
Inhibitory Response to CK II Inhibitor Silmitasertib and CDKs Inhibitor Dinaciclib Is Related to Genetic Differences in Pancreatic Ductal Adenocarcinoma Cell Lines


 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
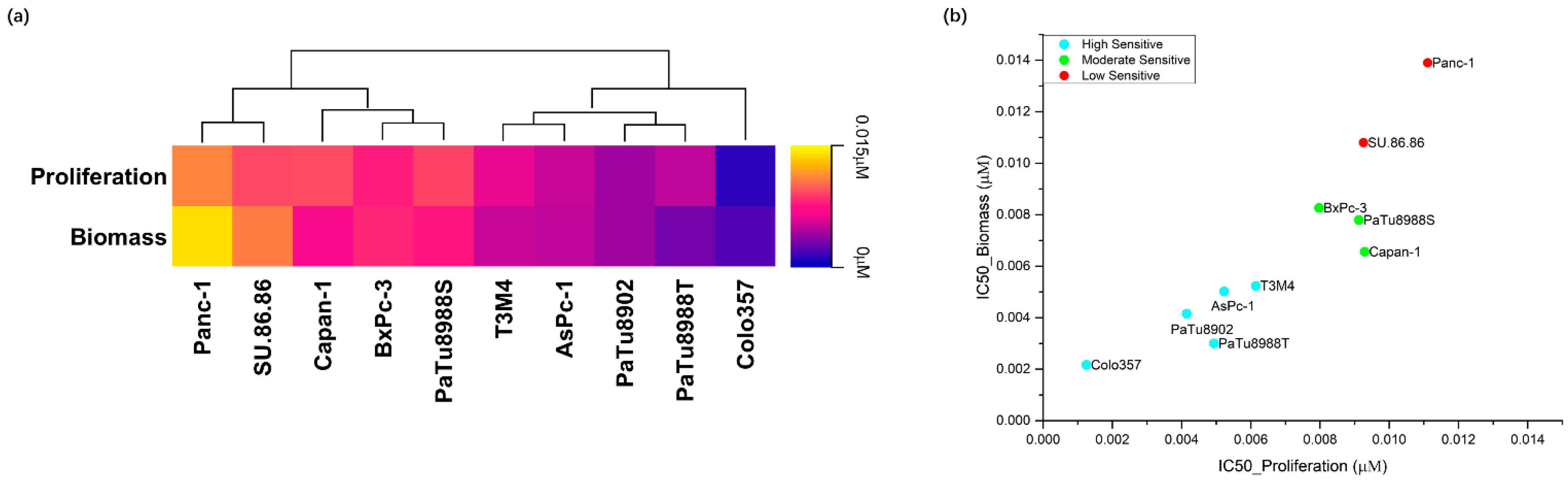
2.1. Effects of Silmitasertib and Dinaciclib on Cell Proliferation, Biomass, and Metabolic Activity
2.2. Silmitasertib and Dinaciclib Induced Cell Deaths in PDAC Cell Lines
2.3. Expression and Genetic Variants of Silmitasertib or Dinaciclib Target Genes
2.4. KRAS and TP53 Gene Variants Were Observed in PDAC Cell Lines
2.4.1. KRAS Variants and Expression in PDAC Cell Lines
2.4.2. KRAS and Inhibitor Response
2.4.3. TP53 Variants and Expression in PDAC Cell Lines
2.4.4. TP53 and Inhibitor Response
3. Discussion
4. Materials and Methods
4.1. Kinase Inhibitors
4.2. Cell Lines and Cell Culture
4.3. Cell Viability Assays
4.3.1. Proliferation
4.3.2. Metabolic Activity
4.3.3. Biomass Quantification
4.4. Identification of IC50
4.5. Apoptosis and Necrosis Analyses
4.6. Nucleic Acid Extraction
4.7. Whole Exome Sequencing
4.8. Variant Calling Filtering Strategy
4.9. Gene Expression Analyses
4.10. Response-Based Clustering Strategy
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT, AKT | Protein kinase B |
| Ala | Alanine |
| Arg | Arginine |
| Asp | Aspartate |
| CDK, CDK | Cyclin-dependent kinase |
| CK2, CSNK2 | Casein kinase II |
| CV | Crystal violet |
| Cys | Cysteine |
| DMSO | Dimethyl sulfoxide |
| DP | Depth of coverage |
| DRB | 5,6-Dichloro-1-ß-D-ribofuranosylbenzimidazole |
| ERK | Extracellular regulated kinase |
| FCS | Fetal calf serum |
| Fs | Frameshift |
| Gln | Glutamine |
| Gly | Glycine |
| GTEx | The genotype-tissue expression |
| His | Histidine |
| IC50 | Half maximal inhibitory concentration |
| Indel | Insertion/deletion |
| JAK | Janus kinase |
| JNK | C-Jun N-terminal kinase |
| KRAS, KRAS | Kirsten’s rat sarcoma viral oncogene homolog |
| MEK | Mitogen-activated protein kinase kinase |
| MKK4 | Dual-specificity mitogen-activated protein kinase kinase 4 |
| OD | Optical density |
| PBS | Phosphate buffer saline |
| PDAC | Pancreatic ductal adenocarcinoma |
| PI | Propidium iodide |
| PI3K | Phosphoinositide 3-kinase |
| PIP3 | Phosphatidylinositol 3,4,5-trisphosphate |
| PTEN | Phosphatase and tensin homolog |
| qual | Variant confidence |
| Rb | Retinoblastoma |
| RNA-seq | RNA sequencing |
| Ser | Serine |
| STAT | Signal transducer and activator of transcription |
| TCGA | The Cancer Genome Atlas Program |
| Thr | Threonine |
| P53, TP53 | Tumor protein p53 |
| TPM | Transcripts per kilobase million |
| UTR | Untranslated region |
| VAF | Variant allele frequency |
| Val | Valine |
| WES | Whole exome sequencing |
| WST-1 | Water soluble tetrazolium-1 |
