
Preparation of Duplex Sequencing Libraries for Archival Paraffin-Embedded Tissue Samples Using Single-Strand-Specific Nuclease P1

 , , , ,
, , , ,
Abstract
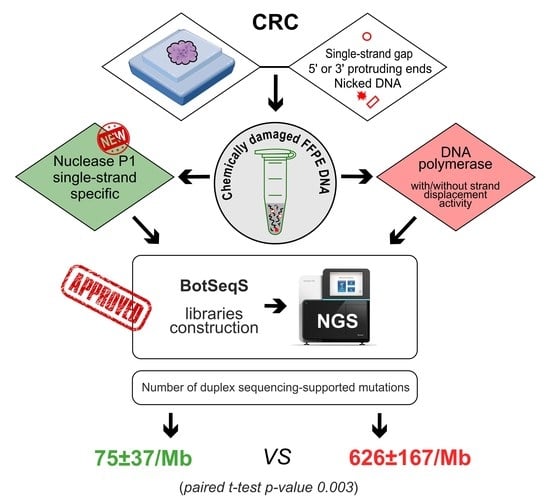
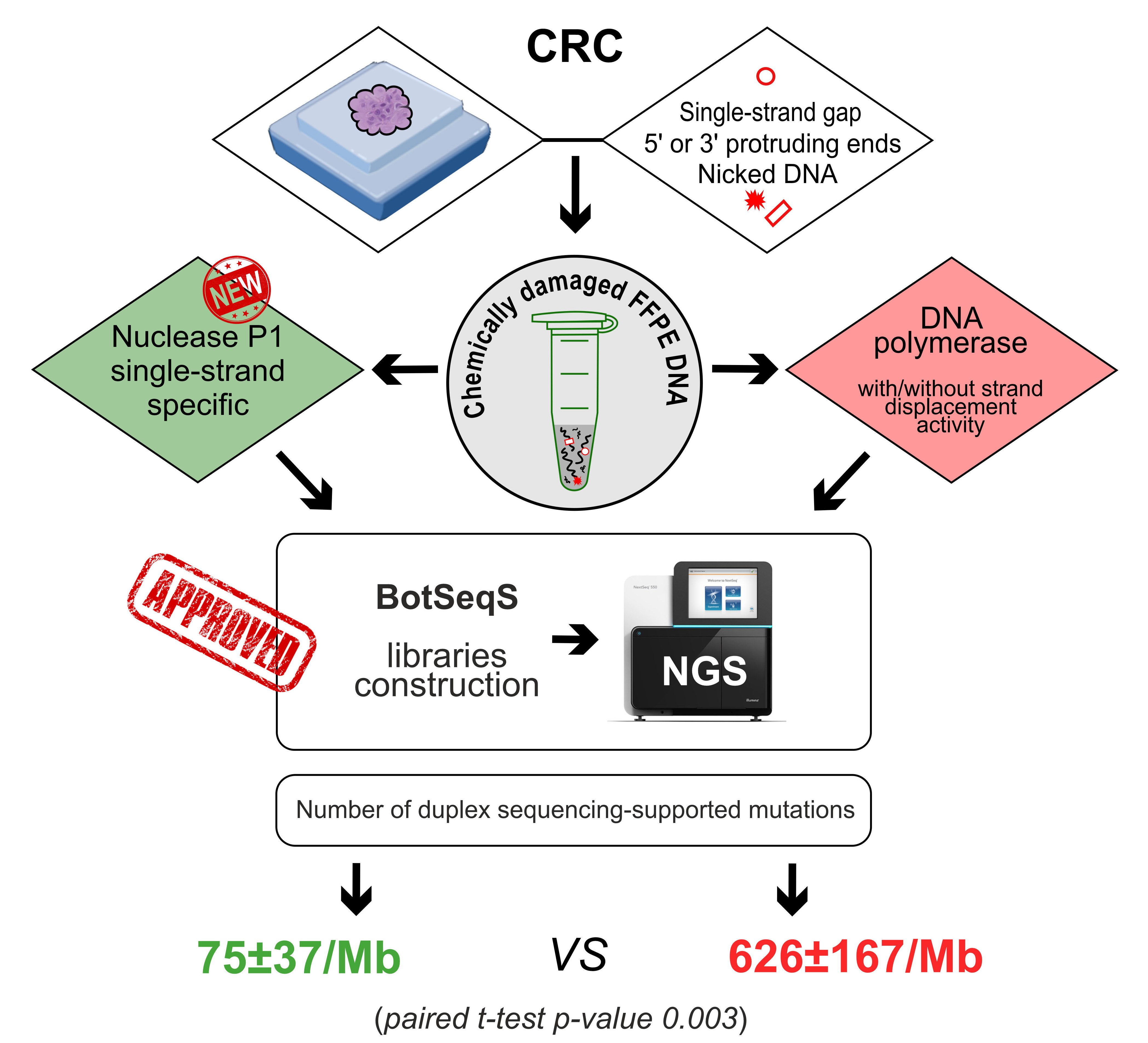
:
1. Introduction
2. Results
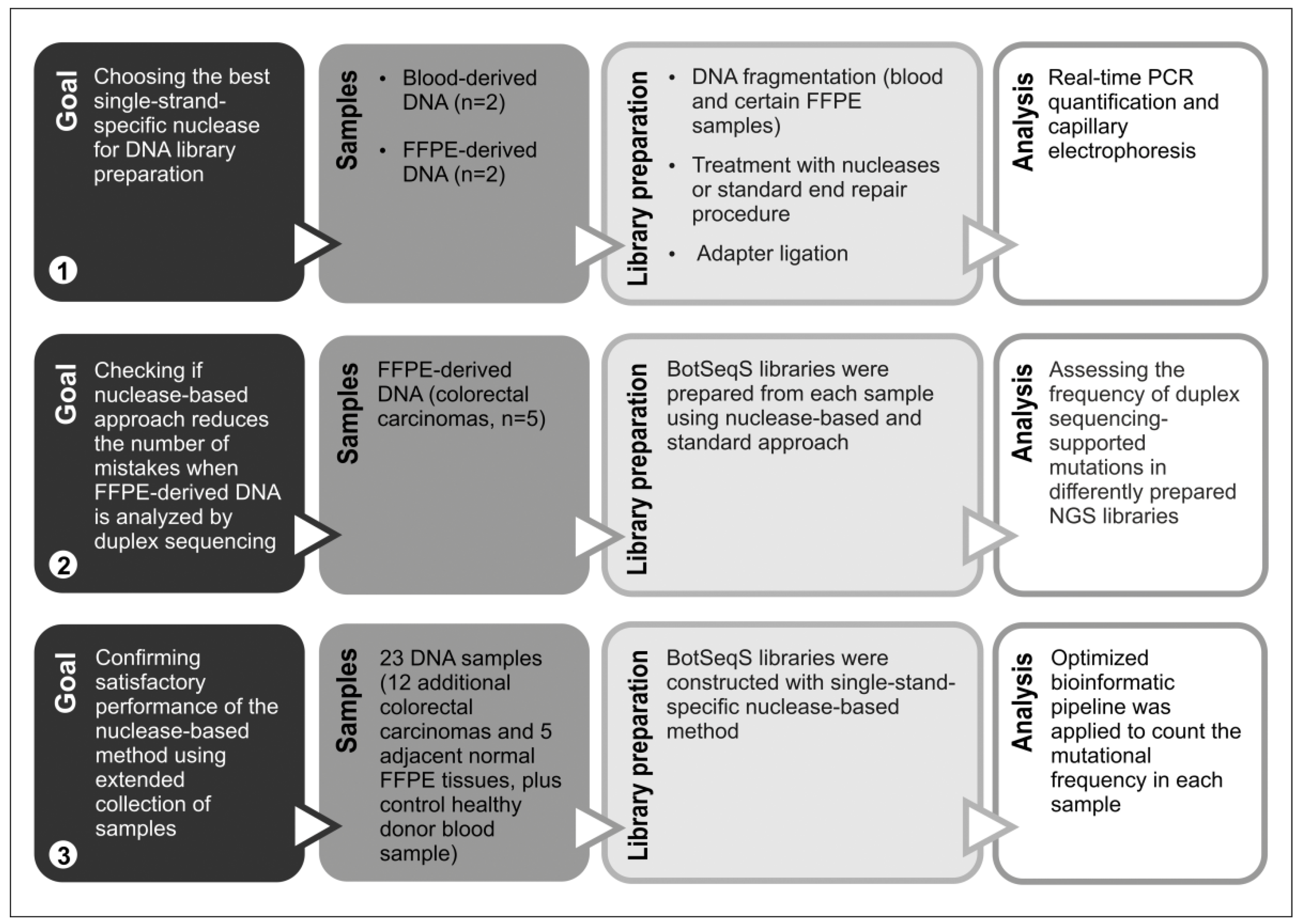
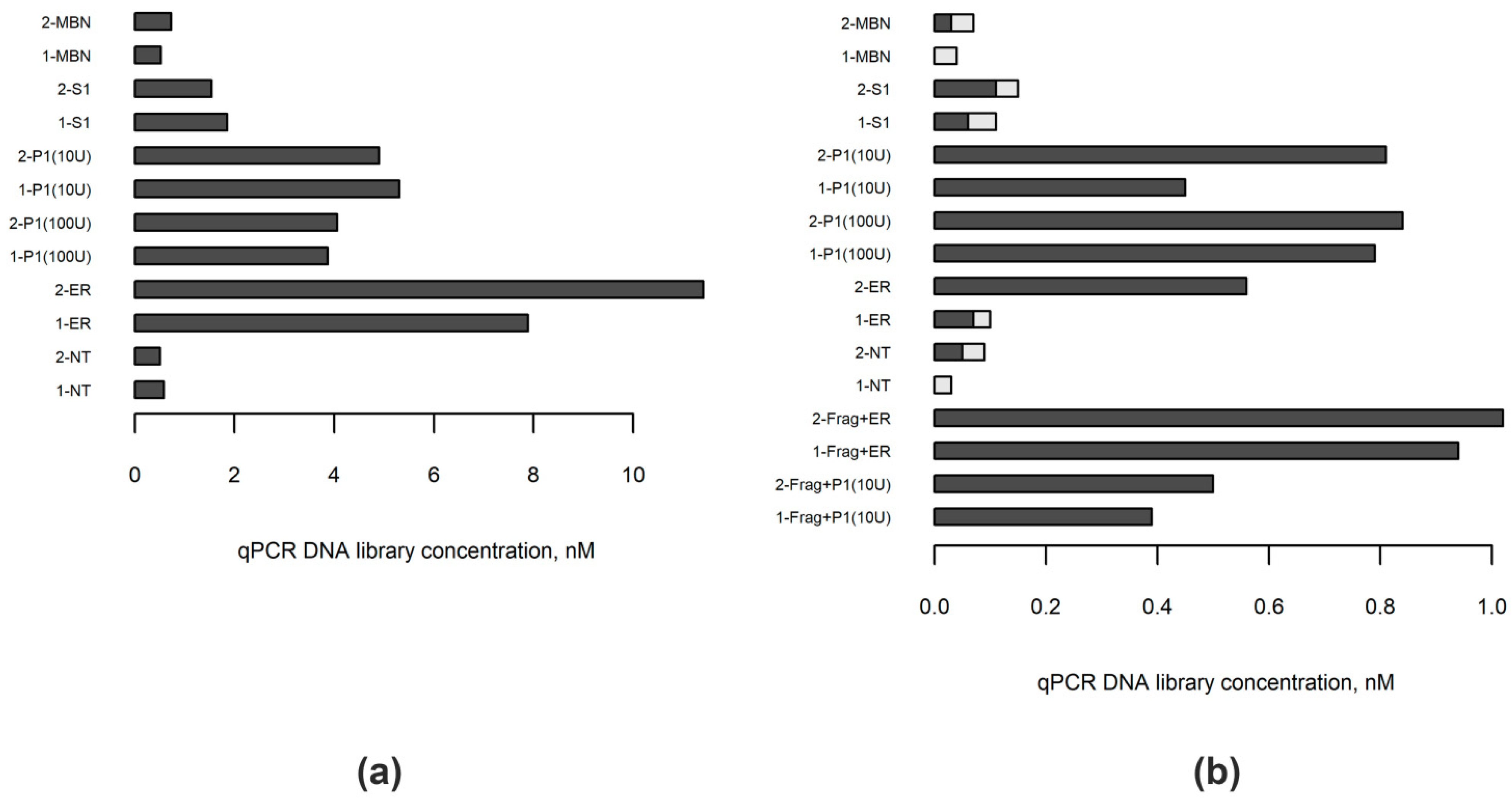
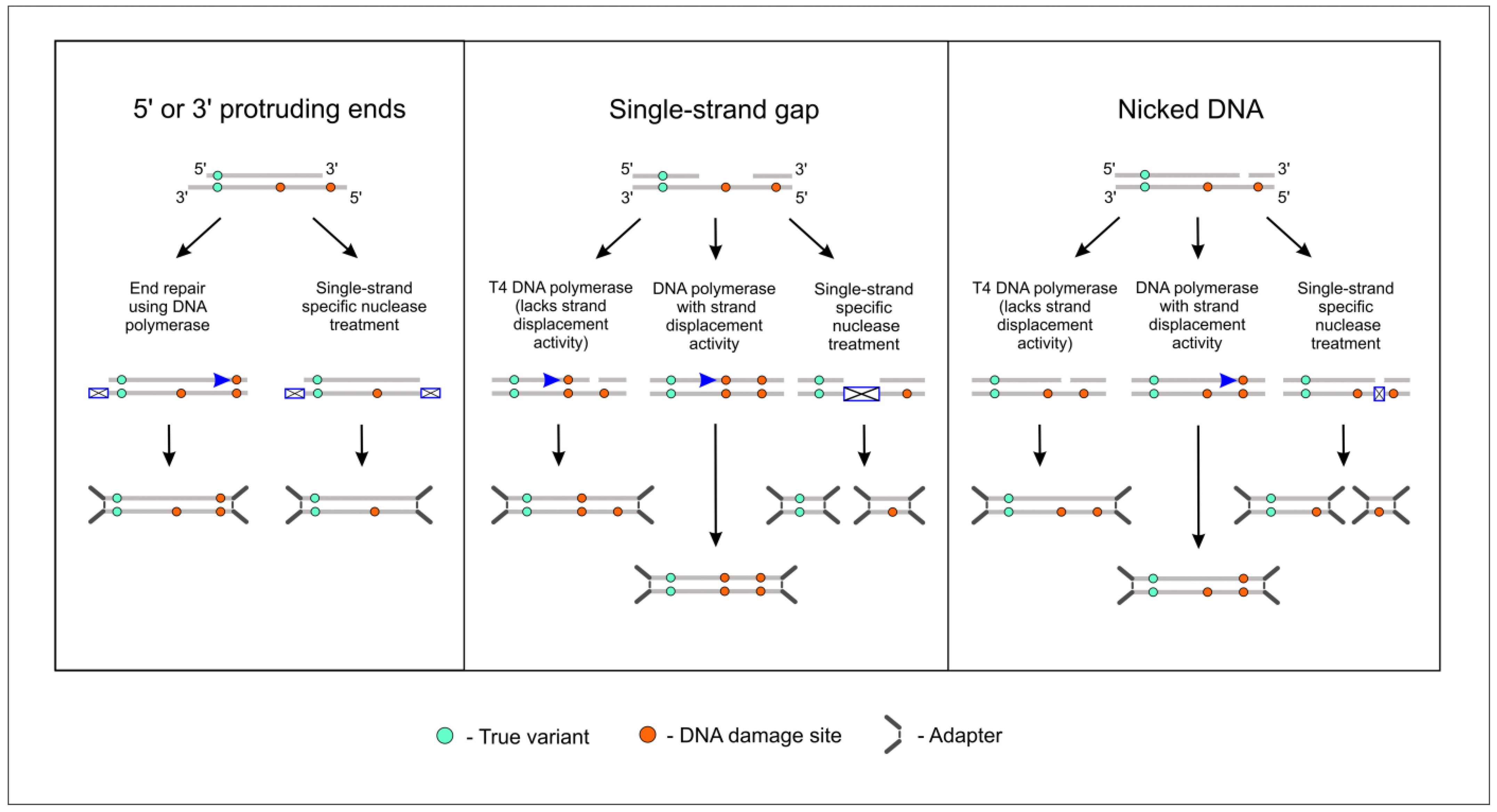
2.1. Choosing Enzyme to Replace DNA Polymerase in the End Repair Process
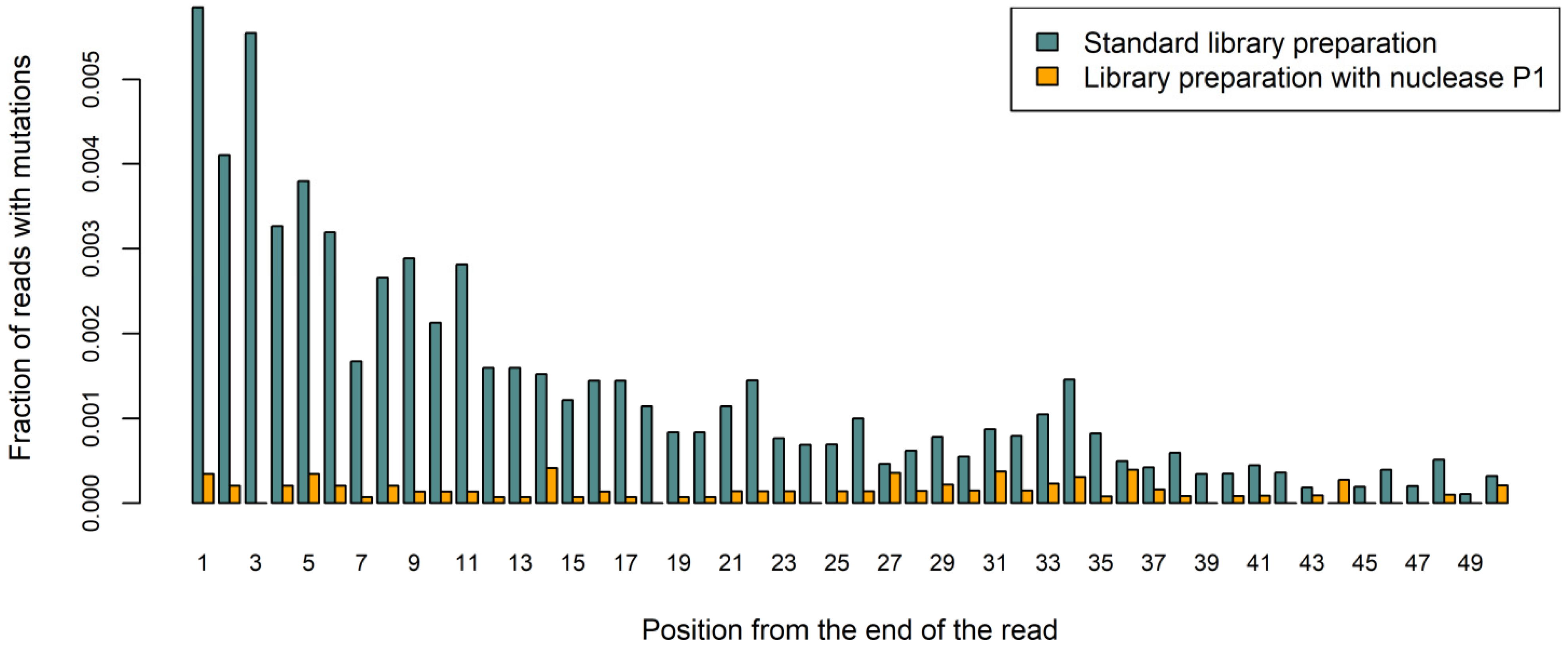
2.2. Comparing the DNA Libraries Prepared from FFPE Samples Using Standard and Nuclease P1-Based Method
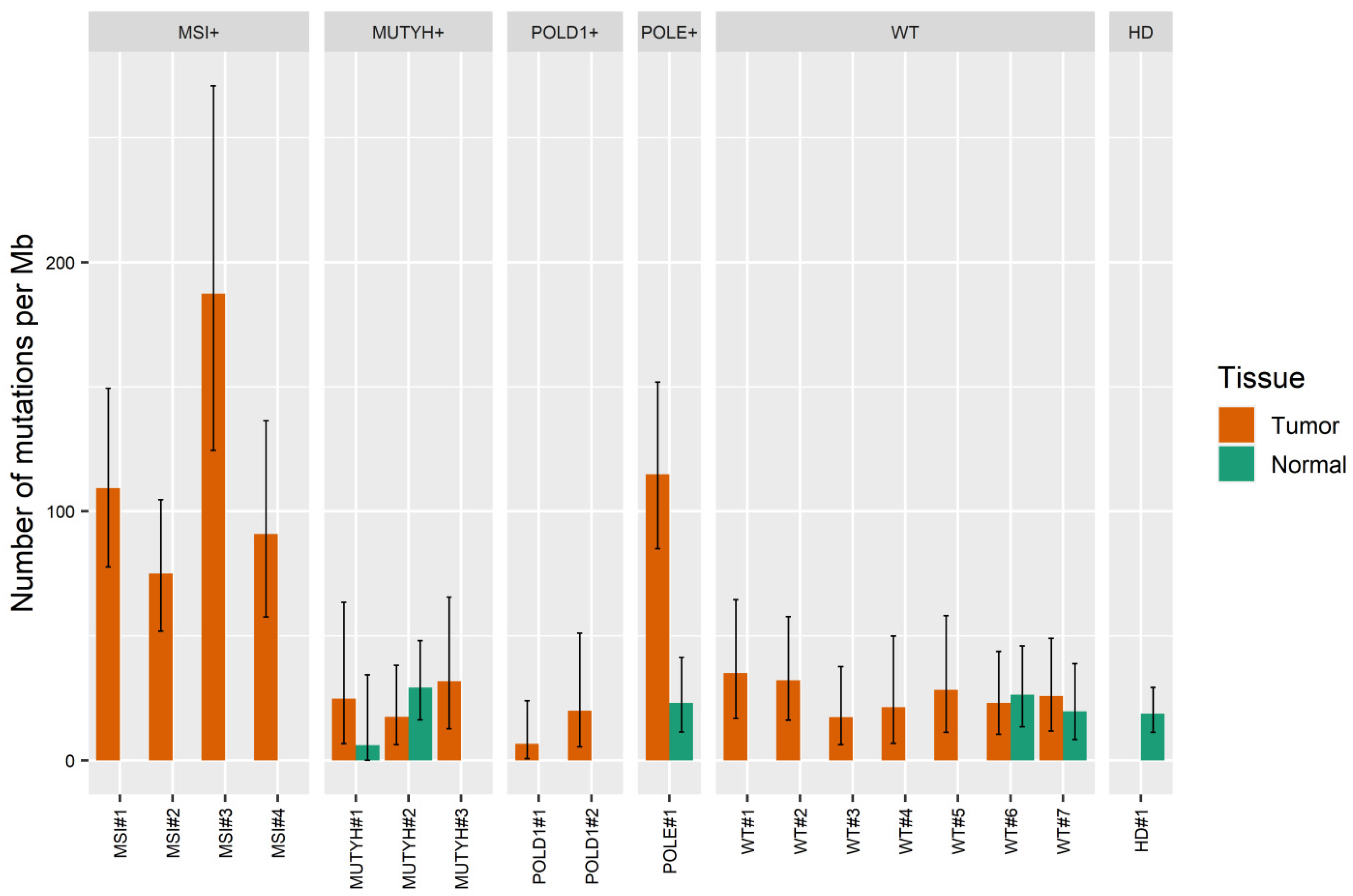
2.3. Quantifying the Number of Mutations in NGS Libraries Prepared with Nuclease P1 from Colorectal Carcinoma Samples with or without Hypermutator Phenotype
3. Discussion
4. Materials and Methods
4.1. Samples and DNA Extraction
4.2. Preparations of NGS Libraries from FFPE- and Blood-Derived DNA Using Standard and Single-Strand-Specific Nuclease-Based Methods
