
The Potential Role of Phenolic Acids from Salvia miltiorrhiza and Cynara scolymus and Their Derivatives as JAK Inhibitors: An In Silico Study


Abstract
:1. Introduction
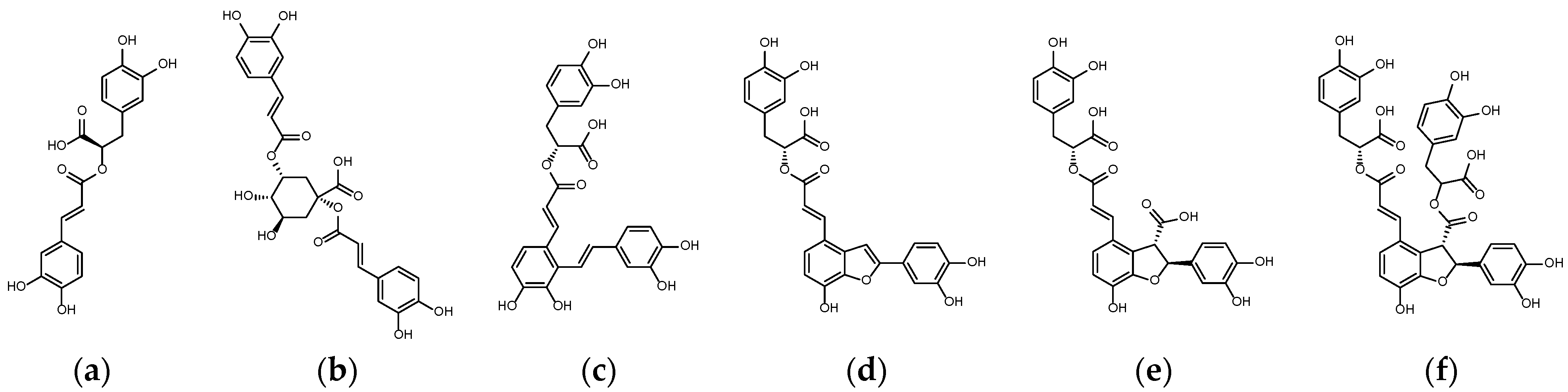
1.1. Natural Substances Involved in JAK-STAT Pathway
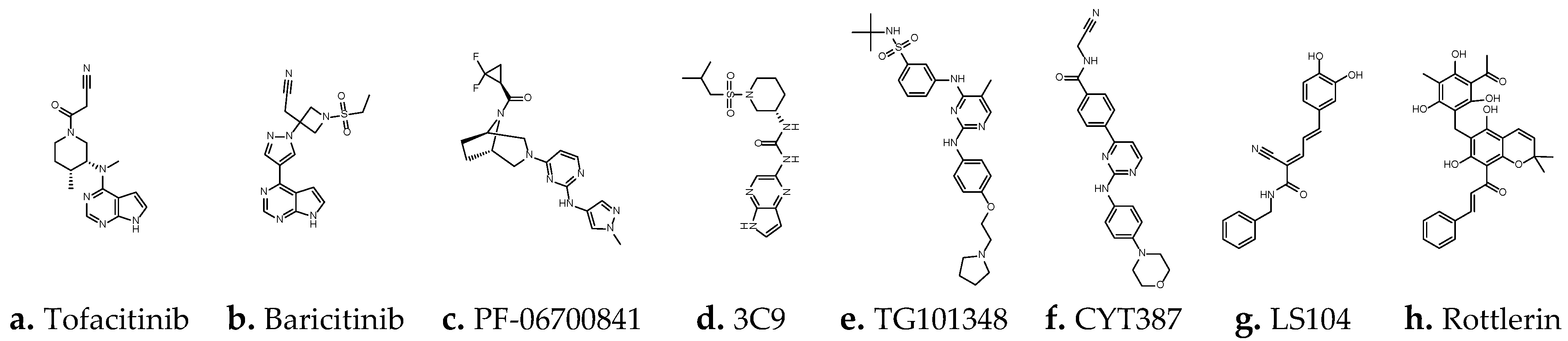
1.2. JAK Family and JAK Inhibitors
2. Results
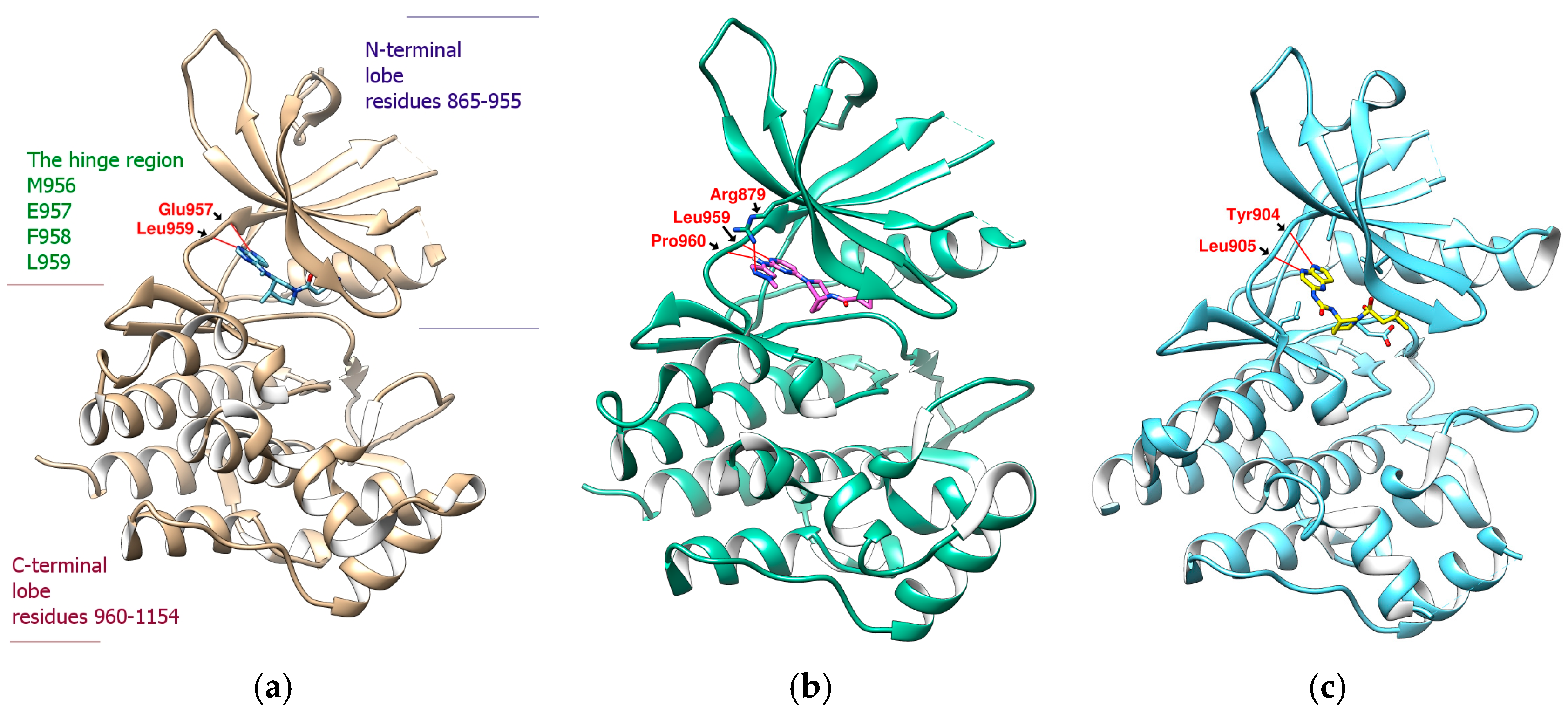
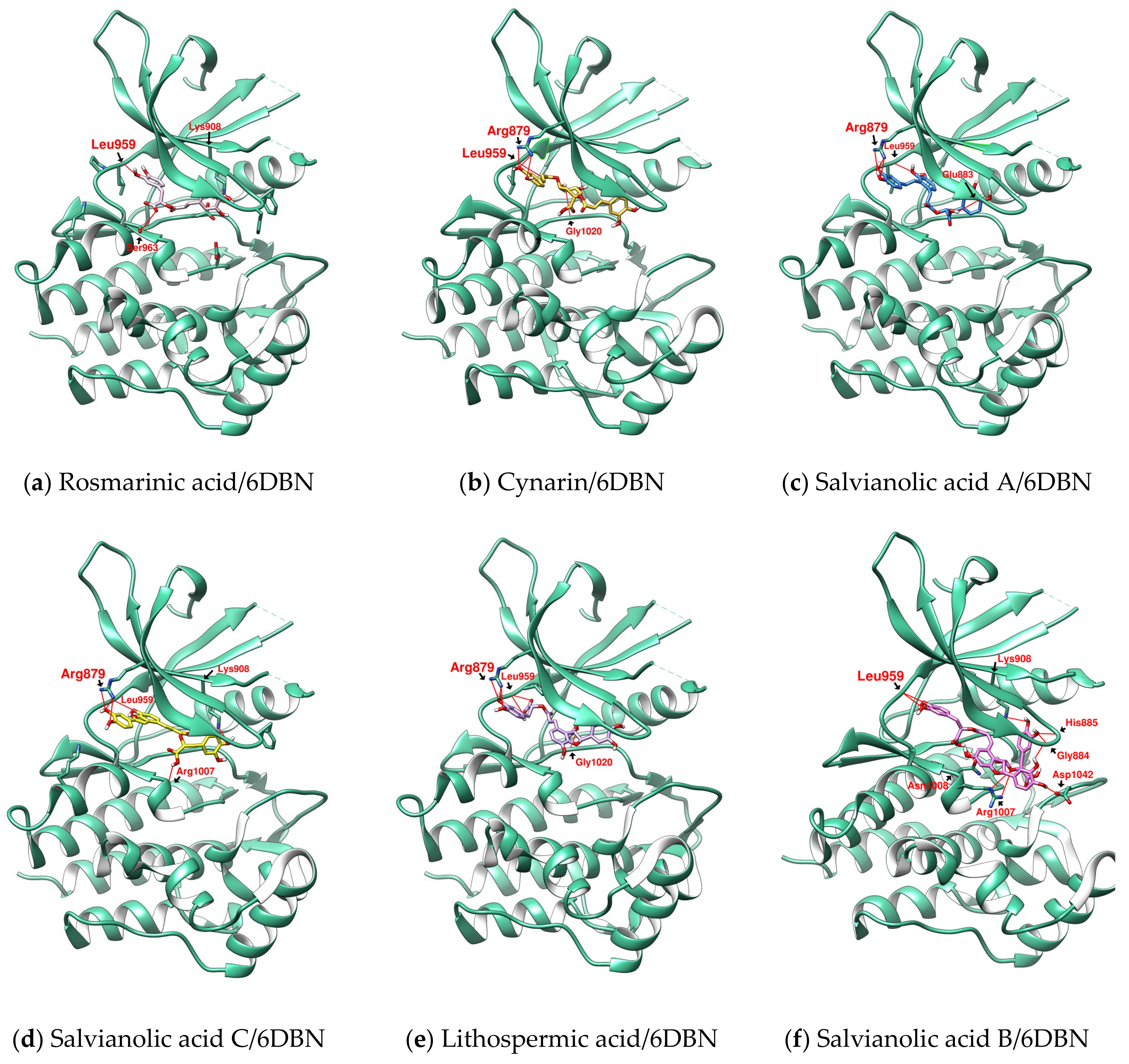
2.1. Binding Affinity and Configuration of Six Phenolic Acids from S. miltiorrhiza and Artichoke
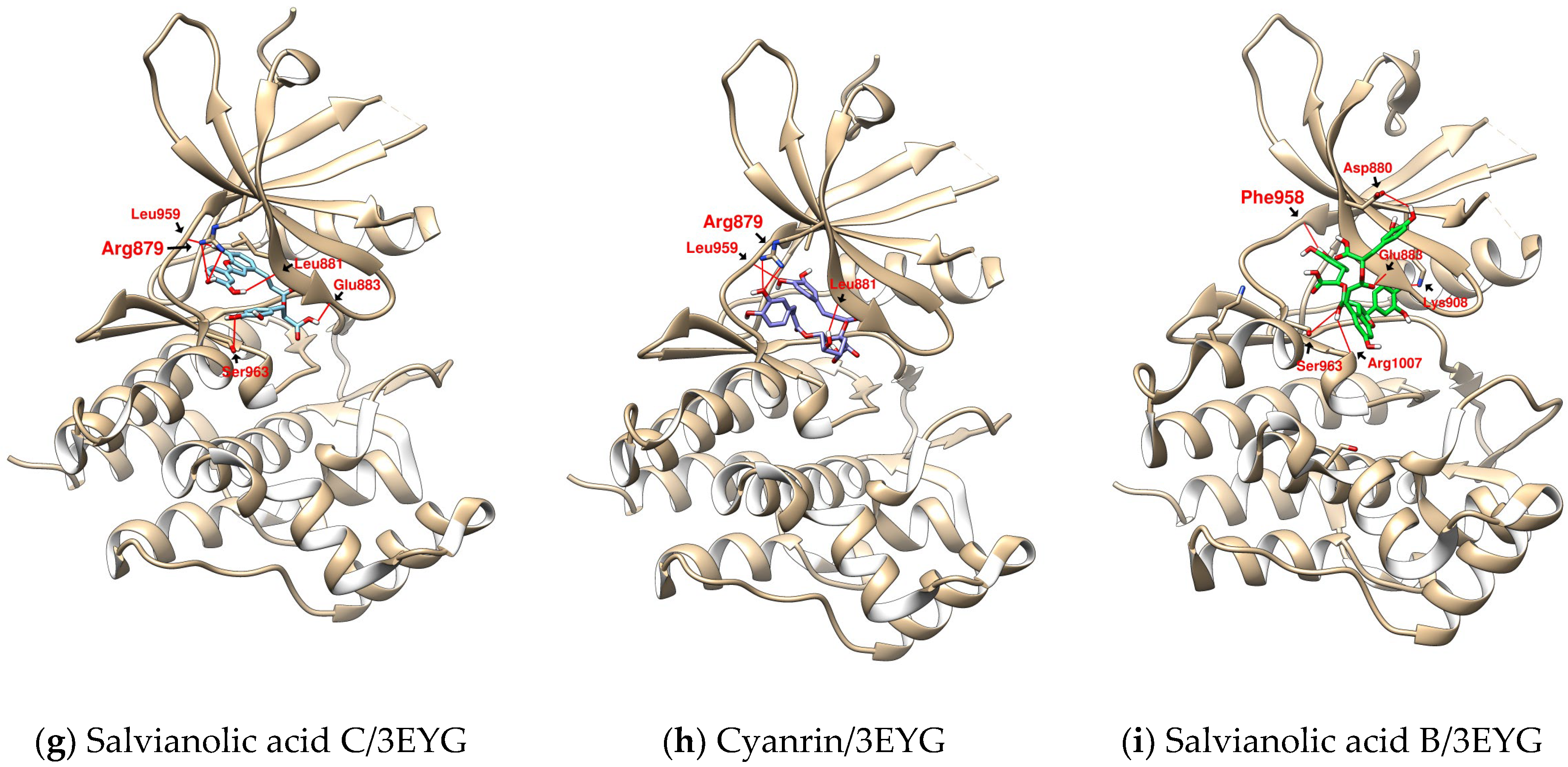
2.2. Further Analysis of the Characteristics of the Docking Configuration and Conformation
3. Discussion
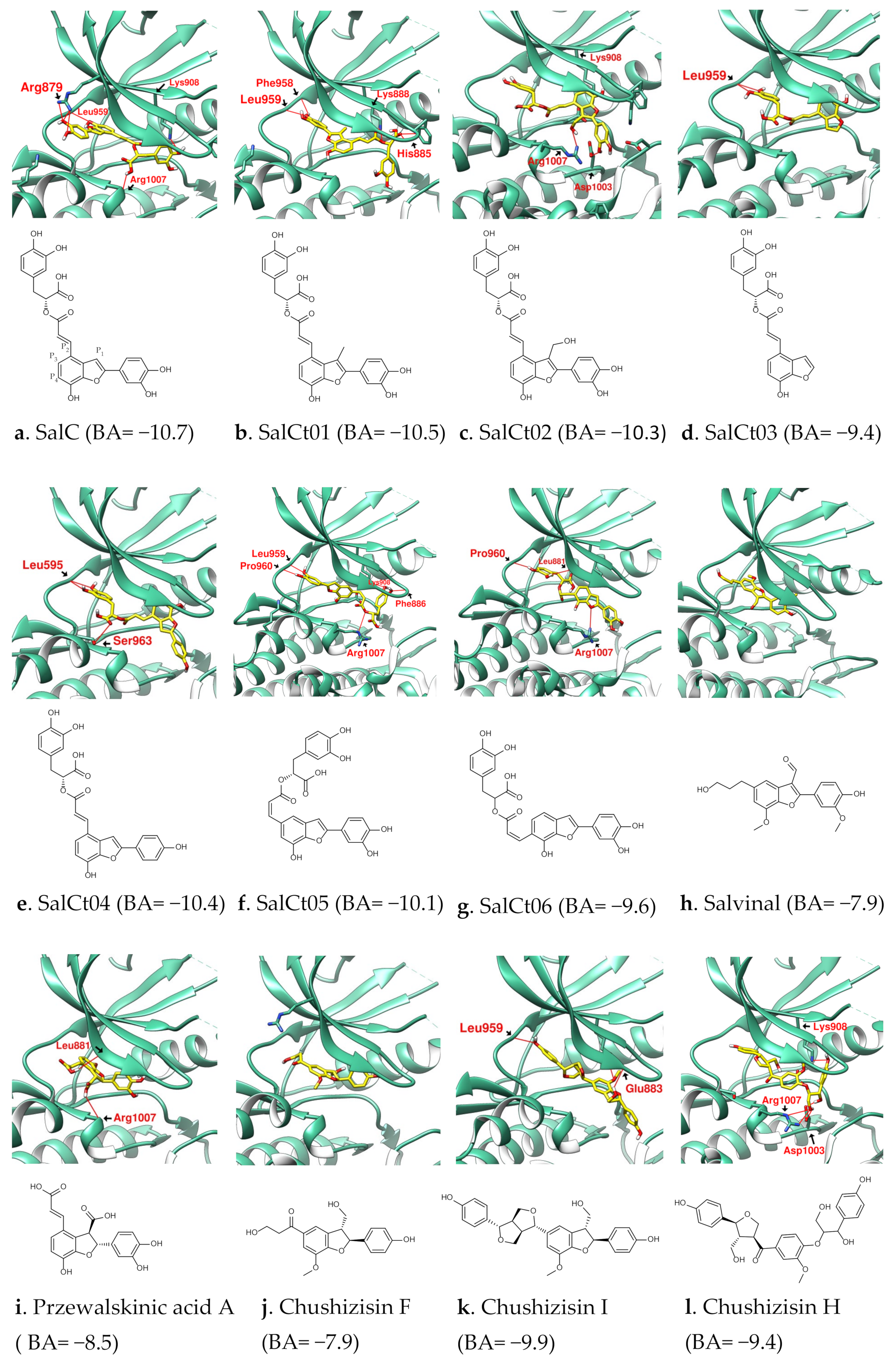
3.1. Analysis of the Structural Characteristics of SalC as a Potential JAK Inhibitor
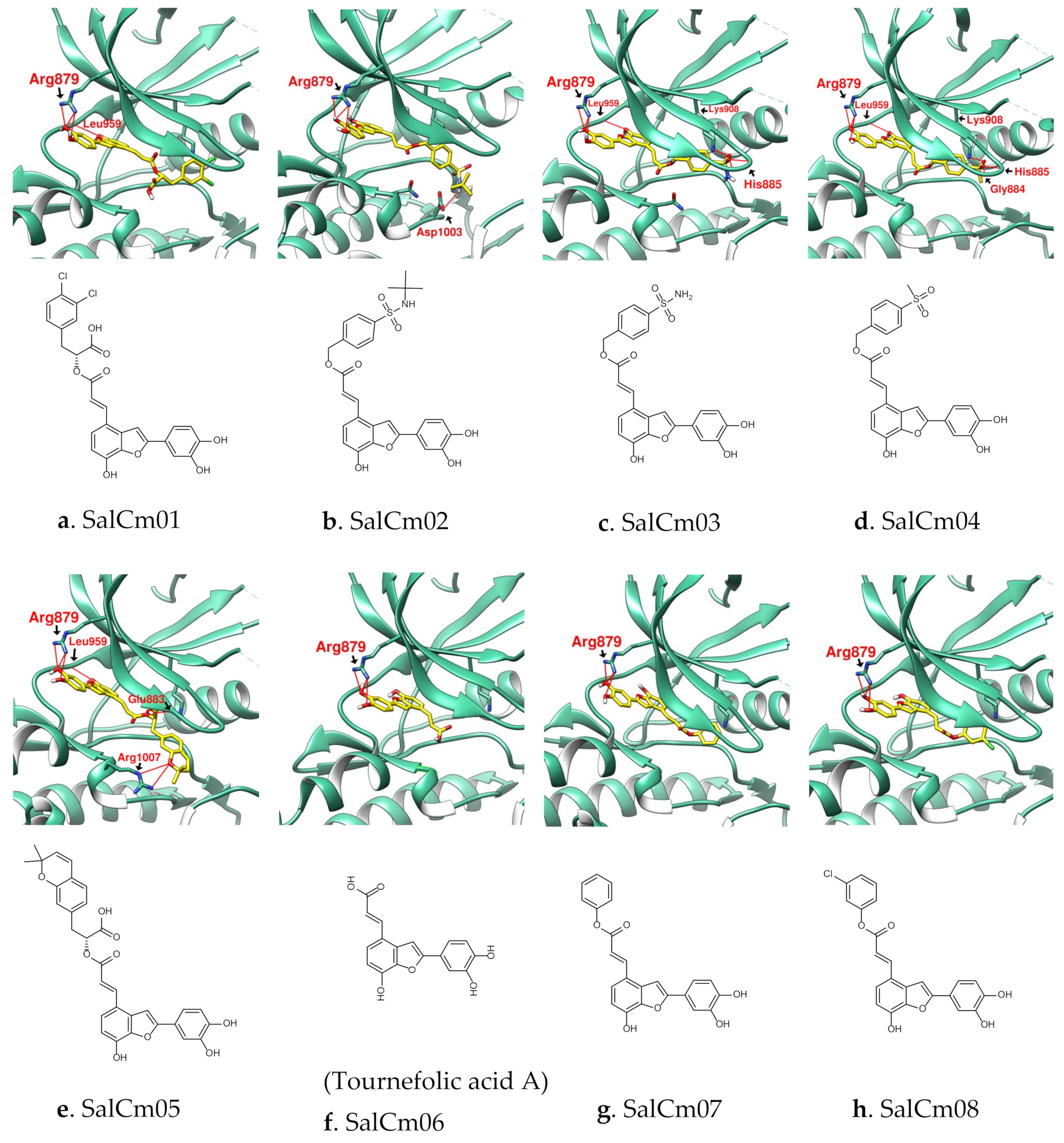
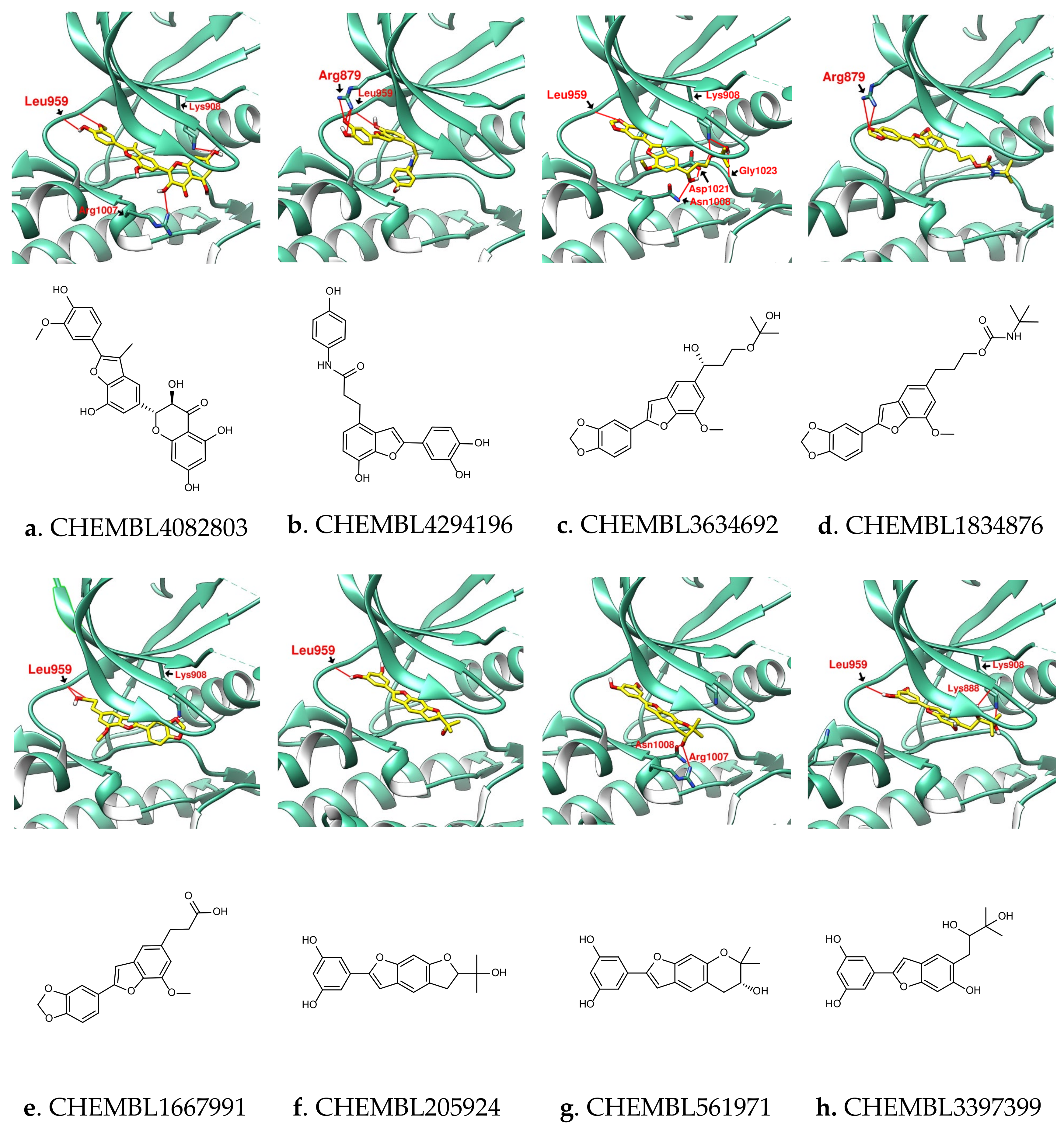
3.2. Design of SalC, RosA, CY and SalA Derivatives with Higher Binding Affinity
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kisseleva, T.; Bhattacharya, S.; Braunstein, J.; Schindler, C.W. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 2002, 285, 1–24. [Google Scholar] [CrossRef]
- Schwartz, D.M.; Bonelli, M.; Gadina, M.; O’Shea, J.J. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nat. Rev. Rheumatol. 2016, 12, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Xin, P.; Xu, X.; Deng, C.; Liu, S.; Wang, Y.; Zhou, X.; Ma, H.; Wei, D.; Sun, S. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int. Immunopharmacol. 2020, 80, 106210. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.; Mysler, E.; Hall, S.; Kivitz, A.J.; Moots, R.J.; Luo, Z.; DeMasi, R.; Soma, K.; Zhang, R.; Takiya, L.; et al. Efficacy and safety of tofacitinib monotherapy, tofacitinib with methotrexate, and adalimumab with methotrexate in patients with rheumatoid arthritis (ORAL Strategy): A phase 3b/4, double-blind, head-to-head, randomised controlled trial. Lancet 2017, 390, 457–468. [Google Scholar] [CrossRef]
- Gadina, M.; Johnson, C.; Schwartz, D.; Bonelli, M.; Hasni, S.; Kanno, Y.; Changelian, P.; Laurence, A.; O’Shea, J.J. Translational and clinical advances in JAK-STAT biology: The present and future of jakinibs. J. Leukoc. Biol. 2018, 104, 499–514. [Google Scholar] [CrossRef]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D.M. JAK-STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef]
