
In Vitro Angiogenesis Inhibition and Endothelial Cell Growth and Morphology


Abstract
:1. Introduction
2. Results
2.1. Angiogenesis Inhibitors’ Effects on Endothelial Cell Morphology and Network Formation
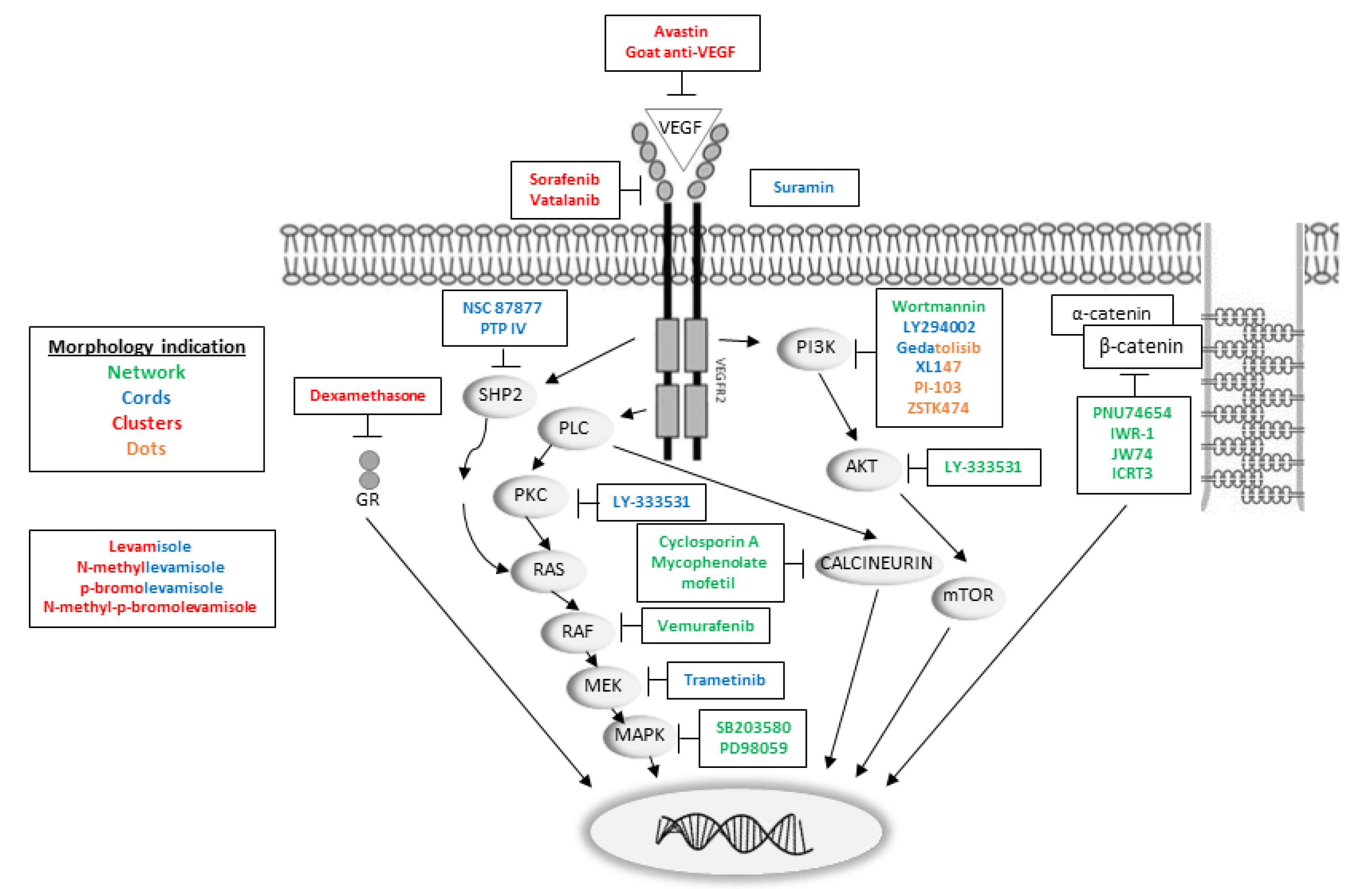
2.2. VEGF and VEGFR Inhibitors
2.3. Levamisole and Its Derivatives
2.4. VEGFR Downstream Inhibitors
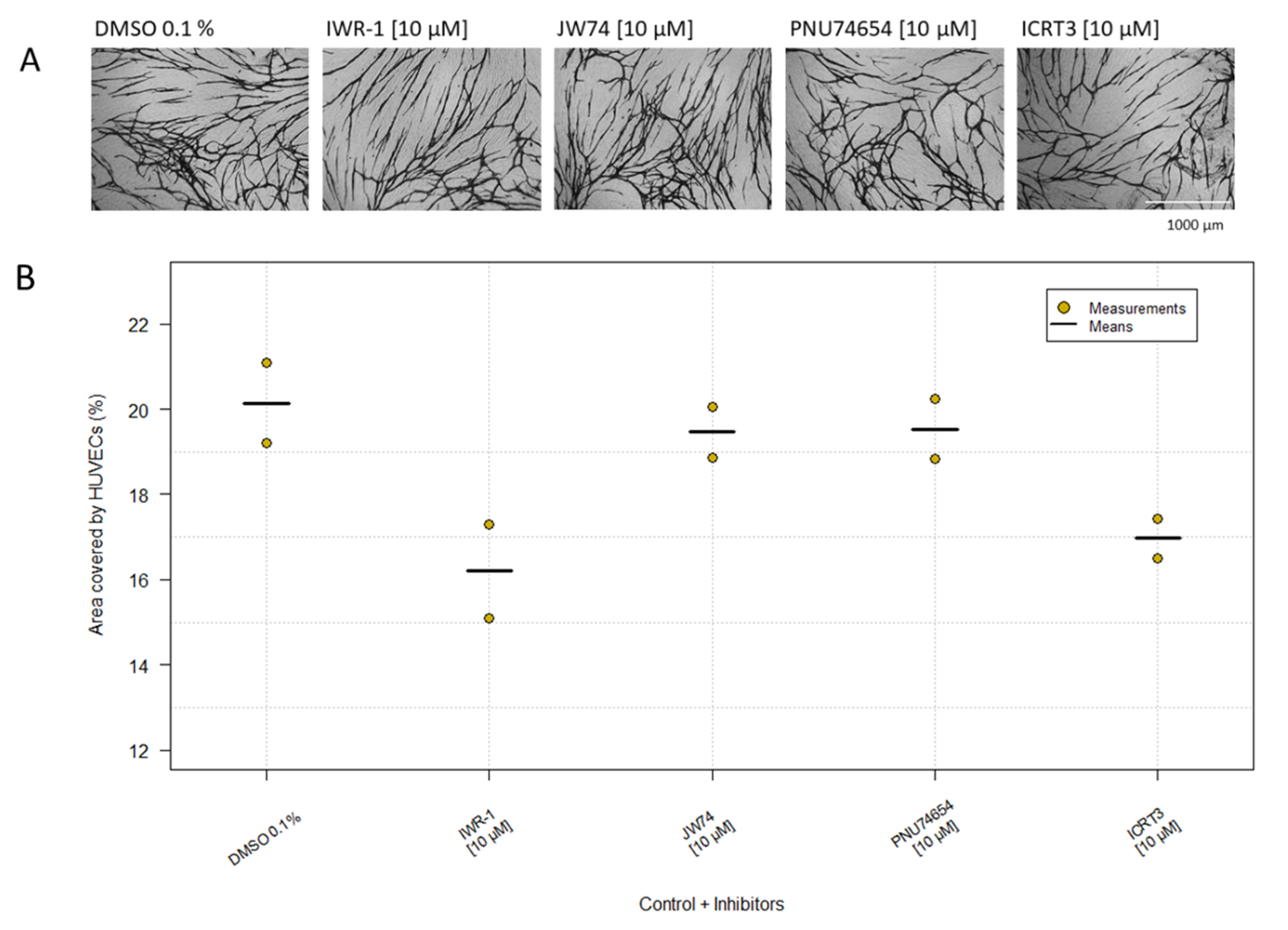
2.5. β-Catenin Inhibitors
2.6. Combinations of Angiogenesis Inhibitors
2.7. Nuclear Receptor Ligands and Miscellaneous Compounds
3. Discussion
3.1. Inhibition of VEGF and VEGFR Signaling
3.2. Miscellaneous Inhibitors
3.3. Combinations of Inhibitors
3.4. Vitamin D and Dexamethasone
3.5. Weakness of the Assay
4. Materials and Methods
4.1. Materials
4.2. Chemical Syntheses
4.3. Methods
4.3.1. Cell Preparation
4.3.2. Co-Culture Assay
4.3.3. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Deyell, M.; Garris, C.S.; Laughney, A.M. Cancer metastasis as a non-healing wound. Br. J. Cancer 2021, 124, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef]
- Aguilar-Cazares, D.; Chavez-Dominguez, R.; Carlos-Reyes, A.; Lopez-Camarillo, C.; Hernadez de la Cruz, O.N.; Lopez-Gonzalez, J.S. Contribution of Angiogenesis to Inflammation and Cancer. Front. Oncol. 2019, 9, 1399. [Google Scholar] [CrossRef] [Green Version]
- Schito, L. Hypoxia-Dependent Angiogenesis and Lymphangiogenesis in Cancer. Adv. Exp. Med. Biol. 2019, 1136, 71–85. [Google Scholar] [PubMed]
- Abou Khouzam, R.; Brodaczewska, K.; Filipiak, A.; Zeinelabdin, N.A.; Buart, S.; Szczylik, C.; Kieda, C.; Chouaib, S. Tumor Hypoxia Regulates Immune Escape/Invasion: Influence on Angiogenesis and Potential Impact of Hypoxic Biomarkers on Cancer Therapies. Front. Immunol. 2021, 11, 613114. [Google Scholar] [CrossRef]
- Friis, T.; Engel, A.M.; Bendiksen, C.D.; Larsen, L.S.; Houen, G. Influence of levamisole and other angiogenesis inhibitors on angiogenesis and endothelial cell morphology in vitro. Cancers 2013, 5, 762–785. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Patenaude, A.; Parker, J.; Karsan, A. Involvement of endothelial progenitor cells in tumor vascularization. Microvasc. Res. 2010, 79, 217–223. [Google Scholar] [CrossRef]
- Huang, X.L.; Khan, M.I.; Wang, J.; Ali, R.; Ali, S.W.; Zahra, Q.U.; Kazmi, A.; Lolai, A.; Huang, Y.L.; Hussain, A.; et al. Role of receptor tyrosine kinases mediated signal transduction pathways in tumor growth and angiogenesis-New insight and futuristic vision. Int. J. Biol. Macromol. 2021, 180, 739–752. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genet. Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laramée, M.; Chabot, C.; Cloutier, M.; Stenne, R.; Holgado-Madruga, M.; Wong, A.J.; Royal, I. The scaffolding adapter Gab1 mediates vascular endothelial growth factor signaling and is required for endothelial cell migration and capillary formation. J. Biol. Chem. 2007, 282, 7758–7769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fresno Vara, J.A.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; González-Barón, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef]
- Fan, Q.W.; Knight, Z.A.; Goldenberg, D.D.; Yu, W.; Mostov, K.E.; Stokoe, D.; Shokat, K.M.; Weiss, W.A. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 2006, 9, 341–349. [Google Scholar] [CrossRef] [Green Version]
