
Soluble Epoxide Hydrolase as a Therapeutic Target for Neuropsychiatric Disorders


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Depression
3. ASD and Schizophrenia
4. Parkinson’s Disease
5. Stroke
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheng, L.; Liu, J.; Chen, Z. The histaminergic system in neuropsychiatric disorders. Biomolecules 2021, 11, 1345. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.J.; O’Donovan, M.C. The genetics of neuropsychiatric disorders. Brain Neurosci. Adv. 2018, 2, 2398212818799271. [Google Scholar] [CrossRef] [PubMed]
- Kessler, R.C.; Amminger, G.P.; Aguilar-Gaxiola, S.; Alonso, J.; Lee, S.; Ustun, T.B. Age of onset of mental disorders: A review of recent literature. Curr. Opin. Psychiatry 2007, 20, 359. [Google Scholar] [CrossRef] [PubMed]
- GBD 2019 Diseases and Injuries Collaborators. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- GBD 2019 Diseases and Injuries Collaborators. Global, regional, and national burden of 12 mental disorders in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Psychiatry 2022, 9, 137–150. [Google Scholar] [CrossRef]
- COVID-19 Mental Disorders Collaborators. Global prevalence and burden of depressive and anxiety disorders in 204 countries and territories in 2020 due to the COVID-19 pandemic. Lancet 2021, 398, 1700–1712. [Google Scholar] [CrossRef]
- Gooch, C.L.; Pracht, E.; Borenstein, A.R. The burden of neurological disease in the United States: A summary report and call to action. Ann. Neurol. 2017, 81, 479–484. [Google Scholar] [CrossRef]
- Hashimoto, K.; Malchow, B.; Falkai, P.; Schmitt, A. Glutamate modulators as potential therapeutic drugs in schizophrenia and affective disorders. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, 367–377. [Google Scholar] [CrossRef]
- Hashimoto, K. Targeting of NMDA receptors in new treatments for schizophrenia. Expert Opin. Ther. Targets 2014, 18, 1049–1063. [Google Scholar] [CrossRef]
- Hashimoto, K. Recent advances in the early intervention in schizophrenia: Future direction from preclinical findings. Curr. Psychiatry Rep. 2019, 21, 75. [Google Scholar] [CrossRef]
- LeWitt, P.A.; Chaudhuri, K.R. Unmet needs in Parkinson disease: Motor and non-motor. Park. Relat. Disord. 2020, 80 (Suppl. 1), S7–S12. [Google Scholar] [CrossRef] [PubMed]
- Grosso Jasutkar, H.; Oh, S.E.; Mouradian, M.M. Therapeutics in the pipeline targeting alpha-synuclein for Parkinson’s disease. Pharmacol. Rev. 2022, 74, 207–237. [Google Scholar] [CrossRef] [PubMed]
- Mongan, D.; Healy, C.; Jones, H.J.; Zammit, S.; Cannon, M.; Cotter, D.R. Plasma polyunsaturated fatty acids and mental disorders in adolescence and early adulthood: Cross-sectional and longitudinal associations in a general population cohort. Transl. Psychiatry 2021, 11, 321. [Google Scholar] [CrossRef] [PubMed]
- Healy-Stoffel, M.; Levant, B. N-3 (Omega-3) fatty acids: Effects on brain dopamine systems and potential role in the etiology and treatment of neuropsychiatric disorders. CNS Neurol. Disord. Drug Targets 2018, 17, 216–232. [Google Scholar] [CrossRef]
- Morgese, M.G.; Schiavone, S.; Mhillaj, E.; Bove, M.; Tucci, P.; Trabace, L. N-3 PUFA diet enrichment prevents amyloid beta-induced depressive-like phenotype. Pharmacol. Res. 2018, 129, 526–534. [Google Scholar] [CrossRef]
- Dagnino-Subiabre, A. Stress and Western diets increase vulnerability to neuropsychiatric disorders: A common mechanism. Nutr. Neurosci. 2021, 24, 624–634. [Google Scholar] [CrossRef]