- Bose, S.; Banerjee, S.; Mondal, A.; Chakraborty, U.; Pumarol, J.; Croley, C.R.; Bishayee, A. Targeting the JAK/STAT Signaling Pathway Using Phytocompounds for Cancer Prevention and Therapy. Cells 2020, 9, 1451. [Google Scholar] [CrossRef]
- Vadapalli, J.; Vanam, A.; Motohashi, N.; Gollapudi, R. Integrated in Silico Docking and MOMA Simulation Methods Reveal Rottlerin as a Potent Janus kinase 2 (JAK2) Inhibitor. Biomed. J. Sci. Tech. Res. 2018, 11, 8216–8225. [Google Scholar]
- Jiang, R.W.; Lau, K.M.; Hon, P.M.; Mak, T.C.; Woo, K.S.; Fung, K.P. Chemistry and biological activities of caffeic acid derivatives from Salvia miltiorrhiza. Curr. Med. Chem. 2005, 12, 237–246. [Google Scholar] [CrossRef]
- Zhou, L.; Zuo, Z.; Chow, M.S. Danshen: An overview of its chemistry, pharmacology, pharmacokinetics, and clinical use. J. Clin. Pharmacol. 2005, 45, 1345–1359. [Google Scholar] [CrossRef]
- Lattanzio, V.; Kroon, P.A.; Linsalata, V.; Cardinali, A. Globe artichoke: A functional food and source of nutraceutical ingredients. J. Funct. Foods 2009, 1, 131–144. [Google Scholar] [CrossRef]
- Topal, M.; Gocer, H.; Topal, F.; Kalin, P.; Köse, L.P.; Gülçin, İ.; Çakmak, K.C.; Küçük, M.; Durmaz, L.; Gören, A.C.; et al. Antioxidant, antiradical, and anticholinergic properties of cynarin purified from the Illyrian thistle (Onopordum illyricum L.). J. Enzyme Inhib. Med. Chem. 2016, 31, 266–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.M.; Xu, S.W.; Liu, P.Q. Salvia miltiorrhizaBurge (Danshen): A golden herbal medicine in cardiovascular therapeutics. Acta Pharmacol. Sin. 2018, 39, 802–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.S.; Luo, X.Y.; Jiang, H.; Xing, Y.; Yang, M.H.; Yuan, G.H.; Tang, Z.; Wang, H. Salvia miltiorrhiza injection restores apoptosis of fibroblast-like synoviocytes cultured with serum from patients with rheumatoid arthritis. Mol. Med. Rep. 2015, 11, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.Y.; Im, D.S. Anti-allergic effects of salvianolic acid A and tanshinone IIA from Salvia miltiorrhiza determined using in vivo and in vitro experiments. Int. Immunopharmacol. 2019, 67, 69–77. [Google Scholar] [CrossRef]
