
ASTool: An Easy-to-Use Tool to Accurately Identify Alternative Splicing Events from Plant RNA-Seq Data


Abstract
:1. Introduction
2. Results and Discussion
2.1. ASTool Can Accurately Identify AS Events
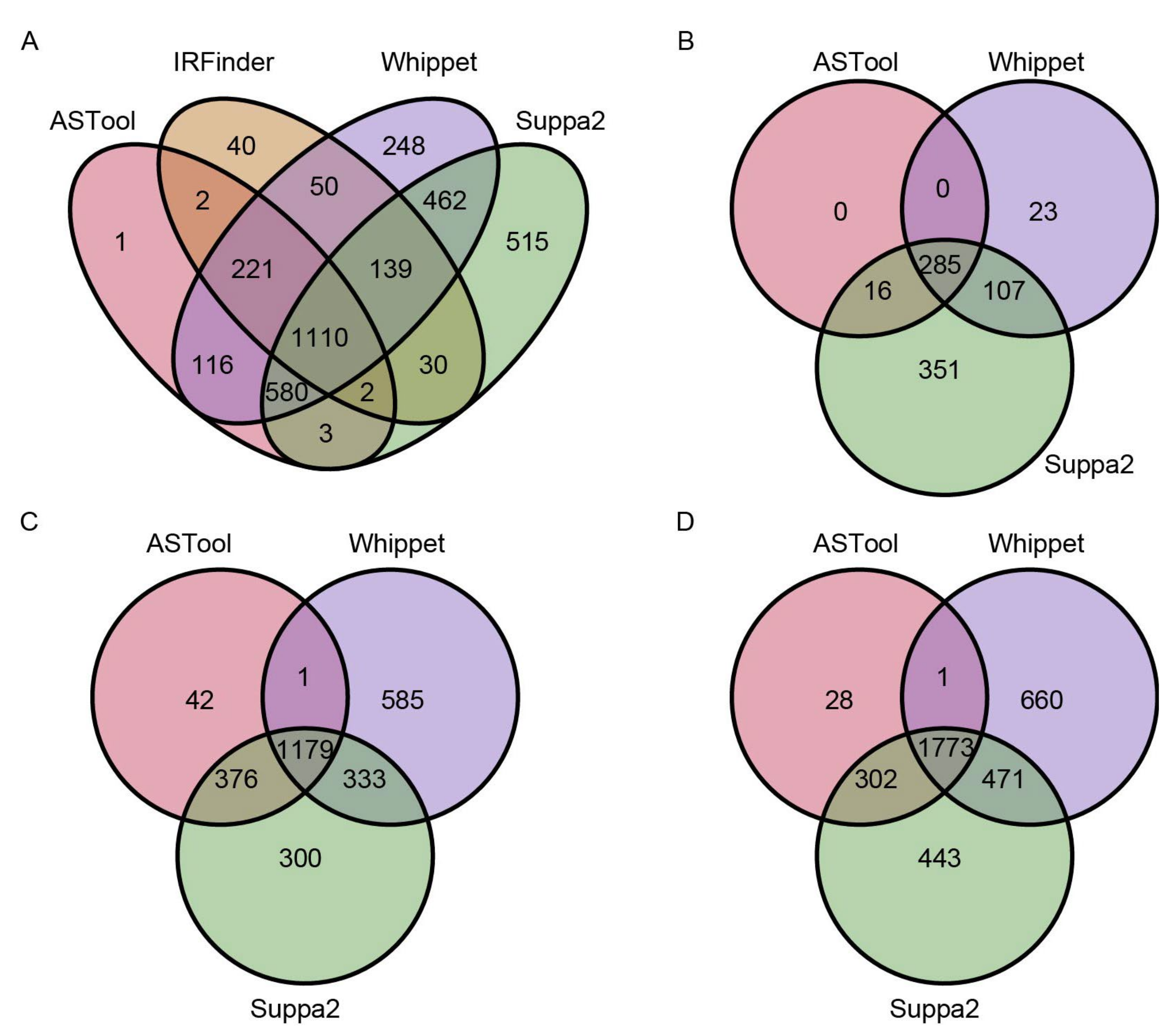
2.2. AS Events Detected by ASTool Are Consistent with Other Tools
2.3. ASTool Can Detect and Visualize Novel IR Events
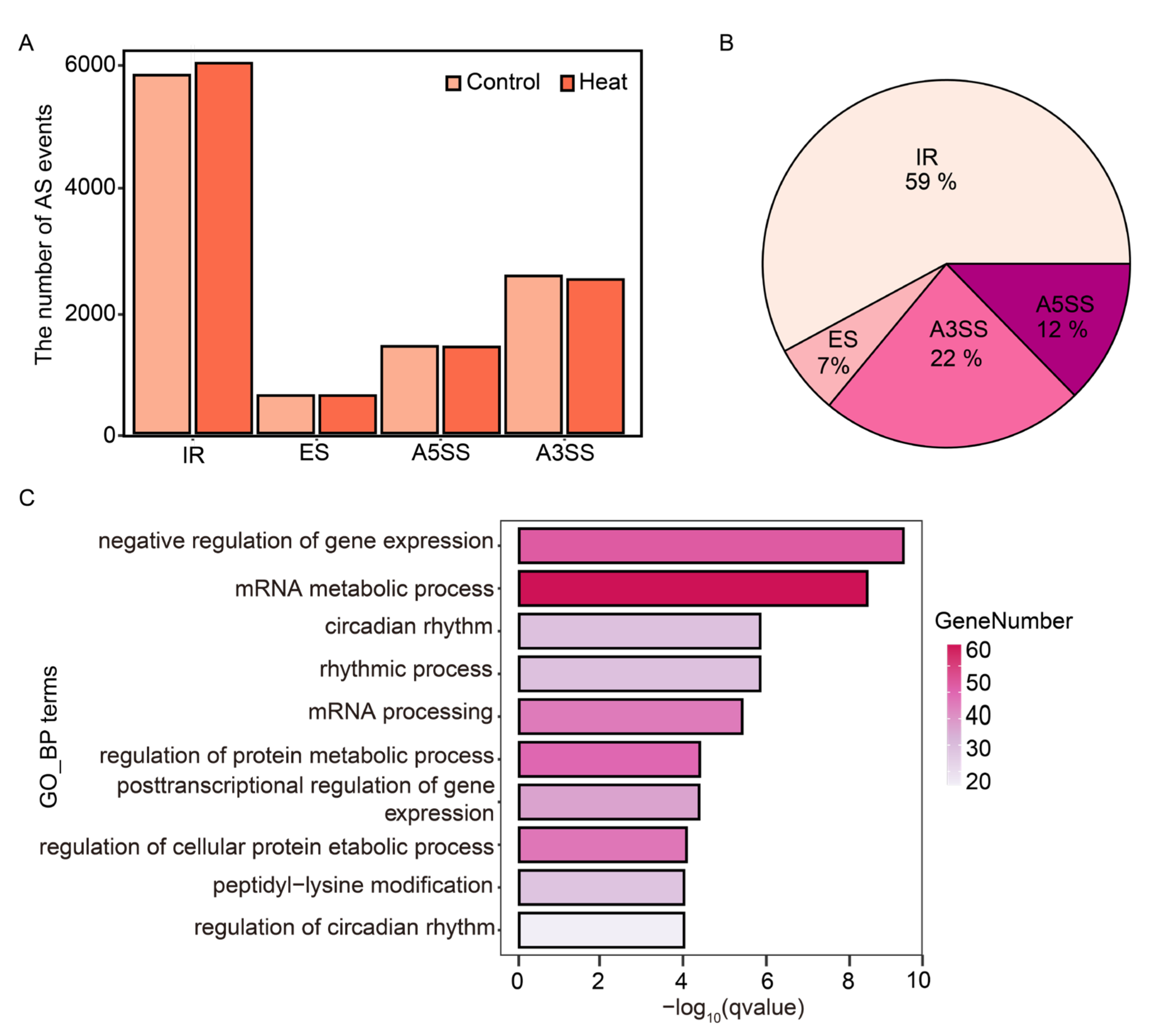
2.4. Using ASTool to Analyze Arabidopsis AS Events in Response to Heat Stress
3. Methods
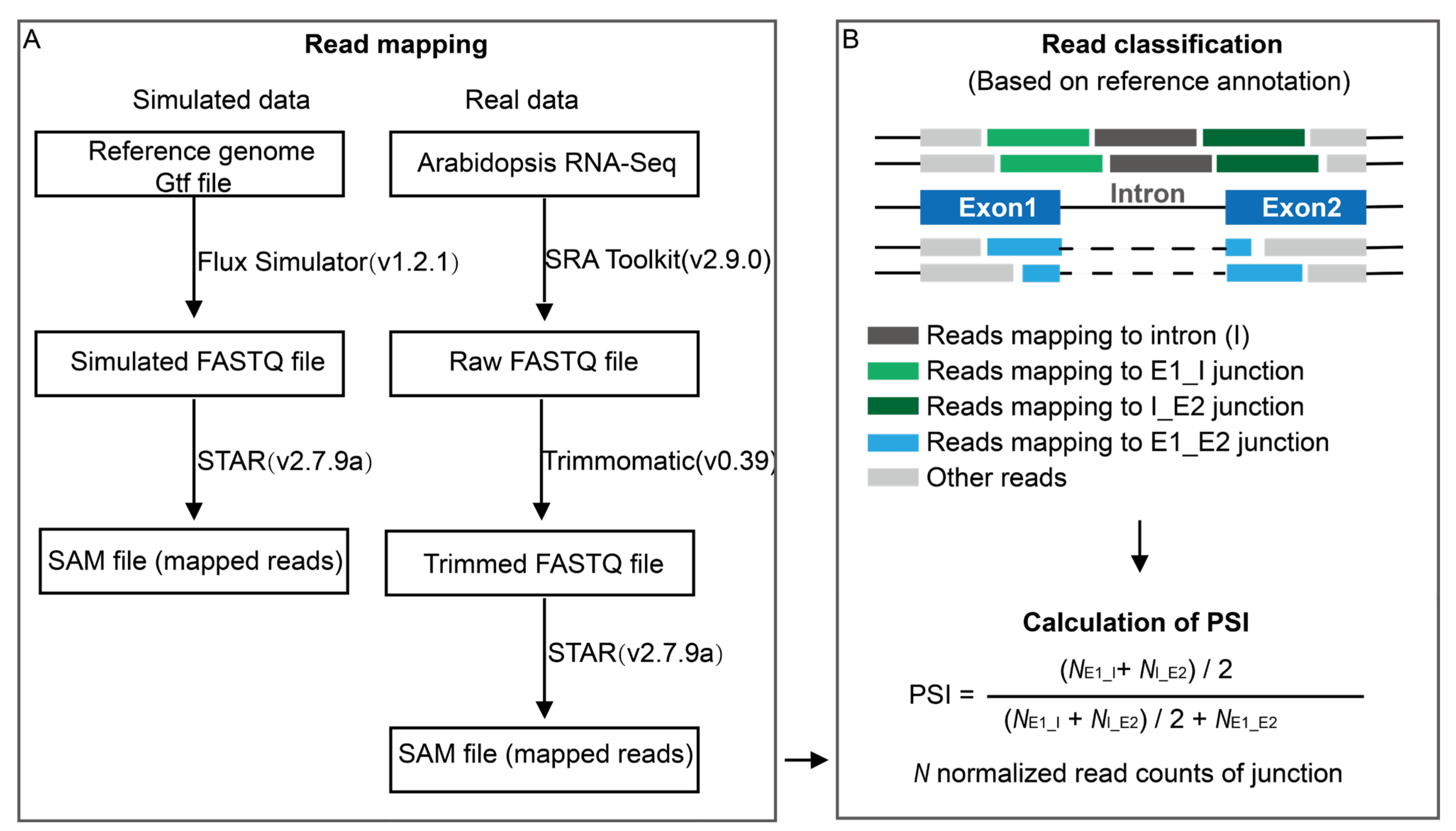
3.1. Simulated RNA-Seq Data and Pre-Processing
3.2. Real RNA-Seq Data and Pre-Processing
3.3. Estimation of PSI
3.4. Comparison with Existing Tools
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Yamada, M.; Han, X.; Ohler, U.; Benfey, P.N. High-resolution expression map of the Arabidopsis root reveals alternative splicing and lincRNA regulation. Dev. Cell 2016, 39, 508–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Haak, D.C.; Fukao, T.; Grene, R.; Hua, Z.; Ivanov, R.; Perrella, G.; Li, S. Multilevel regulation of abiotic stress responses in plants. Front. Plant Sci. 2017, 8, 1564. [Google Scholar] [CrossRef] [PubMed]
- Staiger, D.; Brown, J.W. Alternative splicing at the intersection of biological timing, development, and stress responses. Plant Cell 2013, 25, 3640–3656. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Hou, H.; Song, H.; Lin, K.; Zhang, Z.; Hu, J.; Pang, E. The comparison of alternative splicing among the multiple tissues in cucumber. BMC Plant Biol. 2018, 18, 5. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Li, P.; Jin, G.; Gui, D.; Liu, L.; Zhang, C. Temporal regulation of alternative splicing events in rice memory under drought stress. Plant Divers 2022, 44, 116–125. [Google Scholar] [CrossRef]