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Klompmaker, S.; de Rooij, T.; Korteweg, J.J.; van Dieren, S.; van Lienden, K.P.; van Gulik, T.M.; Busch, O.R.; Besselink, M.G. Systematic review of outcomes after distal pancreatectomy with coeliac axis resection for locally advanced pancreatic cancer. Br. J. Surg. 2016, 103, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Kyriazanos, I.D.; Tsoukalos, G.G.; Papageorgiou, G.; Verigos, K.E.; Miliadis, L.; Stoidis, C.N. Local recurrence of pancreatic cancer after primary surgical intervention: How to deal with this devastating scenario? Surg. Oncol. 2011, 20, e133–e142. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.D.; Zhao, Y.; Zhang, M.; He, R.Z.; Shi, X.H.; Guo, X.J.; Shi, C.J.; Peng, F.; Wang, M.; Shen, M.; et al. Inhibition of Autophagy by Deguelin Sensitizes Pancreatic Cancer Cells to Doxorubicin. Int. J. Mol. Sci. 2017, 18, 370. [Google Scholar] [CrossRef] [Green Version]
- Lovecek, M.; Skalicky, P.; Chudacek, J.; Szkorupa, M.; Svebisova, H.; Lemstrova, R.; Ehrmann, J.; Melichar, B.; Yogeswara, T.; Klos, D.; et al. Different clinical presentations of metachronous pulmonary metastases after resection of pancreatic ductal adenocarcinoma: Retrospective study and review of the literature. World J. Gastroenterol. 2017, 23, 6420–6428. [Google Scholar] [CrossRef] [PubMed]
- Tempero, M.A. NCCN Guidelines Updates: Pancreatic Cancer. J. Natl. Compr. Cancer Netw. 2019, 17, 603–605. [Google Scholar] [CrossRef]
- Litchfield, D.W. Protein kinase CK2: Structure, regulation and role in cellular decisions of life and death. Biochem. J. 2003, 369, 1–15. [Google Scholar] [CrossRef]
- Ruzzene, M.; Bertacchini, J.; Toker, A.; Marmiroli, S. Cross-talk between the CK2 and AKT signaling pathways in cancer. Adv. Biol. Regul. 2017, 64, 1–8. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Qin, H.; Frank, S.J.; Deng, L.; Litchfield, D.W.; Tefferi, A.; Pardanani, A.; Lin, F.T.; Li, J.; Sha, B.; et al. A CK2-dependent mechanism for activation of the JAK-STAT signaling pathway. Blood 2011, 118, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Schevzov, G.; Kee, A.J.; Wang, B.; Sequeira, V.B.; Hook, J.; Coombes, J.D.; Lucas, C.A.; Stehn, J.R.; Musgrove, E.A.; Cretu, A.; et al. Regulation of cell proliferation by ERK and signal-dependent nuclear translocation of ERK is dependent on Tm5NM1-containing actin filaments. Mol. Biol. Cell 2015, 26, 2475–2490. [Google Scholar] [CrossRef] [PubMed]
- Kreutzer, J.N.; Ruzzene, M.; Guerra, B. Enhancing chemosensitivity to gemcitabine via RNA interference targeting the catalytic subunits of protein kinase CK2 in human pancreatic cancer cells. BMC Cancer 2010, 10, 440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giroux, V.; Iovanna, J.; Dagorn, J.C. Probing the human kinome for kinases involved in pancreatic cancer cell survival and gemcitabine resistance. FASEB J. 2006, 20, 1982–1991. [Google Scholar] [CrossRef] [PubMed]