- Tesei, A.; Crippa, A.; Ceccarelli, S.B.; Mauri, M.; Molteni, M.; Agostoni, C.; Nobile, M. The potential relevance of docosahexaenoic acid and eicosapentaenoic acid to the etiopathogenesis of childhood neuropsychiatric disorders. Eur. Child Adolesc. Psychiatry 2017, 26, 1011–1030. [Google Scholar] [CrossRef]
- Daray, F.M.; Grendas, L.N.; Rodante, D.E.; Errasti, A.E.; Cases, G.G.; Moix, C.F.; Uicich, R.E.; GimÉNez, M.I.; Puppo, S.; Fasolino, G.H.; et al. Polyunsaturated fatty acids as predictors of future suicide attempt. Prostaglandins Leukot. Essent. Fat. Acids 2021, 165, 102247. [Google Scholar] [CrossRef]
- Di Miceli, M.; Bosch-Bouju, C.; Layé, S. PUFA and their derivatives in neurotransmission and synapses: A new hallmark of synaptopathies. Proc. Nutr. Soc. 2020, 79, 388–403. [Google Scholar] [CrossRef]
- Chen, W.; Chen, Y.; Wu, R.; Guo, G.; Liu, Y.; Zeng, B.; Liao, X.; Wang, Y.; Wang, X. DHA alleviates diet-induced skeletal muscle fiber remodeling via FTO/m6A/DDIT4/PGC1α signaling. BMC Biol. 2022, 20, 39. [Google Scholar] [CrossRef]
- Díaz, M.; Mesa-Herrera, F.; Marín, R. DHA and its elaborated modulation of antioxidant defenses of the brain: Implications in aging and AD neurodegeneration. Antioxidants 2021, 10, 907. [Google Scholar] [CrossRef] [PubMed]
- Funk Colin, D. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, S.K.; Bazinet, R.P. The emerging role of docosahexaenoic acid in neuroinflammation. Curr. Opin. Investig. Drugs 2008, 9, 735–743. [Google Scholar] [PubMed]
- Borsini, A.; Stangl, D.; Jeffries, A.R.; Pariante, C.M.; Thuret, S. The role of omega-3 fatty acids in preventing glucocorticoid-induced reduction in human hippocampal neurogenesis and increase in apoptosis. Transl. Psychiatry 2020, 10, 219. [Google Scholar] [CrossRef]
- Giacobbe, J.; Benoiton, B.; Zunszain, P.; Pariante, C.M.; Borsini, A. The anti-inflammatory role of omega-3 polyunsaturated fatty acids metabolites in pre-clinical models of psychiatric, neurodegenerative, and neurological disorders. Front. Psychiatry 2020, 11, 122. [Google Scholar] [CrossRef]
- Morisseau, C.; Hammock, B.D. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 37–58. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, K. Role of soluble epoxide hydrolase in metabolism of PUFAs in psychiatric and neurological disorders. Front. Pharmacol. 2019, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- Imig, J.D.; Cervenka, L.; Neckar, J. Epoxylipids and soluble epoxide hydrolase in heart diseases. Biochem. Pharmacol. 2022, 195, 114866. [Google Scholar] [CrossRef]
- Capozzi, M.E.; Hammer, S.S.; McCollum, G.W.; Penn, J.S. Epoxygenated fatty acids inhibit retinal vascular inflammation. Sci. Rep. 2016, 6, 39211. [Google Scholar] [CrossRef]
- Yanai, R.; Mulki, L.; Hasegawa, E.; Takeuchi, K.; Sweigard, H.; Suzuki, J.; Gaissert, P.; Vavvas, D.G.; Sonoda, K.H.; Rothe, M.; et al. Cytochrome P450-generated metabolites derived from ω-3 fatty acids attenuate neovascularization. Proc. Natl. Acad. Sci. USA 2014, 111, 9603–9608. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, L.R.; Fleming, I. Role of cytochrome P450-derived, polyunsaturated fatty acid mediators in diabetes and the metabolic syndrome. Prostaglandins Other Lipid Mediat. 2020, 148, 106407. [Google Scholar] [CrossRef] [PubMed]