- Dong, C.; He, F.; Berkowitz, O.; Liu, J.; Cao, P.; Tang, M.; Shi, H.; Wang, W.; Li, Q.; Shen, Z.; et al. Alternative splicing plays a critical role in maintaining mineral nutrient homeostasis in Rice (Oryza sativa). Plant Cell 2018, 30, 2267–2285. [Google Scholar] [CrossRef] [Green Version]
- Mandadi, K.K.; Scholthof, K.B. Genome-wide analysis of alternative splicing landscapes modulated during plant-virus interactions in Brachypodium distachyon. Plant Cell 2015, 27, 71–85. [Google Scholar] [CrossRef] [Green Version]
- Calixto, C.P.G.; Guo, W.; James, A.B.; Tzioutziou, N.A.; Entizne, J.C.; Panter, P.E.; Knight, H.; Nimmo, H.G.; Zhang, R.; Brown, J.W.S. Rapid and dynamic alternative splicing impacts the Arabidopsis cold response transcriptome. Plant Cell 2018, 30, 1424–1444. [Google Scholar] [CrossRef] [Green Version]
- Vitoriano, C.B.; Calixto, C.P.G. Reading between the Lines: RNA-seq data mining reveals the alternative message of the Rice leaf transcriptome in response to heat stress. Plants 2021, 10, 1647. [Google Scholar] [CrossRef]
- Cecchini, N.M.; Torres, J.R.; Lopez, I.L.; Cobo, S.; Nota, F.; Alvarez, M.E. Alternative splicing of an exitron determines the subnuclear localization of the Arabidopsis DNA glycosylase MBD4L under heat stress. Plant J. 2022. [Google Scholar] [CrossRef]
- Martin, G.; Marquez, Y.; Mantica, F.; Duque, P.; Irimia, M. Alternative splicing landscapes in Arabidopsis thaliana across tissues and stress conditions highlight major functional differences with animals. Genome Biol. 2021, 22, 35. [Google Scholar] [CrossRef]
- Syed, N.H.; Kalyna, M.; Marquez, Y.; Barta, A.; Brown, J.W. Alternative splicing in plants—Coming of age. Trends Plant Sci. 2012, 17, 616–623. [Google Scholar] [CrossRef] [Green Version]
- Keren, H.; Lev-Maor, G.; Ast, G. Alternative splicing and evolution: Diversification, exon definition and function. Nat. Rev. Genet. 2010, 11, 345–355. [Google Scholar] [CrossRef]