- Hamacher, R.; Saur, D.; Fritsch, R.; Reichert, M.; Schmid, R.M.; Schneider, G. Casein kinase II inhibition induces apoptosis in pancreatic cancer cells. Oncol. Rep. 2007, 18, 695–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [Green Version]
- Clinicaltrials. Available online: https://www.clinicaltrials.gov/ (accessed on 1 October 2021).
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Hunter, T.; Pines, J. Cyclins and cancer. II: Cyclin D and CDK inhibitors come of age. Cell 1994, 79, 573–582. [Google Scholar] [CrossRef]
- Bregman, D.B.; Pestell, R.G.; Kidd, V.J. Cell cycle regulation and RNA polymerase II. Front. Biosci. 2000, 5, D244–D257. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Sicinski, P. A kinase of many talents: Non-neuronal functions of CDK5 in development and disease. Open Biol. 2020, 10, 190287. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. Cyclin-dependent protein kinase inhibitors including palbociclib as anticancer drugs. Pharmacol. Res. 2016, 107, 249–275. [Google Scholar] [CrossRef]
- Eggers, J.P.; Grandgenett, P.M.; Collisson, E.C.; Lewallen, M.E.; Tremayne, J.; Singh, P.K.; Swanson, B.J.; Andersen, J.M.; Caffrey, T.C.; High, R.R.; et al. Cyclin-dependent kinase 5 is amplified and overexpressed in pancreatic cancer and activated by mutant K-Ras. Clin. Cancer Res. 2011, 17, 6140–6150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldmann, G.; Mishra, A.; Bisht, S.; Karikari, C.; Garrido-Laguna, I.; Rasheed, Z.; Ottenhof, N.A.; Dadon, T.; Alvarez, H.; Fendrich, V.; et al. Cyclin-dependent kinase inhibitor Dinaciclib (SCH727965) inhibits pancreatic cancer growth and progression in murine xenograft models. Cancer Biol. Ther. 2011, 12, 598–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, D.; Latham, V.M., Jr.; Zhang, X.; Shapiro, G.I. Correction: Combined Depletion of Cell Cycle and Transcriptional Cyclin-Dependent Kinase Activities Induces Apoptosis in Cancer Cells. Cancer Res. 2020, 80, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gojo, I.; Zhang, B.; Fenton, R.G. The cyclin-dependent kinase inhibitor flavopiridol induces apoptosis in multiple myeloma cells through transcriptional repression and down-regulation of Mcl-1. Clin. Cancer Res. 2002, 8, 3527–3538. [Google Scholar] [PubMed]
- Chen, R.; Keating, M.J.; Gandhi, V.; Plunkett, W. Transcription inhibition by flavopiridol: Mechanism of chronic lymphocytic leukemia cell death. Blood 2005, 106, 2513–2519. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Liu, G.Z.; Luo, S.Y.; Chen, R.; Zhang, J.X. Cyclin I promotes cisplatin resistance via Cdk5 activation in cervical cancer. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4533–4541. [Google Scholar]
- Zeng, Y.; Liu, Q.; Wang, Y.; Tian, C.; Yang, Q.; Zhao, Y.; Liu, L.; Wu, G.; Xu, S. CDK5 Activates Hippo Signaling to Confer Resistance to Radiation Therapy Via Upregulating TAZ in Lung Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, 758–769. [Google Scholar] [CrossRef]