- Hiesinger, K.; Wagner, K.M.; Hammock, B.D.; Proschak, E.; Hwang, S.H. Development of multitarget agents possessing soluble epoxide hydrolase inhibitory activity. Prostaglandins Other Lipid Mediat. 2019, 140, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Hildreth, K.; Kodani, S.D.; Hammock, B.D.; Zhao, L. Cytochrome P450-derived linoleic acid metabolites EpOMEs and DiHOMEs: A review of recent studies. J. Nutr. Biochem. 2020, 86, 108484. [Google Scholar] [CrossRef]
- Jones, R.D.; Liao, J.; Tong, X.; Xu, D.; Sun, L.; Li, H.; Yang, G.Y. Epoxy-oxylipins and soluble epoxide hydrolase metabolic pathway as targets for NSAID-induced gastroenteropathy and inflammation-associated carcinogenesis. Front. Pharmacol. 2019, 10, 731. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.P.; Zhang, X.Y.; Morisseau, C.; Hwang, S.H.; Zhang, Z.J.; Hammock, B.D.; Ma, X.C. Discovery of soluble epoxide hydrolase inhibitors from chemical synthesis and natural products. J. Med. Chem. 2021, 64, 184–215. [Google Scholar] [CrossRef]
- Abdoli, A.; Taghipour, A.; Pirestani, M.; Mofazzal Jahromi, M.A.; Roustazadeh, A.; Mir, H.; Ardakani, H.M.; Kenarkoohi, A.; Falahi, S.; Karimi, M. Infections, inflammation, and risk of neuropsychiatric disorders: The neglected role of “co-infection”. Heliyon 2020, 6, e05645. [Google Scholar] [CrossRef]
- Institute of Health Metrics and Evaluation, Global Health Datta Exchange. Available online: http://ghdx.healthdata.org/gbd-results-tool?params=gbd-api-2019-permalink/d780dffbe8a381b25e1416884959e88b (accessed on 1 May 2021).
- Hashimoto, K. Soluble epoxide hydrolase: A new therapeutic target for depression. Expert Opin. Ther. Targets 2016, 20, 1149–1151. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, K. Molecular mechanisms of the rapid-acting and long-lasting antidepressant actions of (R)-ketamine. Biochem. Pharmacol. 2020, 177, 113935. [Google Scholar] [CrossRef]
- Hashimoto, K. Inflammatory biomarkers as differential predictors of antidepressant response. Int. J. Mol. Sci. 2015, 16, 7796–7801. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.C.; Yao, W.; Hashimoto, K. Brain-derived neurotrophic factor (BDNF)-TrkB signaling in inflammation-related depression and potential therapeutic targets. Curr. Neuropharmacol. 2016, 14, 721–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troubat, R.; Barone, P.; Leman, S.; Desmidt, T.; Cressant, A.; Atanasova, B.; Brizard, B.; El Hage, W.; Surget, A.; Belzung, C.; et al. Neuroinflammation and depression: A review. Eur. J. Neurosci. 2021, 53, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Carlessi, A.S.; Borba, L.A.; Zugno, A.I.; Quevedo, J.; Réus, G.Z. Gut microbiota–brain axis in depression: The role of neuroinflammation. Eur. J. Neurosci. 2021, 53, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chang, L.; Hashimoto, K. Molecular mechanisms underlying the antidepressant actions of arketamine: Beyond the NMDA receptor. Mol. Psychiatry 2021, 27, 559–573. [Google Scholar] [CrossRef]
- Chang, L.; Wei, Y.; Hashimoto, K. Brain-gut-microbiota axis in depression: A historical overview and future directions. Brain Res. Bull. 2022, 182, 44–56. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, T.; Liao, L.; Fan, X.; Chang, L.; Hashimoto, K. Brain-spleen axis in health and diseases: A review and future perspective. Brain Res. Bull. 2022, 182, 130–140. [Google Scholar] [CrossRef]
- Zhao, G.; Tu, L.; Li, X.; Yang, S.; Chen, C.; Xu, X.; Wang, P.; Wang, D.W. Delivery of AAV2-CYP2J2 protects remnant kidney in the 5/6-nephrectomized rat via inhibition of apoptosis and fibrosis. Hum. Gene Ther. 2012, 23, 688–699. [Google Scholar] [CrossRef]