- Marquez, Y.; Brown, J.W.; Simpson, C.; Barta, A.; Kalyna, M. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res. 2012, 22, 1184–1195. [Google Scholar] [CrossRef] [Green Version]
- John, S.; Olas, J.J.; Mueller-Roeber, B. Regulation of alternative splicing in response to temperature variation in plants. J. Exp. Bot. 2021, 72, 6150–6163. [Google Scholar] [CrossRef]
- Ganie, S.A.; Reddy, A.S.N. Stress-induced changes in alternative splicing landscape in Rice: Functional significance of splice isoforms in stress tolerance. Biology 2021, 10, 309. [Google Scholar] [CrossRef]
- Reddy, A.S.; Marquez, Y.; Kalyna, M.; Barta, A. Complexity of the alternative splicing landscape in plants. Plant Cell 2013, 25, 3657–3683. [Google Scholar] [CrossRef] [Green Version]
- Jacob, A.G.; Smith, C.W.J. Intron retention as a component of regulated gene expression programs. Hum. Genet 2017, 136, 1043–1057. [Google Scholar] [CrossRef] [Green Version]
- Katz, Y.; Wang, E.T.; Airoldi, E.M.; Burge, C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 2010, 7, 1009–1015. [Google Scholar] [CrossRef]
- Shen, S.; Park, J.W.; Lu, Z.X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [Green Version]
- Middleton, R.; Gao, D.; Thomas, A.; Singh, B.; Au, A.; Wong, J.J.; Bomane, A.; Cosson, B.; Eyras, E.; Rasko, J.E.; et al. IRFinder: Assessing the impact of intron retention on mammalian gene expression. Genome Biol. 2017, 18, 51. [Google Scholar] [CrossRef] [Green Version]
- Sterne-Weiler, T.; Weatheritt, R.J.; Best, A.J.; Ha, K.C.H.; Blencowe, B.J. Efficient and accurate quantitative profiling of alternative splicing patterns of any complexity on a laptop. Mol. Cell 2018, 72, 187–200. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Trincado, J.L.; Entizne, J.C.; Hysenaj, G.; Singh, B.; Skalic, M.; Elliott, D.J.; Eyras, E. SUPPA2: Fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018, 19, 40. [Google Scholar] [CrossRef] [Green Version]