4.3. Quantification of DNA Libraries by Real-Time PCR
4.4. Preparation of BotSeqS Libraries
4.5. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schmitt, M.W.; Kennedy, S.R.; Salk, J.J.; Fox, E.J.; Hiatt, J.B.; Loeb, L.A. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 14508–14513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Q.; Xu, C.; Kim, D.; Lewis, M.; DiCarlo, J.; Wang, Y. Targeted Single Primer Enrichment Sequencing with Single End Duplex-UMI. Sci. Rep. 2019, 9, 4810. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhang, Y.; Wang, D.; Liu, F.; Fu, Y.; Xiang, S.; Su, L.; Li, J.; Dai, H.; Huang, B. SinoDuplex: An Improved Duplex Sequencing Approach to Detect Low-frequency Variants in Plasma cfDNA Samples. Genom. Proteom. Bioinform. 2020, 18, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Valentine, C.C.; Young, R.R.; Fielden, M.R.; Kulkarni, R.; Williams, L.N.; Li, T.; Minocherhomji, S.; Salk, J.J. Direct quantification of in vivo mutagenesis and carcinogenesis using duplex sequencing. Proc. Natl. Acad. Sci. USA 2020, 117, 33414–33425. [Google Scholar] [CrossRef]
- Hoang, M.L.; Kinde, I.; Tomasetti, C.; McMahon, K.W.; Rosenquist, T.A.; Grollman, A.P.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. Genome-wide quantification of rare somatic mutations in normal human tissues using massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2016, 113, 9846–9851. [Google Scholar] [CrossRef] [Green Version]
- You, X.; Thiruppathi, S.; Liu, W.; Cao, Y.; Naito, M.; Furihata, C.; Honma, M.; Luan, Y.; Suzuki, T. Detection of genome-wide low-frequency mutations with Paired-End and Complementary Consensus Sequencing (PECC-Seq) revealed end-repair-derived artifacts as residual errors. Arch. Toxicol. 2020, 94, 3475–3485. [Google Scholar] [CrossRef]
- Abascal, F.; Harvey, L.M.R.; Mitchell, E.; Lawson, A.R.J.; Lensing, S.V.; Ellis, P.; Russell, A.J.C.; Alcantara, R.E.; Baez-Ortega, A.; Wang, Y.; et al. Somatic mutation landscapes at single-molecule resolution. Nature 2021, 593, 405–410. [Google Scholar] [CrossRef]