- Lin, Y.; Yan, Y.; Zhang, H.; Chen, Y.; He, Y.; Wang, S.; Fang, L.; Lv, Y.; Du, G. Salvianolic acid A alleviates renal injury in systemic lupus erythematosus induced by pristane in BALB/c mice. Acta Pharm. Sin. B 2017, 7, 159–166. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Zhang, W.; Wang, J.; Yang, H.; Zhao, X.; Zhou, Q.; Wang, H.; Li, L.; Du, G. Activation of Nrf2 signaling by salvianolic acid C attenuates NF-κB mediated inflammatory response both in vivo and in vitro. Int. Immunopharmacol. 2018, 63, 299–310. [Google Scholar] [CrossRef]
- Li, S.; Wang, R.; Song, F.; Chen, P.; Gu, Y.; Chen, C.; Yuan, Y. Salvianolic acid A suppresses CCl4-induced liver fibrosis through regulating the Nrf2/HO-1, NF-κB/IκBα, p38 MAPK, and JAK1/STAT3 signaling pathways. Drug Chem. Toxicol. 2022, 1–10. [Google Scholar] [CrossRef]
- Chen, S.C.; Lin, Y.L.; Huang, B.; Wang, D.L.; Cheng, J.J. Salvianolic acid B suppresses IFN-γ-induced JAK/STAT1 activation in endothelial cells. Thromb. Res. 2011, 128, 560–564. [Google Scholar] [CrossRef]
- Ben Salem, M.; Affes, H.; Ksouda, K.; Dhouibi, R.; Sahnoun, Z.; Hammami, S.; Zeghal, K.M. Pharmacological Studies of Artichoke Leaf Extract and Their Health Benefits. Plant Foods Hum. Nutr. 2015, 70, 441–453. [Google Scholar] [CrossRef]
- Hueza, I.M.; Gotardo, A.T.; da Silva Mattos, M.I.; Górniak, S.L. Immunomodulatory effect of Cynara scolymus (artichoke) in rats. Phytother. Res. 2019, 33, 167–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, G.C.; Chuang, P.H.; Chang, K.C.; Jan, P.S.; Hwang, P.I.; Wu, H.B.; Yi, M.; Zhou, H.X.; Chen, H.M. Blocking effect of an immuno-suppressive agent, cynarin, on CD28 of T-cell receptor. Pharm. Res. 2009, 26, 375–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandino, G.; Lombardo, S.; Mauromicale, G. Globe artichoke leaves and floral stems as a source of bioactive compounds. Ind. Crops Prod. 2013, 44, 44–49. [Google Scholar] [CrossRef]