- Suehiro, J.; Kanki, Y.; Makihara, C.; Schadler, K.; Miura, M.; Manabe, Y.; Aburatani, H.; Kodama, T.; Minami, T. Genome-wide approaches reveal functional vascular endothelial growth factor (VEGF)-inducible nuclear factor of activated T cells (NFAT) c1 binding to angiogenesis-related genes in the endothelium. J. Biol. Chem. 2014, 289, 29044–29059. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef]
- Dang, A.; Jagan Mohan Venkateshwara Rao, P.; Kishore, R.; Vallish, B.N. Real world safety of bevacizumab in cancer patients: A systematic literature review of case reports. Int. J. Risk Saf. Med. 2020, 32, 163–173. [Google Scholar] [CrossRef]
- Morabito, A.; De Maio, E.; Di Maio, M.; Normanno, N.; Perrone, F. Tyrosine kinase inhibitors of vascular endothelial growth factor receptors in clinical trials: Current status and future directions. Oncologist 2006, 11, 753–764. [Google Scholar] [CrossRef]
- Hojjat-Farsangi, M. Small-molecule inhibitors of the receptor tyrosine kinases: Promising tools for targeted cancer therapies. Int. J. Mol. Sci. 2014, 15, 13768–13801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, R.; Fetterly, G.; Lugade, A.; Thanavala, Y. Sorafenib: A clinical and pharmacologic review. Expert Opin. Pharmacother. 2010, 11, 1943–1955. [Google Scholar] [CrossRef] [PubMed]
- Gotink, K.J.; Verheul, H.M. Anti-angiogenic tyrosine kinase inhibitors: What is their mechanism of action? Angiogenesis 2010, 13, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Walker, T.; Mitchell, C.; Park, M.A.; Yacoub, A.; Graf, M.; Rahmani, M.; Houghton, P.J.; Voelkel-Johnson, C.; Grant, S.; Dent, P. Sorafenib and vorinostat kill colon cancer cells by CD95-dependent and -independent mechanisms. Mol. Pharmacol. 2009, 76, 342–355. [Google Scholar] [CrossRef] [Green Version]
- Dragovich, T.; Laheru, D.; Dayyani, F.; Bolejack, V.; Smith, L.; Seng, J.; Burris, H.; Rosen, P.; Hidalgo, M.; Ritch, P.; et al. Phase II trial of vatalanib in patients with advanced or metastatic pancreatic adenocarcinoma after first-line gemcitabine therapy (PCRT O4-001). Cancer Chemother. Pharmacol. 2014, 74, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, P.; Mulkey, F.; Hasserjian, R.P.; Sanford, B.L.; Vij, R.; Hurd, D.D.; Odenike, O.M.; Bloomfield, C.D.; Owzar, K.; Stone, R.M.; et al. Alliance for Clinical Trials in Oncology. A phase II study of the oral VEGF receptor tyrosine kinase inhibitor vatalanib (PTK787/ZK222584) in myelodysplastic syndrome: Cancer and Leukemia Group B study 10105 (Alliance). Investig. New Drugs 2013, 31, 1311–1320. [Google Scholar] [CrossRef] [Green Version]
- Steeghs, N.; Nortier, J.W.; Gelderblom, H. Small molecule tyrosine kinase inhibitors in the treatment of solid tumors: An update of recent developments. Ann. Surg. Oncol. 2007, 14, 942–953. [Google Scholar] [CrossRef]
- Friis, T.; Kjaer Sørensen, B.; Engel, A.M.; Rygaard, J.; Houen, G. A quantitative ELISA-based co-culture angiogenesis and cell proliferation assay. APMIS 2003, 111, 658–668. [Google Scholar] [CrossRef]
- Friis, T.; Hansen, A.B.; Houen, G.; Engel, A.M. Influence of angiogenesis inhibitors on endothelial cell morphology in vitro. APMIS 2006, 114, 211–224. [Google Scholar] [CrossRef]