- Morin, C.; Sirois, M.; Echave, V.; Gomes, M.M.; Rousseau, E. EET displays anti-inflammatory effects in TNF-α–stimulated human bronchi. Am. J. Respir. Cell Mol. Biol. 2008, 38, 192–201. [Google Scholar] [CrossRef]
- Ren, Q.; Ma, M.; Ishima, T.; Morisseau, C.; Yang, J.; Wagner, K.M.; Zhang, J.C.; Yang, C.; Yao, W.; Dong, C.; et al. Gene deficiency and pharmacological inhibition of soluble epoxide hydrolase confers resilience to repeated social defeat stress. Proc. Natl. Acad. Sci. USA 2016, 113, E1944–E1952. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Ishima, T.; Qu, Y.; Shan, J.; Chang, L.; Wei, Y.; Zhang, J.; Pu, Y.; Fujita, Y.; Tan, Y.; et al. Ingestion of Faecalibaculum rodentium causes depression-like phenotypes in resilient Ephx2 knock-out mice: A role of brain-gut-microbiota axis via the subdiaphragmatic vagus nerve. J. Affect. Disord. 2021, 292, 565–573. [Google Scholar] [CrossRef]
- Qin, X.H.; Wu, Z.; Dong, J.H.; Zeng, Y.N.; Xiong, W.C.; Liu, C.; Wang, M.Y.; Zhu, M.Z.; Chen, W.J.; Zhang, Y.; et al. Liver soluble epoxide hydrolase regulates behavioral and cellular effects of chronic stress. Cell Rep. 2019, 29, 3223–3234.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Tan, Y.; Chang, L.; Hammock, B.D.; Hashimoto, K. Increased expression of soluble epoxide hydrolase in the brain and liver from patients with major psychiatric disorders: A role of brain–liver axis. J. Affect. Disord. 2020, 270, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Ren, Q.; Zhang, J.C.; Chen, Q.X.; Hashimoto, K. Altered expression of BDNF, BDNF pro-peptide and their precursor proBDNF in brain and liver tissues from psychiatric disorders: Rethinking the brain-liver axis. Transl. Psychiatry 2017, 7, e1128. [Google Scholar] [CrossRef] [PubMed]
- Marowsky, A.; Burgener, J.; Falck, J.R.; Fritschy, J.M.; Arand, M. Distribution of soluble and microsomal epoxide hydrolase in the mouse brain and its contribution to cerebral epoxyeicosatrienoic acid metabolism. Neuroscience 2009, 163, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Cao, X.; Zeng, Y.; Qin, X.; Zhu, M.; Ren, J.; Wu, Z.; Huang, Q.; Zhang, Y.; Wang, M.; et al. Astrocytic epoxyeicosatrienoic acid signaling in the medial prefrontal cortex modulates depressive-like behaviors. J. Neurosci. 2019, 39, 4606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Lu, C.L.; Huang, J.; Fan, J.; Guo, F.; Mo, J.W.; Huang, W.Y.; Kong, P.L.; Li, X.W.; Sun, L.R.; et al. A distinct metabolically defined central nucleus circuit bidirectionally controls anxiety-related behaviors. J. Neurosci. 2022, 42, 2356–2370. [Google Scholar] [CrossRef]
- Wu, Q.; Cai, H.; Song, J.; Chang, Q. The effects of sEH inhibitor on depression-like behavior and neurogenesis in male mice. J. Neurosci. Res. 2017, 95, 2483–2492. [Google Scholar] [CrossRef]
- Wu, Q.; Song, J.; Meng, D.; Chang, Q. TPPU, a sEH inhibitor, attenuates corticosterone-induced PC12 cell injury by modulation of BDNF-TrkB pathway. J. Mol. Neurosci. 2019, 67, 364–372. [Google Scholar] [CrossRef]
- Peng, W.; Shen, Y.; Wang, Q.; Ding, J.; Wang, X. TPPU Pre-treatment rescues dendritic spine loss and alleviates depressive behaviours during the latent period in the lithium chloride-pilocarpine-induced status epilepticus rat model. Brain Sci. 2021, 11, 1465. [Google Scholar] [CrossRef]