- Zhou, Y.; Fu, X.; Guan, Y.; Gong, M.; He, K.; Huang, B. 1,3-Dicaffeoylquinic acid targeting 14-3-3 tau suppresses human breast cancer cell proliferation and metastasis through IL6/JAK2/PI3K pathway. Biochem. Pharmacol. 2020, 172, 113752. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.K.; Bamert, R.S.; Patel, O.; Wang, C.; Walden, P.M.; Wilks, A.F.; Fantino, E.; Rossjohn, J.; Lucet, I.S. Dissecting specificity in the Janus kinases: The structures of JAK-specific inhibitors complexed to the JAK1 and JAK2 protein tyrosine kinase domains. J. Mol. Biol. 2009, 387, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Fensome, A.; Ambler, C.M.; Arnold, E.; Banker, M.E.; Brown, M.F.; Chrencik, J.; Clark, J.D.; Dowty, M.E.; Efremov, I.V.; Flick, A.; et al. Dual Inhibition of TYK2 and JAK1 for the Treatment of Autoimmune Diseases: Discovery of ((S)-2,2-Difluorocyclopropyl)((1 R,5 S)-3-(2-((1-methyl-1 H-pyrazol-4-yl)amino)pyrimidin-4-yl)-3,8-diazabicyclo[3.2.1]octan-8-yl)methanone (PF-06700841). J. Med. Chem. 2018, 61, 8597–8612. [Google Scholar] [CrossRef] [PubMed]
- De Vicente, J.; Lemoine, R.; Bartlett, M.; Hermann, J.C.; Hekmat-Nejad, M.; Henningsen, R.; Jin, S.; Kuglstatter, A.; Li, H.; Lovey, A.J.; et al. Scaffold hopping towards potent and selective JAK3 inhibitors: Discovery of novel C-5 substituted pyrrolopyrazines. Bioorg. Med. Chem. Lett. 2014, 24, 4969–4975. [Google Scholar] [CrossRef]
- Mullally, A.; Hood, J.; Harrison, C.; Mesa, R. Fedratinib in myelofibrosis. Blood Adv. 2020, 4, 1792–1800. [Google Scholar] [CrossRef]
- Harrison, C.N.; Vannucchi, A.M.; Platzbecker, U.; Cervantes, F.; Gupta, V.; Lavie, D.; Passamonti, F.; Winton, E.F.; Dong, H.; Kawashima, J.; et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): A randomised, open-label, phase 3 trial. Lancet Haematol. 2018, 5, e73–e81. [Google Scholar] [CrossRef]
- Lipka, D.B.; Hoffmann, L.S.; Heidel, F.; Markova, B.; Blum, M.C.; Breitenbuecher, F.; Kasper, S.; Kindler, T.; Levine, R.L.; Huber, C.; et al. LS104, a non-ATP-competitive small-molecule inhibitor of JAK2, is potently inducing apoptosis in JAK2V617F-positive cells. Mol. Cancer Ther. 2008, 7, 1176–1184. [Google Scholar] [CrossRef] [Green Version]