- Kazi, A.; Chen, L.; Xiang, S.; Vangipurapu, R.; Yang, H.; Beato, F.; Fang, B.; Williams, T.M.; Husain, K.; Underwood, P.; et al. Global Phosphoproteomics Reveal CDK Suppression as a Vulnerability to KRas Addiction in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 4012–4024. [Google Scholar] [CrossRef]
- Vassilev, L.T. Cell cycle synchronization at the G2/M phase border by reversible inhibition of CDK1. Cell Cycle 2006, 5, 2555–2556. [Google Scholar] [CrossRef] [Green Version]
- Le Tourneau, C.; Faivre, S.; Laurence, V.; Delbaldo, C.; Vera, K.; Girre, V.; Chiao, J.; Armour, S.; Frame, S.; Green, S.R.; et al. Phase I evaluation of seliciclib (R-roscovitine), a novel oral cyclin-dependent kinase inhibitor, in patients with advanced malignancies. Eur. J. Cancer 2010, 46, 3243–3250. [Google Scholar] [CrossRef]
- Mita, M.M.; Mita, A.C.; Moseley, J.L.; Poon, J.; Small, K.A.; Jou, Y.M.; Kirschmeier, P.; Zhang, D.; Zhu, Y.; Statkevich, P.; et al. Phase 1 safety, pharmacokinetic and pharmacodynamic study of the cyclin-dependent kinase inhibitor dinaciclib administered every three weeks in patients with advanced malignancies. Br. J. Cancer 2017, 117, 1258–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A.; Cicenas, J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers 2017, 9, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, R.F.; Gordon, E.M.; Anderson, W.F.; Parekh, D. Gene therapy for primary and metastatic pancreatic cancer with intraperitoneal retroviral vector bearing the wild-type p53 gene. Surgery 1998, 124, 143–150; discussion 150–151. [Google Scholar] [CrossRef]
- Boeck, S.; Jung, A.; Laubender, R.P.; Neumann, J.; Egg, R.; Goritschan, C.; Ormanns, S.; Haas, M.; Modest, D.P.; Kirchner, T.; et al. KRAS mutation status is not predictive for objective response to anti-EGFR treatment with erlotinib in patients with advanced pancreatic cancer. J. Gastroenterol. 2013, 48, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Ormanns, S.; Siveke, J.T.; Heinemann, V.; Haas, M.; Sipos, B.; Schlitter, A.M.; Esposito, I.; Jung, A.; Laubender, R.P.; Kruger, S.; et al. pERK, pAKT and p53 as tissue biomarkers in erlotinib-treated patients with advanced pancreatic cancer: A translational subgroup analysis from AIO-PK0104. BMC Cancer 2014, 14, 624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, H.; Kohno, T.; Ueno, H.; Hiraoka, N.; Kondo, S.; Saito, M.; Shimada, Y.; Ichikawa, H.; Kato, M.; Shibata, T.; et al. Utility of Assessing the Number of Mutated KRAS, CDKN2A, TP53, and SMAD4 Genes Using a Targeted Deep Sequencing Assay as a Prognostic Biomarker for Pancreatic Cancer. Pancreas 2017, 46, 335–340. [Google Scholar] [CrossRef] [PubMed]
- EBML. Available online: https://www.ebi.ac.uk/gxa/home (accessed on 10 April 2022).