- Hansen, A.N.; Bendiksen, C.D.; Sylvest, L.; Friis, T.; Staerk, D.; Jørgensen, F.S.; Olsen, C.A.; Houen, G. Synthesis and antiangiogenic activity of N-alkylated levamisole derivatives. PLoS ONE 2012, 7, e45405. [Google Scholar] [CrossRef] [Green Version]
- Friis, T.; Engel, A.M.; Klein, B.M.; Rygaard, J.; Houen, G. Levamisole inhibits angiogenesis in vitro and tumor growth in vivo. Angiogenesis 2005, 8, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Aznar, A.; Muhl, L.; Gaengel, K. VEGF Receptor Tyrosine Kinases: Key Regulators of Vascular Function. Curr. Top. Dev. Biol. 2017, 123, 433–482. [Google Scholar] [PubMed]
- Corti, F.; Simons, M. Modulation of VEGF receptor 2 signaling by protein phosphatases. Pharmacol. Res. 2017, 115, 107–123. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Yazici, Y.D.; Calzada, G.; Wang, Z.Y.; Younes, M.N.; Jasser, S.A.; El-Naggar, A.K.; Myers, J.N. Sorafenib inhibits the angiogenesis and growth of orthotopic anaplastic thyroid carcinoma xenografts in nude mice. Mol. Cancer Res. 2007, 6, 1785–1792. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [Green Version]
- Gedaly, G.; Angulo, P.; Hundley, J.; Daily, M.F.; Chen, C.; Koch, A.; Evers, B.M. PI-103 and sorafenib inhibit hepatocellular carcinoma cell proliferation by blocking Ras/Raf/MAPK and PI3K/AKT/mTOR pathways. Anticancer. Res. 2010, 30, 4951–4958. [Google Scholar]
- Scott, E.N.; Meinhardt, G.; Jacques, C.; Laurent, D.; Thomas, A.L. Vatalanib: The clinical development of a tyrosine kinase inhibitor of angiogenesis in solid tumours. Expert Opin. Investig. Drugs 2007, 16, 367–379. [Google Scholar] [CrossRef]
- Los, M.; Roodhart, J.M.; Voest, E.E. Target practice: Lessons from phase III trials with bevacizumab and vatalanib in the treatment of advanced colorectal cancer. Oncologist 2007, 12, 443–450. [Google Scholar] [CrossRef] [Green Version]
- Wood, J.M.; Bold, G.; Buchdunger, E.; Cozens, R.; Ferrari, S.; Frei, J.; Hofmann, F.; Mestan, J.; Mett, H.; O’Reilly, T.; et al. PTK787/ZK 222584, a Novel and Potent Inhibitor of Vascular Endothelial Growth Factor Receptor Tyrosine Kinases, Impairs Vascular Endothelial Growth Factor-induced Responses and Tumor Growth after Oral Administration. Cancer Res. 2000, 60, 2178–2189. [Google Scholar] [PubMed]
- Sylvest, L.; Bendiksen, C.D.; Houen, G. Phosphatase inhibitors with anti-angiogenic effect in vitro. APMIS 2010, 118, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Sung, S.S.; Yip, M.L.; Lawrence, H.R.; Ren, Y.; Guida, W.C.; Sebti, S.M.; Lawrence, N.J.; Wu, J. Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Mol. Pharmacol. 2006, 5, 17626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrera, S.; Senra, J.; Acosta, M.I.; Althubiti, M.; Hammond, E.M.; de Verdier, P.J.; Macip, S. The role of the HIF-1alpha transcription factor in increased cell division at physiological oxygen tensions. PLoS ONE 2014, 9, e97938. [Google Scholar] [CrossRef] [Green Version]
- Spagnolo, F.; Ghiorzo, P.; Orgiano, L.; Pastorino, L.; Picasso, V.; Tornari, E.; Ottaviano, V.; Queirolo, P. BRAF-mutant melanoma: Treatment approaches, resistance mechanisms, and diagnostic strategies. OncoTargets Ther. 2015, 8, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.H.; Jiang, G.; Zheng, J.Z.; Lu, Z.; Hunter, T.; Vogt, P.K. Phosphatidylinositol 3-kinase signaling controls levels of hypoxia-inducible factor 1. Cell Growth Differ. 2001, 12, 363–369. [Google Scholar]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 2, 51. [Google Scholar] [CrossRef] [Green Version]