- Haan, C.; Rolvering, C.; Raulf, F.; Kapp, M.; Drückes, P.; Thoma, G.; Behrmann, I.; Zerwes, H.-G. Jak1 has a dominant role over Jak3 in signal transduction through γc-containing cytokine receptors. Chem. Biol. 2011, 18, 314–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Han, J.; Li, H.-f.; Fan, L.; Liu, A.-h.; Guo, D.-a. Analysis on the stability of total phenolic acids and salvianolic acid B from Salvia miltiorrhiza by HPLC and HPLC-MSn. Nat. Prod. Commun. 2008, 3, 669–679. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.N.; Zhang, X.; Xu, W.Z.; Ma, X.N.; Jia, Z.; Zheng, Y.M.; You, S. Studies on the stability of salvianolic acid B as potential drug material. Phytochem. Anal. 2011, 22, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Simon, J.E.; Aviles, I.F.; He, K.; Zheng, Q.Y.; Tadmor, Y. Analysis of antioxidative phenolic compounds in artichoke (Cynara scolymus L.). J. Agric. Food Chem. 2003, 51, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Zhang, L.; Sun, Z.B.; Cheng, X.H.; Zhang, Y.; Zou, H.B. Salvia miltiorrhiza prevents deep vein thrombosis via antioxidative effects in endothelial cells. Mol. Med. Rep. 2015, 11, 3593–3600. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Gao, Z.G.; Wu, Y.; Stevens, R.C.; Jacobson, K.A.; Zhao, S. Salvianolic acids from antithrombotic Traditional Chinese Medicine Danshen are antagonists of human P2Y(1) and P2Y(12) receptors. Sci. Rep. 2018, 8, 8084. [Google Scholar] [CrossRef]
- Verden, A.; Dimbil, M.; Kyle, R.; Overstreet, B.; Hoffman, K.B. Analysis of Spontaneous Postmarket Case Reports Submitted to the FDA Regarding Thromboembolic Adverse Events and JAK Inhibitors. Drug Saf. 2018, 41, 357–361. [Google Scholar] [CrossRef]
- Taylor, P.C.; Weinblatt, M.E.; Burmester, G.R.; Rooney, T.P.; Witt, S.; Walls, C.D.; Issa, M.; Salinas, C.A.; Saifan, C.; Zhang, X.; et al. Cardiovascular Safety During Treatment With Baricitinib in Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 1042–1055. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.; Wu, M.; Zhang, Y.; Zhao, X.; Yu, J.; Zheng, X. The structure-dependent self-association of five phenolic acids in aqueous solution. Magn. Reson. Chem. 2014, 52, 460–466. [Google Scholar] [CrossRef]
- Zhao, G.R.; Zhang, H.M.; Ye, T.X.; Xiang, Z.J.; Yuan, Y.J.; Guo, Z.X.; Zhao, L.B. Characterization of the radical scavenging and antioxidant activities of danshensu and salvianolic acid B. Food Chem. Toxicol. 2008, 46, 73–81. [Google Scholar] [CrossRef]