- Gojo, I.; Sadowska, M.; Walker, A.; Feldman, E.J.; Iyer, S.P.; Baer, M.R.; Sausville, E.A.; Lapidus, R.G.; Zhang, D.; Zhu, Y.; et al. Clinical and laboratory studies of the novel cyclin-dependent kinase inhibitor dinaciclib (SCH 727965) in acute leukemias. Cancer Chemother. Pharmacol. 2013, 72, 897–908. [Google Scholar] [CrossRef] [Green Version]
- Hwang, D.W.; So, K.S.; Kim, S.C.; Park, K.M.; Lee, Y.J.; Kim, S.W.; Choi, C.M.; Rho, J.K.; Choi, Y.J.; Lee, J.C. Autophagy Induced by CX-4945, a Casein Kinase 2 Inhibitor, Enhances Apoptosis in Pancreatic Cancer Cell Lines. Pancreas 2017, 46, 575–581. [Google Scholar] [CrossRef]
- Subramaniam, D.; Periyasamy, G.; Ponnurangam, S.; Chakrabarti, D.; Sugumar, A.; Padigaru, M.; Weir, S.J.; Balakrishnan, A.; Sharma, S.; Anant, S. CDK-4 inhibitor P276 sensitizes pancreatic cancer cells to gemcitabine-induced apoptosis. Mol. Cancer Ther. 2012, 11, 1598–1608. [Google Scholar] [CrossRef] [Green Version]
- Criscitiello, C.; Viale, G.; Esposito, A.; Curigliano, G. Dinaciclib for the treatment of breast cancer. Expert Opin. Investig. Drugs 2014, 23, 1305–1312. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- di Magliano, M.P.; Logsdon, C.D. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 2013, 144, 1220–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grochola, L.F.; Taubert, H.; Greither, T.; Bhanot, U.; Udelnow, A.; Wurl, P. Elevated transcript levels from the MDM2 P1 promoter and low p53 transcript levels are associated with poor prognosis in human pancreatic ductal adenocarcinoma. Pancreas 2011, 40, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Kotler, E.; Shani, O.; Goldfeld, G.; Lotan-Pompan, M.; Tarcic, O.; Gershoni, A.; Hopf, T.A.; Marks, D.S.; Oren, M.; Segal, E. A Systematic p53 Mutation Library Links Differential Functional Impact to Cancer Mutation Pattern and Evolutionary Conservation. Mol. Cell 2018, 71, 178–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef]
- Brown, M.S.; Diallo, O.T.; Hu, M.; Ehsanian, R.; Yang, X.; Arun, P.; Lu, H.; Korman, V.; Unger, G.; Ahmed, K.; et al. CK2 modulation of NF-kappaB, TP53, and the malignant phenotype in head and neck cancer by anti-CK2 oligonucleotides in vitro or in vivo via sub-50-nm nanocapsules. Clin. Cancer Res. 2010, 16, 2295–2307. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- SKlearn. Available online: https://scikit-learn.org/stable/index.html (accessed on 25 November 2021).







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Sender, S.; Sekora, A.; Kong, W.; Bauer, P.; Ameziane, N.; Krake, S.; Radefeldt, M.; Al-Ali, R.; Weiss, F.U.; et al. Inhibitory Response to CK II Inhibitor Silmitasertib and CDKs Inhibitor Dinaciclib Is Related to Genetic Differences in Pancreatic Ductal Adenocarcinoma Cell Lines. Int. J. Mol. Sci. 2022, 23, 4409. https://doi.org/10.3390/ijms23084409
Ma Y, Sender S, Sekora A, Kong W, Bauer P, Ameziane N, Krake S, Radefeldt M, Al-Ali R, Weiss FU, et al. Inhibitory Response to CK II Inhibitor Silmitasertib and CDKs Inhibitor Dinaciclib Is Related to Genetic Differences in Pancreatic Ductal Adenocarcinoma Cell Lines. International Journal of Molecular Sciences. 2022; 23(8):4409. https://doi.org/10.3390/ijms23084409
Chicago/Turabian StyleMa, Yixuan, Sina Sender, Anett Sekora, Weibo Kong, Peter Bauer, Najim Ameziane, Susann Krake, Mandy Radefeldt, Ruslan Al-Ali, Frank Ulrich Weiss, and et al. 2022. "Inhibitory Response to CK II Inhibitor Silmitasertib and CDKs Inhibitor Dinaciclib Is Related to Genetic Differences in Pancreatic Ductal Adenocarcinoma Cell Lines" International Journal of Molecular Sciences 23, no. 8: 4409. https://doi.org/10.3390/ijms23084409
APA StyleMa, Y., Sender, S., Sekora, A., Kong, W., Bauer, P., Ameziane, N., Krake, S., Radefeldt, M., Al-Ali, R., Weiss, F. U., Lerch, M. M., Parveen, A., Zechner, D., Junghanss, C., & Murua Escobar, H. (2022). Inhibitory Response to CK II Inhibitor Silmitasertib and CDKs Inhibitor Dinaciclib Is Related to Genetic Differences in Pancreatic Ductal Adenocarcinoma Cell Lines. International Journal of Molecular Sciences, 23(8), 4409. https://doi.org/10.3390/ijms23084409