- Shen, Y.; Peng, W.; Chen, Q.; Hammock, B.D.; Liu, J.; Li, D.; Yang, J.; Ding, J.; Wang, X. Anti-inflammatory treatment with a soluble epoxide hydrolase inhibitor attenuates seizures and epilepsy-associated depression in the LiCl-pilocarpine post-status epilepticus rat model. Brain Behav. Immun. 2019, 81, 535–544. [Google Scholar] [CrossRef]
- Borsini, A.; Nicolaou, A.; Camacho-Muñoz, D.; Kendall, A.C.; Di Benedetto, M.G.; Giacobbe, J.; Su, K.P.; Pariante, C.M. Omega-3 polyunsaturated fatty acids protect against inflammation through production of LOX and CYP450 lipid mediators: Relevance for major depression and for human hippocampal neurogenesis. Mol. Psychiatry 2021, 26, 6773–6788. [Google Scholar] [CrossRef] [PubMed]
- Anita, N.Z.; Forkan, N.; Kamal, R.; Nguyen, M.M.; Yu, D.; Major-Orfao, C.; Wong, S.K.; Lanctôt, K.L.; Herrmann, N.; Oh, P.I.; et al. Serum soluble epoxide hydrolase related oxylipins and major depression in patients with type 2 diabetes. Psychoneuroendocrinology 2021, 126, 105149. [Google Scholar] [CrossRef] [PubMed]
- Swardfager, W.; Hennebelle, M.; Yu, D.; Hammock, B.D.; Levitt, A.J.; Hashimoto, K.; Taha, A.Y. Metabolic/inflammatory/vascular comorbidity in psychiatric disorders; soluble epoxide hydrolase (sEH) as a possible new target. Neurosci. Biobehav. Rev. 2018, 87, 56–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Q. Soluble epoxide hydrolase inhibitor: A novel potential therapeutic or prophylactic drug for psychiatric disorders. Front. Pharmacol. 2019, 10, 420. [Google Scholar] [CrossRef] [PubMed]
- Atone, J.; Wagner, K.; Hashimoto, K.; Hammock, B.D. Cytochrome P450 derived epoxidized fatty acids as a therapeutic tool against neuroinflammatory diseases. Prostaglandins Other Lipid Mediat. 2020, 147, 106385. [Google Scholar] [CrossRef]
- Borsini, A. The role of soluble epoxide hydrolase and its inhibitors in depression. Brain Behav. Immun. Health 2021, 16, 100325. [Google Scholar] [CrossRef]
- Bilbo, S.D.; Block, C.L.; Bolton, J.L.; Hanamsagar, R.; Tran, P.K. Beyond infection—Maternal immune activation by environmental factors, microglial development, and relevance for autism spectrum disorders. Exp. Neurol. 2018, 299, 241–251. [Google Scholar] [CrossRef]
- Brown, A.S.; Meyer, U. maternal immune activation and neuropsychiatric illness: A translational research perspective. Am. J. Psychiatry 2018, 175, 1073–1083. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, M.V.; Moon, H.M.; Su, J.; Palmer, T.D.; Courchesne, E.; Pramparo, T. Maternal immune activation dysregulation of the fetal brain transcriptome and relevance to the pathophysiology of autism spectrum disorder. Mol. Psychiatry 2018, 23, 1001–1013. [Google Scholar] [CrossRef] [Green Version]
- Solek, C.M.; Farooqi, N.; Verly, M.; Lim, T.K.; Ruthazer, E.S. Maternal immune activation in neurodevelopmental disorders. Dev. Dyn. 2018, 247, 588–619. [Google Scholar] [CrossRef]
- Conway, F.; Brown, A.S. Maternal immune activation and related factors in the risk of offspring psychiatric disorders. Front. Psychiatry 2019, 10, 430. [Google Scholar] [CrossRef] [PubMed]