- XD, M.E.; Cao, Y.F.; Che, Y.Y.; Li, J.; Shang, Z.P.; Zhao, W.J.; Qiao, Y.J.; Zhang, J.Y. Danshen: A phytochemical and pharmacological overview. Chin. J. Nat. Med. 2019, 17, 59–80. [Google Scholar] [CrossRef]
- Mei, R.Q.; Wang, Y.H.; Du, G.H.; Liu, G.M.; Zhang, L.; Cheng, Y.X. Antioxidant Lignans from the Fruits of Broussonetia papyrifera. J. Nat. Prod. 2009, 72, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Radadiya, A.; Shah, A. Bioactive benzofuran derivatives: An insight on lead developments, radioligands and advances of the last decade. Eur. J. Med. Chem. 2015, 97, 356–376. [Google Scholar] [CrossRef]
- Miao, Y.-h.; Hu, Y.-h.; Yang, J.; Liu, T.; Sun, J.; Wang, X.-j. Natural source, bioactivity and synthesis of benzofuran derivatives. RSC Adv. 2019, 9, 27510–27540. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.-Q.; Wilkinson, B. Drug discovery beyond the ‘rule-of-five’. Curr. Opin. Biotechnol. 2007, 18, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.-D.; Zhang, G.-P.; Leib, M.; Hu, L.-H. First total synthesis of salvianolic acid C, tournefolic acid A, and tournefolal. Arkivoc 2012, 6, 204–213. [Google Scholar] [CrossRef] [Green Version]
- Zoete, V.; Daina, A.; Bovigny, C.; Michielin, O. SwissSimilarity: A Web Tool for Low to Ultra High Throughput Ligand-Based Virtual Screening. J. Chem. Inf. Model. 2016, 56, 1399–1404. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Martin, Y.C. A bioavailability score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Shapovalov, M.V.; Dunbrack, R.L., Jr. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011, 19, 844–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| JAK1/JAK2 (PDB:3EYG) | JAK1 (PDB:6DBN) | JAK3 (PDB:4QT1) | |
|---|---|---|---|
| Rosmarinic acid | −8.7 | −8.8 | −9.0 |
| Cynarin | −9.6 | −9.7 | −9.8 |
| Salvianolic acid A | −9.7 | −9.8 | −9.9 |
| Salvianolic acid C | −10.7 | −10.7 | −11.0 |
| Lithospermic acid | −9.4 | −10.0 | −10.1 |
| Salvianolic acid B | −9.6 | −10.3 | −10.2 |
| Ligand | Docking Receptor | Closest Distance to Met 956 | Hydrogen Bond the Hinge Region (Length: Å) | Hydrogen Bond N- and C-Terminal Lobes (Length: Å) | Hydrophobic Contacts | π-Interactions (π-Cation and π-π Stacking) |