- Do, H.; Dobrovic, A. Sequence artifacts in DNA from formalin-fixed tissues: Causes and strategies for minimization. Clin. Chem. 2015, 61, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Mathieson, W.; Thomas, G.A. Why Formalin-fixed, Paraffin-embedded Biospecimens Must Be Used in Genomic Medicine: An Evidence-based Review and Conclusion. J. Histochem. Cytochem. 2020, 68, 543–552. [Google Scholar] [CrossRef]
- Desai, N.A.; Shankar, V. Single-strand-specific nucleases. FEMS Microbiol. Rev. 2003, 26, 457–491. [Google Scholar] [CrossRef]
- Chun, S.M.; Sung, C.O.; Jeon, H.; Kim, T.I.; Lee, J.Y.; Park, H.; Kim, Y.; Kim, D.; Jang, S.J. Next-Generation Sequencing Using S1 Nuclease for Poor-Quality Formalin-Fixed, Paraffin-Embedded Tumor Specimens. J. Mol. Diagn. 2018, 20, 802–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haile, S.; Corbett, R.D.; Bilobram, S.; Bye, M.H.; Kirk, H.; Pandoh, P.; Trinh, E.; MacLeod, T.; McDonald, H.; Bala, M.; et al. Sources of erroneous sequences and artifact chimeric reads in next generation sequencing of genomic DNA from formalin-fixed paraffin-embedded samples. Nucleic Acids Res. 2019, 47, e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, H.; Wong, S.Q.; Li, J.; Dobrovic, A. Reducing sequence artifacts in amplicon-based massively parallel sequencing of formalin-fixed paraffin-embedded DNA by enzymatic depletion of uracil-containing templates. Clin. Chem. 2013, 59, 1376–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsubo, Y.; Matsumura, S.; Ikeda, N.; Yamane, M. Single-strand specific nuclease enhances accuracy of error-corrected sequencing and improves rare mutation-detection sensitivity. Arch. Toxicol. 2022, 96, 377–386. [Google Scholar] [CrossRef] [PubMed]