- Kong, D.; Yamori, T. ZSTK474 is an ATP-competitive inhibitor of class I phosphatidylinositol 3 kinase isoforms. Cancer Sci. 2007, 98, 1638–1642. [Google Scholar] [CrossRef]
- Gills, J.J.; Dennis, P.A. Perifosine: Update on a novel Akt inhibitor. Curr. Oncol. Rep. 2009, 11, 102–110. [Google Scholar] [CrossRef]
- Weisz, A.; Koren, B.; Cohen, T.; Neufeld, G.; Kleinberger, T.; Lewis, B.S.; Flugelman, M.Y. Increased vascular endothelial growth factor 165 binding to kinase insert domain-containing receptor after infection of human endothelial cells by recombinant adenovirus encoding the Vegf(165) gene. Circulation 2001, 103, 1887–1892. [Google Scholar] [CrossRef] [Green Version]
- Gagliardi, A.; Hadd, H.; Collins, D.C. Inhibition of Angiogenesis by Suramin. Cancer Res. 1992, 52, 5073–5075. [Google Scholar] [PubMed]
- Cook, J.W.; Sterman, D.H.; Singhal, S.; Smythe, W.R.; Kaiser, L.R. Suramin inhibits the growth of malignant mesothelioma in vitro, and in vivo, in murine flank and intraperitoneal models. Lung Cancer 2006, 42, 263–274. [Google Scholar] [CrossRef]
- Prigozhina, N.L.; Heisel, A.J.; Seldeen, J.R.; Cosford, N.D.; Price, J.H. Amphiphilic suramin dissolves Matrigel, causing an ‘inhibition’ artefact within in vitro angiogenesis assays. Int. J. Exp. Pathol. 2013, 94, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis. Annu. Rev. Med. 2006, 57, 1–18. [Google Scholar] [CrossRef]
- Osuka, K.; Usuda, N.; Aoyama, M.; Yamahata, H.; Takeuchi, M.; Yasuda, M.; Takayasu, M. Expression of the JAK/STAT3/SOCS3 signaling pathway in herniated lumbar discs. Neurosci. Lett. 2014, 569, 55–58. [Google Scholar] [CrossRef]
- Jia, J.; Ye, T.; Cui, P.; Hua, Q.; Zeng, H.; Zhao, D. AP-1 transcription factor mediates VEGF-induced endothelial cell migration and proliferation. Microvasc. Res. 2016, 105, 103–108. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Dodge, M.E.; Tang, W.; Lu, J.; Ma, Z.; Fan, C.W.; Wei, S.; Hao, W.; Kilgore, J.; Williams, N.S.; et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 2009, 5, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Lampugnani, M.G.; Orsenigo, F.; Gagliani, M.C.; Tacchetti, C.; Dejana, E. Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J. Cell Biol. 2006, 174, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, A.A.; Deeb, K.K.; Pike, J.W.; Johnson, C.S.; Trump, D.L. Dexamethasone enhances 1alpha,25-dihydroxyvitamin D3 effects by increasing vitamin D receptor transcription. J. Biol. Chem. 2011, 286, 36228–36237. [Google Scholar] [CrossRef] [Green Version]
- Chung, I.; Han, G.; Seshadri, M.; Gillard, B.M.; Yu, W.D.; Foster, B.A.; Trump, D.L.; Johnson, C.S. Role of vitamin D receptor in the antiproliferative effects of calcitriol in tumor-derived endothelial cells and tumor angiogenesis in vivo. Cancer Res. 2009, 69, 967–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, G.; Chung, I.; Yu, W.D.; Romano, M.; Modzelewski, R.A.; Johnson, C.S.; Trump, D.L. Calcitriol (1,25-dihydroxycholecalciferol) selectively inhibits proliferation of freshly isolated tumor-derived endothelial cells and induces apoptosis. Oncology 2006, 70, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Díaz, L.; Díaz-Muñoz, M.; García-Gaytán, A.C.; Méndez, I. Mechanistic Effects of Calcitriol in Cancer Biology. Nutrients 2015, 7, 5020–5050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborti, C.K. Vitamin D as a promising anticancer agent. Indian J. Pharmacol. 