|---|---|---|---|---|---|---|
| RosA | 6DBN | 4.324 | L959 (3.11) | K908 (3.912) | V889 | K908 |
| S963 (3.759) | L1010 | (π-cation) | ||||
| CY | 6DBN | 2.952 | L959 (3.829) | R879 (2.955) | L881 | K908 |
| G1020 (2.662) | G962 | (π-cation) | ||||
| SalA | 6DBN | 3.716 | L959 (3.391) | R879 (2.979) | L881 | - |
| E883 (2.009) | G962 | |||||
| SalC | 6DBN | 3.77 | L959 (2.782) | R879 (3.074) | L881 | K908 |
| R1007 (2.060) | G884 | (π-cation) | ||||
| K908 (3.300) | G962 | |||||
| L1010 | ||||||
| LSA | 6DBN | 4.179 | L959 (3.074) | R879 (2.995) | L881 | K908 |
| G1020 (2.182) | G962 | (π-cation) | ||||
| SalB | 6DBN | 4.187 | L959 (2.341) | G884 (2.328) | V889 | F886 (π-π) |
| H885 (2.080) | L1010 | |||||
| K908 (3.379) | ||||||
| R1007 (3.621) | ||||||
| N1008 (3.077) | ||||||
| D1042 (1.966) | ||||||
| SalC | 3EYG | 3.849 | L959 (3.050) | R879 (3.943) | L1010 | - |
| L881 (2.068) | A906 | |||||
| E883 (2.359) | L881 | |||||
| S963 (3.672) | V889 | |||||
| G962 | ||||||
| CY | 3EYG | 2.828 | L959 (2.967) | R879 (3.052) | L1010 | - |
| L881 (2.704) | V889 | |||||
| G962 | ||||||
| SalB | 3EYG | 4.237 | F958 (2.490) | K908 (3.158) | V889 | - |
| S963 (3.799) | L1010 | |||||
| R1007 (2.512) | G882 | |||||
| D880 (3.056) | ||||||
| E883 (3.086) |
| Name | SalC | SalCm01 | SalCm02 | SalCm03 | SalCm04 | SalCm05 | SalCm06 | SalCm07 | SalCm08 |
|---|---|---|---|---|---|---|---|---|---|
| Binding score | −10.7 | −11.1 | −10.7 | −11.3 | −11.6 | −12.2 | −9.4 | −10.5 | −10.6 |
| MW (g/mol) | 492.43 | 529.32 | 537.58 | 481.47 | 480.49 | 542.53 | 312.27 | 388.37 | 422.81 |
| GI absorption | Low | Low | Low | Low | Low | Low | High | High | High |
| Lipinski’s rule violation | ④ | ① | ① | - | - | ① | - | - | - |
| Bioavailability score | 0.11 | 0.56 | 0.55 | 0.55 | 0.55 | 0.56 | 0.56 | 0.55 | 0.55 |
| Name | CHEMB-L4082803 | CHEMB- L4294196 | CHEMB- L3634692 | CHEMB- L1834876 | CHEMB-L1667991 | CHEMB- L205924 | CHEMB- L561971 | CHEMB-L3397399 |
|---|---|---|---|---|---|---|---|---|
| Binding score | −11.2 | −9.9 | −9.4 | −9.3 | −9.0 | −9.4 | −9.4 | −9.4 |
| MW (g/mol) | 464.42 | 405.40 | 400.42 | 425.47 | 340.33 | 326.34 | 326.34 | 344.36 |