- Siniscalco, D.; Schultz, S.; Brigida, A.L.; Antonucci, N. Inflammation and neuro-immune dysregulations in autism spectrum disorders. Pharmaceuticals 2018, 11, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matta, S.M.; Hill-Yardin, E.L.; Crack, P.J. The influence of neuroinflammation in autism spectrum disorder. Brain Behav. Immun. 2019, 79, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Gevezova, M.; Sarafian, V.; Anderson, G.; Maes, M. Inflammation and mitochondrial dysfunction in autism spectrum disorder. CNS Neurol. Disord. Drug Targets 2020, 19, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.Y.; Xu, L.l.; Shao, L.; Xia, R.M.; Yu, Z.H.; Ling, Z.X.; Yang, F.; Deng, M.; Ruan, B. Maternal infection during pregnancy and risk of autism spectrum disorders: A systematic review and meta-analysis. Brain Behav. Immun. 2016, 58, 165–172. [Google Scholar] [CrossRef]
- Hashimoto, K. Risk of neuropsychiatric disorders in offspring of COVID-19-infected pregnant women and nutritional intervention. Eur. Arch. Psychiatry Clin. Neurosci. 2021, 271, 387–389. [Google Scholar] [CrossRef]
- Lins, B. Maternal immune activation as a risk factor for psychiatric illness in the context of the SARS-CoV-2 pandemic. Brain Behav. Immun. Health 2021, 16, 100297. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Suzuki, T.; Hashimoto, K. Mechanisms of action of fluvoxamine for COVID-19: A historical review. Mol. Psychiatry 2022, 1–10. [Google Scholar] [CrossRef]
- Guma, E.; Plitman, E.; Chakravarty, M.M. The role of maternal immune activation in altering the neurodevelopmental trajectories of offspring: A translational review of neuroimaging studies with implications for autism spectrum disorder and schizophrenia. Neurosci. Biobehav. Rev. 2019, 104, 141–157. [Google Scholar] [CrossRef]
- Haddad, F.L.; Patel, S.V.; Schmid, S. Maternal immune activation by poly(I:C) as a preclinical model for neurodevelopmental disorders: A focus on autism and schizophrenia. Neurosci. Biobehav. Rev. 2020, 113, 546–567. [Google Scholar] [CrossRef]
- Ma, M.; Ren, Q.; Yang, J.; Zhang, K.; Xiong, Z.; Ishima, T.; Pu, Y.; Hwang, S.H.; Toyoshima, M.; Iwayama, Y.; et al. Key role of soluble epoxide hydrolase in the neurodevelopmental disorders of offspring after maternal immune activation. Proc. Natl. Acad. Sci. USA 2019, 116, 7083–7088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Ehrenstein, O.S.; Ling, C.; Cui, X.; Cockburn, M.; Park, A.S.; Yu, F.; Wu, J.; Ritz, B. Prenatal and infant exposure to ambient pesticides and autism spectrum disorder in children: Population based case-control study. BMJ 2019, 364, l962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pu, Y.; Yang, J.; Chang, L.; Qu, Y.; Wang, S.; Zhang, K.; Xiong, Z.; Zhang, J.; Tan, Y.; Wang, X.; et al. Maternal glyphosate exposure causes autism-like behaviors in offspring through increased expression of soluble epoxide hydrolase. Proc. Natl. Acad. Sci. USA 2020, 117, 11753–11759. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Ma, L.; Shan, J.; Wan, X.; Hammock, B.D.; Hashimoto, K. Autism-like behaviors in male juvenile offspring after maternal glyphosate exposure. Clin. Psychopharmacol. Neurosci. 2021, 19, 554–558. [Google Scholar] [CrossRef]
- Hashimoto, K.; Hammock, B.D. Reply to Reeves and Dunn: Risk for autism in offspring after maternal glyphosate exposure. Proc. Natl. Acad. Sci. USA 2021, 118, e2016496118. [Google Scholar] [CrossRef]
- Ma, M.; Ren, Q.; Fujita, Y.; Ishima, T.; Zhang, J.C.; Hashimoto, K. Effects of AS2586114, a soluble epoxide hydrolase inhibitor, on hyperlocomotion and prepulse inhibition deficits in mice after administration of phencyclidine. Pharmacol. Biochem. Behav. 2013, 110, 98–103. [Google Scholar] [CrossRef]