2011, 43, 113–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouillon, R.; Eelen, G.; Verlinden, L.; Mathieu, C.; Carmeliet, G.; Verstuyf, A. Vitamin D and cancer. J. Steroid Biochem. Mol. Biol. 2006, 102, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.D.; Papadmitrakopoulos, F.; Burgess, D.J. Concurrent delivery of dexamethasone and VEGF for localized inflammation control and angiogenesis. J. Control. Release 2007, 117, 68–79. [Google Scholar] [CrossRef]
- Luo, J.C.; Shin, V.Y.; Liu, E.S.; Ye, Y.N.; Wu, W.K.; So, W.H.; Chang, F.Y.; Cho, C.H. Dexamethasone delays ulcer healing by inhibition of angiogenesis in rat stomachs. Eur. J. Pharmacol. 2004, 485, 275–281. [Google Scholar] [CrossRef]
- Chai, M.; Gu, C.; Shen, Q.; Liu, J.; Zhou, Y.; Jin, Z.; Xiong, W.; Zhou, Y.; Tan, W. Hypoxia alleviates dexamethasone-induced inhibition of angiogenesis in cocultures of HUVECs and rBMSCs via HIF-1alpha. Stem Cell Res. Ther. 2020, 11, 343. [Google Scholar] [CrossRef]
- Fan, Z.; Sehm, T.; Rauh, M.; Buchfelder, M.; Eyupoglu, I.Y.; Savaskan, N.E. Dexamethasone alleviates tumor-associated brain damage and angiogenesis. PLoS ONE 2014, 9, e93264. [Google Scholar]
- Almawi, W.Y.; Melemedjian, O.K. Molecular mechanisms of glucocorticoid antiproliferative effects: Antagonism of transcription factor activity by glucocorticoid receptor. J. Leukoc. Biol. 2002, 71, 9–15. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Inhibitors | Concentration | HUVEC Morphology | Inhibitory Rate 0–5 | GFP-HUVEC Morphology | Inhibitory Rate 0–5 |
|---|---|---|---|---|---|---|
| Control | DMSO | 0.1% | Network | 0 | Network | 0 |
| VEGF-A | Avastin | 2.5 mg/mL | Clusters | - | NT | - |
| 250 µg/mL | Clusters + short cords | - | Clusters + short cords | - | ||
| 25 µg/mL | Clusters + short cords | - | Clusters + short cords | - | ||
| Goat anti-VEGF | 40 µg/mL | Clusters + short cords | - | NT | - | |
| 5 µg/mL | Clusters +long cords | - | NT | - | ||
| VEGF/Neuropilin/PDGF/bFGF | Suramin | 100 µM | Short cords | - | Short cords | - |
| 50 µM | Short cords | - | NT | - | ||
| 10 µM | Network | 2 | Network | 2 | ||
| VEGFR-2 | Sorafenib | 1 µM | Clusters | - | Clusters | - |
| 100 nM | Network | 2 | Network | - | ||
| 10 nM | Network | 1 | NT | - | ||
| 1 nM | Network | 1 | NT | - | ||
| Vatalanib | 10 µM | Clusters | - | Dead | - | |
| 1 µM | Clusters | - | Clusters | - | ||
| 100 nM | Clusters + short cords | - | Clusters | - | ||
| 10 nM | Clusters + long cords | - | Clusters | - | ||
| β-catenin | IWR-1 | 10 µM | Network | 0 | Network | 0 |
| PNU-74654 | 10 µM | Network | 0 | Network | 0 | |
| JW74 | 10 µM | Network | 0 | Network | 0 | |
| ICRT3 | 10 µM | Network | 0 | Network | 0 | |
| Alkaline phosphatase | Levamisole | 1 mM | Oval clusters + long cords | - | Oval clusters + network | - |
| P-bromo-levamisole | 1 mM | Dead | - | Dead | - | |
| 100 µM | Network | 2 | Long cords | - | ||
| 10 µM | Network | 0 | Network | 0 | ||
| N-methyl-levamisole | 1 mM | Dead | - | Dots | - | |
| 100 µM | Oval clusters + short cords | - | Clusters + short cords | - | ||
