 1005760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Chen, X.; Ren, H.; Zhang, Z.; Zhang, H.; Wang, J.; Wang, X.; Huang, R. ERF protein JERF1 that transcriptionally modulates the expression of abscisic acid biosynthesis-related gene enhances the tolerance under salinity and cold in tobacco. Planta 2007, 226, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, D. Recovering the original intentions of risk assessment and management of genetically modified organisms in the European Union. Front. Bioeng. Biotechnol. 2018, 6, 52. [Google Scholar] [CrossRef] [PubMed]
- Klumper, W.; Qaim, M. A meta-analysis of the impacts of genetically modified crops. PLoS ONE 2014, 9, 111629. [Google Scholar]
- Manachini, B.; Bazan, G.; Schicchi, R. Potential impact of genetically modified Lepidoptera-resistant Brassica napus in biodiversity hotspots: Sicily as a theoretical model. Insect. Sci. 2018, 25, 562–580. [Google Scholar] [CrossRef] [PubMed]
- Robaey, Z. Gone with the wind: Conceiving of moral responsibility in the case of GMO contamination. Sci. Eng. Ethics 2016, 22, 889–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.L.; Li, Y.H.; Wu, K.M. Risk assessment and ecological effects of transgenic Bacillus thuringiensis crops on non-target organisms. J. Integr. Plant Biol. 2011, 53, 520–538. [Google Scholar] [CrossRef] [PubMed]
- Schiemann, J. Session VII: Risk management and monitoring. Environ. Biosaf. Res. 2006, 5, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, S.; Christiaens, O.; Giddings, K.; Jones, H.; Mezzetti, B.; Moronta-Barrios, F.; Perry, J.N.; Sweet, J.B.; Taning, C.N.T.; Smagghe, G.; et al. Biosafety of GM crop plants expressing dsRNA: Data requirements and EU regulatory considerations. Front. Plant. Sci. 2020, 11, 940. [Google Scholar] [CrossRef]
- Ayesh, G.; Pankaj, K.; Ajay, K.T.; Dinesh, K.S. REVIEW: In vitro plant regeneration studies and their potential applications in Populus spp.: A review. Isr. J. Plant Sci. 2016, 63, 77–84. [Google Scholar]
- Watanabe, S.; Kojima, K.; Ide, Y.; Sasaki, S. Effects of saline and osmotic stress on proline and sugar accumulation in Populus euphratica in vitro. Plant Cell Tissue Organ Cult. 2000, 63, 199–206. [Google Scholar] [CrossRef]
- Wang, S.J.; Chen, B.H.; Li, H.Q. Euphrates Poplar Forest; China Enviromental Science Press: Beijing, China, 1996. [Google Scholar]
- Li, B.; Duan, H.; Li, J.; Deng, X.W.; Yin, W.; Xia, X. Global identification of miRNAs and targets in Populus euphratica under salt stress. Plant Mol. Biol. 2013, 81, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Qin, Y.; Duan, H.; Yin, W.; Xia, X. Genome-wide characterization of new and drought stress responsive microRNAs in Populus euphratica. J. Exp. Bot. 2011, 62, 3765–3779. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Dun, H.; Lian, C.; Zhang, X.; Yin, W.; Xia, X. The role of peu-miR164 and its target PeNAC genes in response to abiotic stress in Populus euphratica. Plant Physiol. Biochem. PPB 2017, 115, 418–438. [Google Scholar] [CrossRef]
- Ma, J.; Lu, J.; Xu, J.; Duan, B.; He, X.; Liu, J. Genome-wide identification of WRKY genes in the desert poplar Populus euphratica and Adaptive Evolution of the genes in response to salt stress. Evol. Bioinform. Online 2015, 11 (Suppl. 1), 47–55. [Google Scholar] [CrossRef] [PubMed]
- Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. AP2/ERF family transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta 2012, 1819, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Gu, R.S.; Liu, Q.L.; Pe, D.; Jiang, X.N. Understanding saline and osmotic tolerance of Populus euphratica suspended cells. Plant Cell Tissue Organ Cult. 2004, 78, 261–265. [Google Scholar] [CrossRef]
- Zhang, S.; Dai, J.; Jiang, H. Research progress in molecular biology of Populus euphratica Oliv. World For. Res. 2019, 32, 22–27. [Google Scholar]