- Kimura, Y.; Takeoka, Y.; Inoue, M.; Maeda, M.; Fujiyama, K. Double-knockout of putative endo-beta-N-acetylglucosaminidase (ENGase) genes in Arabidopsis thaliana: Loss of ENGase activity induced accumulation of high-mannose type free N-glycans bearing N,N′-acetylchitobiosyl unit. Biosci. Biotechnol. Biochem. 2011, 75, 1019–1021. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Pandey, S.; Assmann, S.M. Arabidopsis extra-large G proteins (XLGs) regulate root morphogenesis. Plant J. 2008, 53, 248–263. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Davis, A.M.; Ronald, J.; Ma, Z.; Wilkinson, A.J.; Philippou, K.; Shindo, T.; Queitsch, C.; Davis, S.J. HSP90 Contributes to entrainment of the Arabidopsis circadian clock via the morning loop. Genetics 2018, 210, 1383–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griebel, T.; Zacher, B.; Ribeca, P.; Raineri, E.; Lacroix, V.; Guigo, R.; Sammeth, M. Modelling and simulating generic RNA-Seq experiments with the flux simulator. Nucleic Acids Res. 2012, 40, 10073–10083. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Achuthan, P.; Akanni, W.; Allen, J.; Amode, M.R.; Armean, I.M.; Bennett, R.; Bhai, J.; Billis, K.; Boddu, S.; et al. Ensembl 2019. Nucleic Acids Res. 2019, 47, D745–D751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.S.; Yang, H.; Ding, L.; Song, Z.T.; Ma, H.; Chang, F.; Liu, J.X. Tissue-specific transcriptomics reveals an important role of the unfolded protein response in maintaining fertility upon heat stress in Arabidopsis. Plant Cell 2017, 29, 1007–1023. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Leinonen, R.; Sugawara, H.; Shumway, M.; International Nucleotide Sequence Database Collaboration. The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Braunschweig, U.; Barbosa-Morais, N.L.; Pan, Q.; Nachman, E.N.; Alipanahi, B.; Gonatopoulos-Pournatzis, T.; Frey, B.; Irimia, M.; Blencowe, B.J. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res. 2014, 24, 1774–1786. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tools | Single Thread (min) | Ten Threads (min) |
|---|---|---|
| ASTool | 160.4 | 23.5 |
| Whippet | 32.0 | NA a |
| IRFinder | 154.4 | 31.3 |
| Suppa2 | 16.1 | 15.4 |
| Classification | Tools | Detection of Four Main AS Types | Detection of Non-Strict AS Events | Detection of Novel IR Events |
|---|---|---|---|---|
| Exon-based | ASTool | √ | √ | √ |
| Whippet | √ | √ | × | |
| IRFinder | × | √ | √ | |
| Transcript-based | Suppa2 | √ | × | × |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, H.; Guo, X.; Wang, T.; Zhang, Z. ASTool: An Easy-to-Use Tool to Accurately Identify Alternative Splicing Events from Plant RNA-Seq Data. Int. J. Mol. Sci. 2022, 23, 4079. https://doi.org/10.3390/ijms23084079
Qi H, Guo X, Wang T, Zhang Z. ASTool: An Easy-to-Use Tool to Accurately Identify Alternative Splicing Events from Plant RNA-Seq Data. International Journal of Molecular Sciences. 2022; 23(8):4079. https://doi.org/10.3390/ijms23084079
Chicago/Turabian StyleQi, Huan, Xiaokun Guo, Tianpeng Wang, and Ziding Zhang. 2022. "ASTool: An Easy-to-Use Tool to Accurately Identify Alternative Splicing Events from Plant RNA-Seq Data" International Journal of Molecular Sciences 23, no. 8: 4079. https://doi.org/10.3390/ijms23084079
APA StyleQi, H., Guo, X., Wang, T., & Zhang, Z. (2022). ASTool: An Easy-to-Use Tool to Accurately Identify Alternative Splicing Events from Plant RNA-Seq Data. International Journal of Molecular Sciences, 23(8), 4079. https://doi.org/10.3390/ijms23084079