- McDonough, S.J.; Bhagwate, A.; Sun, Z.; Wang, C.; Zschunke, M.; Gorman, J.A.; Kopp, K.J.; Cunningham, J.M. Use of FFPE-derived DNA in next generation sequencing: DNA extraction methods. PLoS ONE 2019, 14, e0211400. [Google Scholar] [CrossRef] [Green Version]
- Viel, A.; Bruselles, A.; Meccia, E.; Fornasarig, M.; Quaia, M.; Canzonieri, V.; Policicchio, E.; Urso, E.D.; Agostini, M.; Genuardi, M.; et al. A Specific Mutational Signature Associated with DNA 8-Oxoguanine Persistence in MUTYH-defective Colorectal Cancer. EBioMedicine 2017, 20, 39–49. [Google Scholar] [CrossRef]
- Briggs, S.; Tomlinson, I. Germline and somatic polymerase ε and δ mutations define a new class of hypermutated colorectal and endometrial cancers. J. Pathol. 2013, 230, 148–153. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Ouyang, W.; Zhao, W.; Shao, L.; Li, B.; Liu, B.; Wang, D.; Han-Zhang, H.; Zhang, Z.; Shao, L.; et al. Distinctive genomic characteristics in POLE/POLD1-mutant cancers can potentially predict beneficial clinical outcomes in patients who receive immune checkpoint inhibitor. Ann. Transl. Med. 2021, 9, 129. [Google Scholar] [CrossRef]
- Keshinro, A.; Vanderbilt, C.; Kim, J.K.; Firat, C.; Chen, C.T.; Yaeger, R.; Ganesh, K.; Segal, N.H.; Gonen, M.; Shia, J.; et al. Tumor-Infiltrating Lymphocytes, Tumor Mutational Burden, and Genetic Alterations in Microsatellite Unstable, Microsatellite Stable, or Mutant POLE/POLD1 Colon Cancer. JCO Precis. Oncol. 2021, 5, 817–826. [Google Scholar] [CrossRef]
- Müllenbach, R.; Lagoda, P.J.; Welter, C. An efficient salt-chloroform extraction of DNA from blood and tissues. Trends Genet. 1989, 5, 391. [Google Scholar]
- NEBNext Technical Guide for Illumina. Available online: https://international.neb.com/products/-/media/nebus/files/brochures/nebnextillumina.pdf?rev=253e68f9011147ecab29d91a727588ac&hash=C0DC098E1B292B40636BD4B64EF70B1D (accessed on 19 October 2021).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard. Available online: https://broadinstitute.github.io/picard/ (accessed on 19 October 2021).