- Yang, C.; Wei, H. Designing microarray and RNA-Seq experiments for greater systems biology discovery in modern plant genomics. Mol. Plant 2015, 8, 196–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buschmann, H.; Green, P.; Sambade, A.; Doonan, J.H.; Lloyd, C.W. Cytoskeletal dynamics in interphase, mitosis and cytokinesis analysed through Agrobacterium-mediated transient transformation of tobacco BY-2 cells. New Phytol. 2011, 190, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, S.Y.; Ohta, M.; Usui, A.; Shinshi, H.; Ohme-Takagi, M. Arabidopsis ethylene-responsive element binding factors act as transcriptional activators or repressors of GCC box-mediated gene expression. Plant Cell 2000, 12, 393–404. [Google Scholar]
- Xu, Z.S.; Xia, L.Q.; Chen, M.; Cheng, X.G.; Zhang, R.Y.; Li, L.C.; Zhao, Y.X.; Lu, Y.; Ni, Z.Y.; Liu, L. Isolation and molecular characterization of the Triticum aestivum L. ethylene-responsive factor 1 (TaERF1) that increases multiple stress tolerance. Plant Mol. Biol. 2007, 65, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Peng, T.; Dai, W. Critical cis-acting elements and interacting transcription factors: Key players associated with abiotic stress responses in plants. Plant Mol. Biol. Report. 2014, 32, 303–317. [Google Scholar] [CrossRef]
- Wen, S.S.; Ge, X.L.; Wang, R.; Yang, H.F.; Bai, Y.E.; Guo, Y.H.; Zhang, J.; Lu, M.Z.; Zhao, S.T.; Wang, L.Q. An efficient agrobacterium-mediated transformation method for hybrid poplar 84K (Populus alba x P. glandulosa) using calli as explants. Int. J. Mol. Sci. 2022, 23, 2216. [Google Scholar] [CrossRef] [PubMed]
- Rajput, V.D.; Chen, Y.; Ayup, M. Effects of high salinity on physiological and anatomical indices in the early stages of Populus euphratica growth. Russ. J. Plant Physiol. 2015, 62, 229–236. [Google Scholar] [CrossRef]
- Tian, Q.; Chen, J.; Wang, D.; Wang, H.; Liu, C.; Wang, S.; Xia, X.; Yin, W. Overexpression of a Populus euphratica CBF4 gene in poplar confers tolerance to multiple stresses. Plant Cell Tissue Organ Cult. 2016, 128, 391–407. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, X.; Yao, J.; Zhao, N.; Chen, S. Overexpression mechanism of PeREM1.3 from Populus euphratica enhancing salt tolerance in transgenic tobacco. Beijing Linye Daxue Xuebao/J. Beijing For. Univ. 2019, 41, 1–9. [Google Scholar]
- Weiss, J.N.; Karma, A.; MacLellan, W.R.; Deng, M.; Rau, C.D.; Rees, C.M.; Wang, J.; Wisniewski, N.; Eskin, E.; Horvath, S.; et al. “Good enough solutions” and the genetics of complex diseases. Circ. Res. 2012, 111, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Farber, C.R. Systems-level analysis of genome-wide association data. G3 Bethesda Md. 2013, 3, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Sun, Z.; Li, J.; Cheng, D.; Luo, C.; Dai, C. Characterization of miRNA160/164 and their targets expression of beet (Beta vulgaris) seedlings under the salt tolerance. Plant Mol. Biol. Report. 2018, 36, 790–799. [Google Scholar] [CrossRef]
- Sakamoto, H.; Matsuda, O.; Iba, K. ITN1, a novel gene encoding an ankyrin-repeat protein that affects the ABA-mediated production of reactive oxygen species and is involved in salt-stress tolerance in Arabidopsis thaliana. Plant J. 2008, 56, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Fu, S.; Chen, S.; Zhang, W.; Qi, C. Ethylene response factor BnERF2-like (ERF2. 4) from Brassica napus L. enhances submergence tolerance and alleviates oxidative damage caused by submergence in Arabidopsis thaliana. Crop J. 2016, 4, 199–211. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Du, G.; Xiang, J.; Hu, C.; Li, X.; Wang, W.; Zhu, H.; Qiao, L.; Zhao, C.; Wang, J.; et al. Genome-wide identification of auxin response factor (ARF) gene family and the miR160-ARF18-mediated response to salt stress in peanut (Arachis hypogaea L.). Genomics 2021, 114, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Besseau, S.; Toronen, P.; Sipari, N.; Kollist, H.; Holm, L.; Palva, E.T. Defense-related transcription factors WRKY70 and WRKY54 modulate osmotic stress tolerance by regulating stomatal aperture in Arabidopsis. New Phytol. 2013, 200, 457–472. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.; Jia, W.; Zhang, J. AtMEK1 mediates stress-induced gene expression of CAT1 catalase by triggering H2O2 production in Arabidopsis. J. Exp. Bot. 2007, 58, 2969–2981. [Google Scholar] [CrossRef]