| 10 µM | Network | 1 | Long cords | - | ||
| N-methyl- p-bromo-levamisole | 1 mM | Dead | - | Dead | - | |
| 100 µM | Oval clusters | - | Clusters | - | ||
| 10 µM | Network | 2 | Clusters + short cords | - | ||
| 1 µM | Network | 0 | Network | 0 | ||
| Calcineurin | Cyclosporin A | 10 µM | Network | 0 | Dead | - |
| 1 µM | Network | 0 | Network | 0 | ||
| 100 nM | Network | 0 | Network | 0 | ||
| 10 nM | Network | 0 | Network | 0 | ||
| Mycophenolate mofetil | 10 µM | Short cords | - | Short cords | - | |
| 1 µM | Short cords | - | Short cords | - | ||
| 100 nM | Network | 1 | Short cords | - | ||
| 10 nM | Network | 0 | Short cords | - | ||
| MAPK | SB203580 | 10 µM | Network | 2 | Network | 1 |
| 1 µM | Network | 2 | Network | 1 | ||
| 100 nM | Network | 1 | Network | 1 | ||
| 10 nM | Network | 1 | Network | 1 | ||
| PD98059 1 | 10 µM | Network | 3 | Long cords | - | |
| 1 µM | Network | 2 | Network | 2 | ||
| 100 nM | Network | 1 | Network | 1 | ||
| 10 nM | Network | 1 | Network | 1 | ||
| MEK | Trametinib | 1 µM | Curly short cords | - | Dead | - |
| 100 nM | Short cords | - | Dead | - | ||
| 10 nM | Short cords | - | Dead | - | ||
| 1 nM | Network | 2 | NT | - | ||
| RAF | Vemurafenib | 10 µM | Dead | - | Dead | - |
| 1 µM | Network | 0 | Network | 0 | ||
| 100 nM | Network | 0 | Network | 0 | ||
| 10 nM | Network | 0 | Network | 0 | ||
| PI3K | Gedatolisib 2 | 30 µM | Dead | - | Dead | - |
| 300 nM | Short cords + dots | - | Dead | - | ||
| 30 nM | Network | 1 | Dots | - | ||
| 3 nM | Network | 1 | NT | - | ||
| Wortmannin | 10 µM | Network | 3 | Network | 0 | |
| 1 µM | Network | 2 | Network | 0 | ||
| 100 nM | Network | 2 | Network | 0 | ||
| ZSTK474 | 100 µM | Dead | - | NT | - | |
| 10 µM | Dots | - | NT | - | ||
| 1 µM | Long cords | - | NT | - | ||
| 100 nM | Network | 2 | NT | - | ||
| 10 nM | Network | 1 | NT | - | ||
| 1 nM | Network | 1 | NT | - | ||
| PI-103 2 | 100 µM | Dead | - | NT | - | |
| 10 µM | Dots | - | NT | - | ||
| 1 µM | Short cords + dots | - | NT | - | ||
| 100 nM | Long cords | - | NT | - | ||
| 10 nM | Network | 2 | NT | - | ||
| 1 nM | Network | 1 | NT | - | ||
| XL147 | 50 µM | Dead | - | NT | - | |
| 500 nM | Short cords + dots | - | NT | - | ||
| 50 nM | Network | 0 | NT | - | ||
| 5 nM | Network | 0 | NT | - | ||
| 500 pM | Network | 0 | NT | - | ||
| LY294002 | 100 µM | Dead | - | NT | - | |
| 10 µM | Short cords | - | NT | - | ||
| 1 µM | Network | 1 | NT | - | ||
| 100 nM | Network | 0 | NT | - | ||
| 10 nM | Network | 0 | NT | - | ||
| 1 nM | Network | 0 | NT | - | ||
| SHP1 and SHP2 | PTP IV 3 | 100 µM | Dead | - | NT | - |
| 10 µM | Dead | - | NT | - | ||
| 1 µM | Long cords | - | NT | - | ||
| 100 nM | Network | 1 | NT | - | ||
| 10 nM | Network | 0 | NT | - | ||
| NSC 87877 4 | 100 µM | Short cords | - | NT | - | |
| 10 µM | Network | 1 | NT | - | ||
| 1 µM | Network | 0 | NT | - | ||
| 100 nM | Network | 0 | NT | - | ||
| 10 nM | Network | 0 | NT | - | ||
| 1 nM | Network | 0 | NT | - | ||
| PKC | LY33353 | 50 µM | Dead | - | NT | - |
| 10 µM | Short cords | - | NT | - | ||
| 1 µM | Network | 2 | NT | - | ||
| 100 nM | Network | 0 | NT | - | ||
| 10 nM | Network | 0 | NT | - | ||
| 1 nM | Network | 0 | NT | - | ||
| AKT | Perifosine | 50 nM | Network | 2 | Dead | - |
| 5 nM | Network | 1 | Network | 0 | ||