- Genome Reference Consortium. Human Genome Overview. Available online: https://www.ncbi.nlm.nih.gov/grc/human (accessed on 19 October 2021).
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Pysam. Available online: https://github.com/pysam-developers/pysam (accessed on 19 October 2021).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org (accessed on 5 August 2021).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer International Publishing: New York, NY, USA, 2016; pp. 1–260. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Label | Sex | Age | Main Genotyping Result | Mutations in KRAS, NRAS or BRAF Genes |
|---|---|---|---|---|
| MSI#1 | female | 35 | MSI | No |
| MSI#2 | female | 71 | MSI | BRAF V600E |
| MSI#3 | female | 45 | MSI | No |
| MSI#4 | female | 78 | MSI | KRAS Q61K |
| MUTYH#1 | female | 48 | MUTYH Y179C (ClinVar VCV000005293.37, pathogenic), G396D (ClinVar VCV000005294, pathogenic) | KRAS G12C |
| MUTYH#2 | female | 59 | MUTYH Y179C (ClinVar VCV000005293.37, pathogenic), G396D (ClinVar VCV000005294, pathogenic) | KRAS G12C |
| MUTYH#3 | male | 39 | MUTYH R245H (ClinVar VCV000140877.19, pathogenic), G396D (ClinVar VCV000005294, pathogenic) | KRAS G12C |
| POLD1#1 | male | 42 | POLD1 A516V (dbSNP rs752755096, rare SNP, significance unknown) | BRAF V600E |
| POLD1#2 | female | 51 | POLD1 V538I (ClinVar VCV001025450.1, uncertain significance) | BRAF V600E |
| POLE#1 | male | 33 | POLE S297F (somatic mutation, COSMIC COSM937330, FATHMM prediction: pathogenic) | KRAS A146T |
| WT#1 | male | 68 | KRAS G13D | |
| WT#2 | male | 67 | No | |
| WT#3 | male | 68 | No | |
| WT#4 | female | 66 | KRAS G12V | |
| WT#5 | female | 72 | KRAS G12V | |
| WT#6 | female | 40 | No | |
| WT#7 | male | 69 | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitiushkina, N.V.; Yanus, G.A.; Kuligina, E.S.; Laidus, T.A.; Romanko, A.A.; Kholmatov, M.M.; Ivantsov, A.O.; Aleksakhina, S.N.; Imyanitov, E.N. Preparation of Duplex Sequencing Libraries for Archival Paraffin-Embedded Tissue Samples Using Single-Strand-Specific Nuclease P1. Int. J. Mol. Sci. 2022, 23, 4586. https://doi.org/10.3390/ijms23094586
Mitiushkina NV, Yanus GA, Kuligina ES, Laidus TA, Romanko AA, Kholmatov MM, Ivantsov AO, Aleksakhina SN, Imyanitov EN. Preparation of Duplex Sequencing Libraries for Archival Paraffin-Embedded Tissue Samples Using Single-Strand-Specific Nuclease P1. International Journal of Molecular Sciences. 2022; 23(9):4586. https://doi.org/10.3390/ijms23094586
Chicago/Turabian StyleMitiushkina, Natalia V., Grigory A. Yanus, Ekatherina Sh. Kuligina, Tatiana A. Laidus, Alexandr A. Romanko, Maksim M. Kholmatov, Alexandr O. Ivantsov, Svetlana N. Aleksakhina, and Evgeny N. Imyanitov. 2022. "Preparation of Duplex Sequencing Libraries for Archival Paraffin-Embedded Tissue Samples Using Single-Strand-Specific Nuclease P1" International Journal of Molecular Sciences 23, no. 9: 4586. https://doi.org/10.3390/ijms23094586
APA StyleMitiushkina, N. V., Yanus, G. A., Kuligina, E. S., Laidus, T. A., Romanko, A. A., Kholmatov, M. M., Ivantsov, A. O., Aleksakhina, S. N., & Imyanitov, E. N. (2022). Preparation of Duplex Sequencing Libraries for Archival Paraffin-Embedded Tissue Samples Using Single-Strand-Specific Nuclease P1. International Journal of Molecular Sciences, 23(9), 4586. https://doi.org/10.3390/ijms23094586