- Guan, L.M.; Zhao, J.; Scandalios, J.G. Cis-elements and trans-factors that regulate expression of the maize Cat1 antioxidant gene in response to ABA and osmotic stress: H2O2 is the likely intermediary signaling molecule for the response. Plant J. 2000, 22, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Zhao, L.; Gong, S.; Sun, X.; Li, P.; Qin, L.; Zhou, Y.; Xu, W.; Li, X. A cotton mitogen-activated protein kinase (GhMPK6) is involved in ABA-induced CAT1 expression and H2O2 production. J. Genet. Genom. 2011, 38, 557–565. [Google Scholar] [CrossRef]
- He, Z.; Wu, J.; Sun, X.; Dai, M. The maize clade A PP2C phosphatases play critical roles in multiple abiotic stress responses. Int. J. Mol. Sci. 2019, 20, 3573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.; Hong, Y.; Ren, Z.; Tang, K.; Zhang, H.; Zhu, J.; Zhao, C. A role for PICKLE in the regulation of cold and salt stress tolerance in Arabidopsis. Front. Plant Sci. 2019, 10, 900. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, Y.; Zhang, J.; Ma, X.; Li, Y.; Li, M.; Wang, D.; Kang, M.; Wu, H.; Yang, Y.; et al. Improved genome assembly provides new insights into genome evolution in a desert poplar (Populus euphratica). Mol. Ecol. Resour. 2020, 20, 781–794. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Nat. Preced. 2010, 5, 4282. [Google Scholar]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Luo, T.; Fu, X.; Fan, Q.; Liu, J. Cloning and molecular characterization of a mitogen-activated protein kinase gene from Poncirus trifoliata whose ectopic expression confers dehydration/drought tolerance in transgenic tobacco. J. Exp. Bot. 2011, 62, 5191–5206. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Han, R.H.; Liao, G.L.; Zhang, M.H. A Comparative Study on the Different Extraction Techniques about the Chlorophyll Concentration of Plant Leaf. J. Guangdong Univ. Educ. 2011, 31, 80–83. [Google Scholar]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ge, X.-L.; Zhang, L.; Du, J.-J.; Wen, S.-S.; Qu, G.-Z.; Hu, J.-J. Transcriptome Analysis of Populus euphratica under Salt Treatment and PeERF1 Gene Enhances Salt Tolerance in Transgenic Populus alba × Populus glandulosa. Int. J. Mol. Sci. 2022, 23, 3727. https://doi.org/10.3390/ijms23073727
Ge X-L, Zhang L, Du J-J, Wen S-S, Qu G-Z, Hu J-J. Transcriptome Analysis of Populus euphratica under Salt Treatment and PeERF1 Gene Enhances Salt Tolerance in Transgenic Populus alba × Populus glandulosa. International Journal of Molecular Sciences. 2022; 23(7):3727. https://doi.org/10.3390/ijms23073727
Chicago/Turabian StyleGe, Xiao-Lan, Lei Zhang, Jiu-Jun Du, Shuang-Shuang Wen, Guan-Zheng Qu, and Jian-Jun Hu. 2022. "Transcriptome Analysis of Populus euphratica under Salt Treatment and PeERF1 Gene Enhances Salt Tolerance in Transgenic Populus alba × Populus glandulosa" International Journal of Molecular Sciences 23, no. 7: 3727. https://doi.org/10.3390/ijms23073727
APA StyleGe, X. -L., Zhang, L., Du, J. -J., Wen, S. -S., Qu, G. -Z., & Hu, J. -J. (2022). Transcriptome Analysis of Populus euphratica under Salt Treatment and PeERF1 Gene Enhances Salt Tolerance in Transgenic Populus alba × Populus glandulosa. International Journal of Molecular Sciences, 23(7), 3727. https://doi.org/10.3390/ijms23073727