| GI absorption | Low | High | High | High | High | High | High | High |
| Lipinski’s rule violation | - | - | - | - | - | - | - | - |
| Bioavailability score | 0.55 | 0.55 | 0.55 | 0.55 | 0.56 | 0.55 | 0.55 | 0.55 |
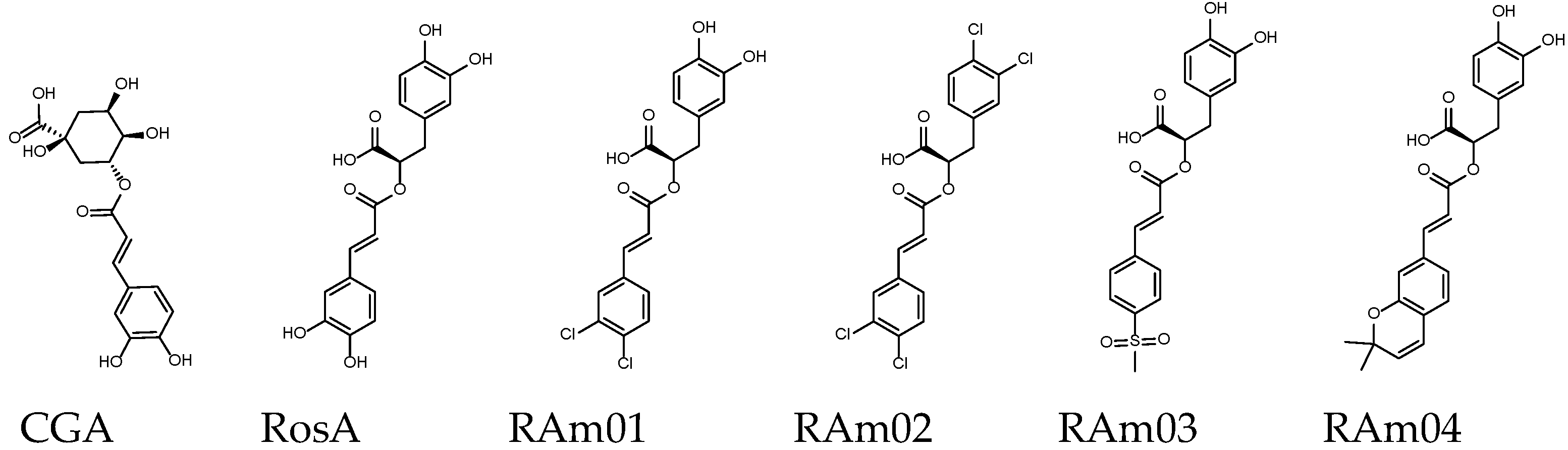
| Name | CGA | RosA | RAm01 | RAm02 | RAm03 | RAm04 |
|---|---|---|---|---|---|---|
| Binding score 1 | −8.8 | −8.8 | −8.9 | −8.9 | −9.3 | −10.0 |
| MW (g/mol) | 354.31 | 360.31 | 397.21 | 434.10 | 443.30 | 410.42 |
| GI absorption [48] | Low | Low | High | High | High | High |
| Lipinski’s rule violation 2 | ④ | - | - | ② | - | - |
| Bioavailability score [49] | 0.11 | 0.56 | 0.56 | 0.85 | 0.56 | 0.56 |
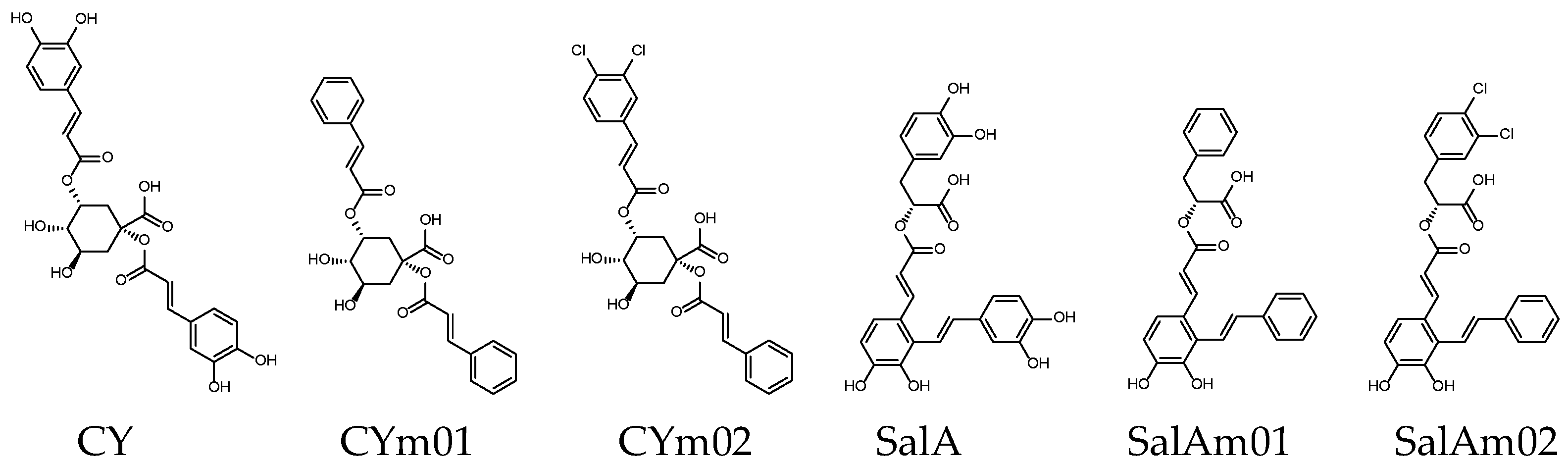
| Name | CY | CYm01 | CYm02 | SalA | SalAm01 | SalAm02 |
|---|---|---|---|---|---|---|
| Binding score 1 | −9.7 | −9.2 | −9.7 | −9.8 | −9.3 | −9.7 |
| MW (g/mol) | 516.45 | 452.45 | 521.34 | 494.45 | 430.45 | 499.34 |
| GI absorption [48] | Low | High | High | Low | High | High |
| Lipinski’s rule violation 2 | ①③④ | - | ① | ④ | - | - |
| Bioavailability score [49] | 0.11 | 0.56 | 0.56 | 0.11 | 0.56 | 0.56 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, H.-J.; Tzen, J.T.C. The Potential Role of Phenolic Acids from Salvia miltiorrhiza and Cynara scolymus and Their Derivatives as JAK Inhibitors: An In Silico Study. Int. J. Mol. Sci. 2022, 23, 4033. https://doi.org/10.3390/ijms23074033
Liao H-J, Tzen JTC. The Potential Role of Phenolic Acids from Salvia miltiorrhiza and Cynara scolymus and Their Derivatives as JAK Inhibitors: An In Silico Study. International Journal of Molecular Sciences. 2022; 23(7):4033. https://doi.org/10.3390/ijms23074033
Chicago/Turabian StyleLiao, Hui-Jun, and Jason T. C. Tzen. 2022. "The Potential Role of Phenolic Acids from Salvia miltiorrhiza and Cynara scolymus and Their Derivatives as JAK Inhibitors: An In Silico Study" International Journal of Molecular Sciences 23, no. 7: 4033. https://doi.org/10.3390/ijms23074033
APA StyleLiao, H. -J., & Tzen, J. T. C. (2022). The Potential Role of Phenolic Acids from Salvia miltiorrhiza and Cynara scolymus and Their Derivatives as JAK Inhibitors: An In Silico Study. International Journal of Molecular Sciences, 23(7), 4033. https://doi.org/10.3390/ijms23074033