| 500 pM | Network | 0 | Network | 0 | ||
| STAT 5 | STAT 5 | 100 µM | Short cords | - | NT | - |
| 10 µM | Network | 1 | NT | - | ||
| 1 µM | Network | 0 | NT | - | ||
| 100 nM | Network | 0 | NT | - | ||
| 10 nM | Network | 0 | NT | - | ||
| JAK-2 | AG-490 | 100 µM | Dead | - | NT | - |
| 10 µM | Network | 1 | NT | - | ||
| 1 µM | Network | 1 | NT | - | ||
| 100 nM | Network | 0 | NT | - | ||
| 10 nM | Network | 0 | NT | - | ||
| JAK 1 & 2 | Ruxolitinib | 100 µM | Dead | - | NT | - |
| 10 µM | Network | 1 | NT | - | ||
| 1 µM | Network | 0 | NT | - | ||
| 100 nM | Network | 0 | NT | - | ||
| 10 nM | Network | 0 | NT | - | ||
| STAT 3 | SH-4-54 | 100 µM | Dead | - | NT | - |
| 10 µM | Dead | - | NT | - | ||
| 1 µM | Network | 1 | NT | - | ||
| 100 nM | Network | 0 | NT | - | ||
| 10 nM | Network | 0 | NT | - | ||
| NSC 74859 | 100 µM | Dead | - | Dots | - | |
| 10 µM | Network | 0 | Network | 0 | ||
| 1 µM | Network | 0 | Network | 0 | ||
| JAK 2 & STAT 3 | WP1066 | 100 µM | Dead | - | NT | - |
| 10 µM | Dead | - | NT | - | ||
| 1 µM | Network | 0 | NT | - | ||
| 100 nM | Network | 0 | NT | - | ||
| 10 nM | Network | 0 | NT | - | ||
| GR | Dexamethasone | 1 mM | Dead | - | NT | - |
| 100 µM | Cluster | - | NT | - | ||
| 10 µM | Cluster | - | NT | - | ||
| 1 µM | Cluster | - | NT | - | ||
| 100 nM | Cluster + cords | - | NT | - | ||
| 10 nM | Cluster + cords | - | NT | - | ||
| 1 nM | Cluster + cords | - | NT | - | ||
| 100 pM | Cluster + cords | - | NT | - | ||
| 10 pM | Cluster + cords | - | NT | - | ||
| 1 pM | Cluster + cords | - | NT | - | ||
| PP1 | Tautomycetin | 1 µM | Network | 0 | NT | - |
| 100 nM | Network | 0 | NT | - | ||
| 10 nM | Network | 0 | NT | - | ||
| 1 nM | Network | 0 | NT | - | ||
| 100 pM | Network | 0 | NT | - | ||
| Histone deacetylase and histone acetyl-transferase | Butyric acid | 1 mM | Short cords | - | NT | - |
| 100 µM | Network | 0 | NT | - | ||
| 10 µM | Network | 0 | NT | - | ||
| 1 µM | Network | 0 | NT | - | ||
| 100 nM | Network | 0 | NT | - | ||
| 10 nM | Network | 0 | NT | - | ||
| Valproic acid | 1 mM | Short cords | - | NT | - | |
| 100 µM | Network | 1 | NT | - | ||
| 10 µM | Network | 0 | NT | - | ||
| 1 µM | Network | 0 | NT | - | ||
| 100 nM | Network | 0 | NT | - | ||
| 10 nM | Network | 0 | NT | - | ||
| Anacardic acid | 100 µM | Dead | - | NT | - | |
| 10 µM | Dead | - | NT | - | ||
| 1 µM | Network | 0 | NT | - | ||
| 100 nM | Network | 0 | NT | - | ||
| 10 nM | Network | 0 | NT | - | ||
| 1 nM | Network | 0 | NT | - | ||
| 100 pM | Network | 0 | NT | - | ||
| Retinoic acidreceptor (RAR) | Retinol | 100 µM | Dead | - | NT | - |
| 10 µM | Network | 0 | NT | - | ||
| 1 µM | Network | 0 | NT | - | ||
| 100 nM | Network | 0 | NT | - | ||
| 10 nM | Network | 0 | NT | - | ||
| 1 nM | Network | 0 | NT | - | ||
| Retinoic acid | 100 µM | Dead | - | NT | - | |
| 10 µM | Network | 0 | NT | - | ||
| 1 µM | Network | 0 | NT | - | ||
| 100 nM | Network | 0 | NT | - | ||
| 10 nM | Network | 0 | NT | - | ||
| VDR | 25-hydroxyvitamin D3 monohydrate | 1 µM | Short cords | - | NT | - |
| 100 nM | Network | 0 | NT | - | ||
| 10 nM | Network | 0 | NT | - | ||
| 1 nM | Network | 0 | NT | - | ||
| 100 pM | Network | 0 | NT | - | ||
| 25-hydroxyvitamin D3 monohydrate + vitamin D-binding protein (DBP) | 1 µM | Short cords | - | NT | - | |
| 100 nM | Network | 0 | NT | - | ||
| 10 nM | Network | 0 | NT | - | ||
| 1 nM | Network | 0 | NT | - | ||
| 100 pM | Network | 0 | NT | - |
| Combinations of Inhibitors | HUVEC Morphology | Inhibitory Rate 0–5 |
|---|---|---|
| LY333531 [10 µM] NSC 87877 [100 µM] Gedatolisib [300 nM] | Dots | - |
| LY294002 [10 µM] LY333531 [10 µM] NSC 87877 [100 µM] | Cords | - |
| Perifosine [50 nM] LY333531 [10 µM] NSC 87877 [100 µM] | Network | 3 |
| NSC 87877 [100 µM] LY294002 [10 µM] | Cords | - |
| NSC 87877 [100 µM] GEDA [30 nM] | Network | 3 |
| LY333531 [10 µM] PTP IV [1 µM] | Cords | - |
| LY294002 [10 µM] LY333531 [10 µM] | Cords | - |
| LY333531 [10 µM] NSC 87877 [100 µM] | Dots | - |
| PTP IV [100 nM] GEDA [30 nM] | Network | 3 |
| Perifosine [50 nM] LY333531 [10 µM] NSC 87877 [100 µM] | Network | 3 |
| Perifosine [50 nM] LY333531 [10 µM] | Network | 2 |
| Suramin [100 µM] AG-490 [10 µM] | Network | 3 |
| Sorafenib [1 µM] STAT 5 [100 µM] | Network | 4 |
| Suramin [100 µM] STAT 5 [100 µM] | Network | 4 |
| Sorafenib [1 µM] NSC 74859 [100 µM] | Cords | - |
| Avastin [250 µg/mL] STAT 5 [100 µM] | Cords | - |
| Suramin [100 µM] NSC 74859 [100 µM] | Cords | - |
| PTP IV [1 µM] NSC 87877 [100 µM] | Cords | - |
| Trametinib [100 nM] LY333531 [10 µM] | Cords | - |
| Sorafenib [1 µM] Vatalanib [1 µM] | Clusters | - |
| Avastin [25 µg/mL] Vatalanib [1 µM] | Clusters | - |
| Poly goat antibody [5 µg/mL] Vatalanib [1 µM] | Clusters | - |
| Vatalanib [1 µM] AG-490 [10 µM] | Clusters | - |
| Vatalanib [1 µM] NSC 74859 [100 µM] | Clusters | - |
| Avastin [250 µg/mL] NSC 74859 [100 µM] | Clusters | - |
| Vatalanib [1 µM] STAT 5 [100 µM] | Clusters | - |
| Avastin [250 µg/mL] AG-490 [10 µM] | Clusters + cords | - |
| NSC 87877 [100 µM] Vatalanib [1 µM] | Clusters + dots | - |
| Myco. Mofetil [10 µM] Sorafenib [1 µM] | Dead | - |
| ZSTK474 [10 µM] PI-103 [10 µM] | Dead | - |
| LY333531 [10 µM] PI-103 [10 µM] | Dead | - |
| LY333531 [10 µM] PI-103 [10 µM] Sorafenib [10 µM] PTP IV [10 µM] | Dead | - |
| LY333531 [10 µM] PI-103 [10 µM] Sorafenib [1 µM] NSC 87877 [100 µM] | Dead | - |
| HUVEC Coverage Area % | HUVEC Coverage Area Grading | Inhibitory Rate 0–5 |
|---|---|---|
| 0–3% | + | 5 |
| 3–6% | ++ | 4 |
| 6–9% | +++ | 3 |
| 9–12% | ++++ | 2 |
| 12–18% | +++++ | 1 |
| >18% | ++++++ | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ljoki, A.; Aslam, T.; Friis, T.; Ohm, R.G.; Houen, G. In Vitro Angiogenesis Inhibition and Endothelial Cell Growth and Morphology. Int. J. Mol. Sci. 2022, 23, 4277. https://doi.org/10.3390/ijms23084277
Ljoki A, Aslam T, Friis T, Ohm RG, Houen G. In Vitro Angiogenesis Inhibition and Endothelial Cell Growth and Morphology. International Journal of Molecular Sciences. 2022; 23(8):4277. https://doi.org/10.3390/ijms23084277
Chicago/Turabian StyleLjoki, Arlinda, Tanzila Aslam, Tina Friis, Ragnhild G. Ohm, and Gunnar Houen. 2022. "In Vitro Angiogenesis Inhibition and Endothelial Cell Growth and Morphology" International Journal of Molecular Sciences 23, no. 8: 4277. https://doi.org/10.3390/ijms23084277
APA StyleLjoki, A., Aslam, T., Friis, T., Ohm, R. G., & Houen, G. (2022). In Vitro Angiogenesis Inhibition and Endothelial Cell Growth and Morphology. International Journal of Molecular Sciences, 23(8), 4277. https://doi.org/10.3390/ijms23084277