- Iyer, M.R.; Kundu, B.; Wood, C.M. Soluble epoxide hydrolase inhibitors: An overview and patent review from the last decade. Expert Opin. Ther. Pat. 2022, 12, 1–19. [Google Scholar] [CrossRef]
- Hashimoto, K. Understanding the link between maternal infections and neurodevelopmental disorders in offspring: The role of abnormalities in metabolism of polyunsaturated fatty acids. Brain Behav. Immun. 2019, 81, 4–5. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Kodani, S.D.; Morisseau, C. Role of epoxy-fatty acids and epoxide hydrolases in the pathology of neuro-inflammation. Biochimie 2019, 159, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J. Parkinson’s disease: Health-related quality of life, economic cost, and implications of early treatment. Am. J. Manag. Care 2010, 16, S87–S93. [Google Scholar] [PubMed]
- Zarriello, S.; Tuazon, J.P.; Corey, S.; Schimmel, S.; Rajani, M.; Gorsky, A.; Incontri, D.; Hammock, B.D.; Borlongan, C.V. Humble beginnings with big goals: Small molecule soluble epoxide hydrolase inhibitors for treating CNS disorders. Prog. Neurobiol. 2019, 172, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Borlongan, C.V. Fatty acid chemical mediator provides insights into the pathology and treatment of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, 6322–6324. [Google Scholar] [CrossRef] [Green Version]
- Dehay, B.; Bourdenx, M.; Gorry, P.; Przedborski, S.; Vila, M.; Hunot, S.; Singleton, A.; Olanow, C.W.; Merchant, K.M.; Bezard, E. Targeting α-synuclein for treatment of Parkinson’s disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2015, 14, 855–866. [Google Scholar] [CrossRef] [Green Version]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Standaert, D.G. Ten unresolved questions about neuroinflammation in Parkinson’s disease. Mov. Disord. 2021, 36, 16–24. [Google Scholar] [CrossRef]
- Pallàs, M.; Vázquez, S.; Sanfeliu, C.; Galdeano, C.; Griñán-Ferré, C. Soluble epoxide hydrolase inhibition to face neuroinflammation in Parkinson’s disease: A new therapeutic strategy. Biomolecules 2020, 10, 703. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Chen, C.; Gong, W.; Li, Y.; Edin, M.L.; Zeldin, D.C.; Wang, D.W. Epoxyeicosatrienoic acids attenuate reactive oxygen species level, mitochondrial dysfunction, caspase activation, and apoptosis in carcinoma cells treated with arsenic trioxide. J. Pharmacol. Exp. Ther. 2011, 339, 451. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Liu, J.; Dong, R.; Zhu, J.; Tao, C.; Zheng, R.; Zhu, S. 14,15-epoxyeicosatrienoic acid promotes production of brain derived neurotrophic factor from astrocytes and exerts neuroprotective effects during ischaemic injury. Neuropathol. App. Neurobiol. 2016, 42, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.M.; Hsu, P.C.; Hung, C.C.; Hu, Y.Y.; Huang, Y.J.; Gan, Y.L.; Lin, C.H.; Shie, F.S.; Chang, W.K.; Kao, L.S. Soluble epoxide hydrolase inhibition attenuates excitotoxicity involving 14,15-epoxyeicosatrienoic acid–mediated astrocytic survival and plasticity to preserve glutamate homeostasis. Mol. Neurobiol. 2019, 56, 8451–8474. [Google Scholar] [CrossRef] [PubMed]
- Sedelis, M.; Schwarting, R.K.W.; Huston, J.P. Behavioral phenotyping of the MPTP mouse model of Parkinson’s disease. Behav. Brain Res. 2001, 125, 109–125. [Google Scholar] [CrossRef]
- Jackson-Lewis, V.; Przedborski, S. Protocol for the MPTP mouse model of Parkinson’s disease. Nat. Protoc. 2007, 2, 141–151. [Google Scholar] [CrossRef]
- Ren, Q.; Ma, M.; Yang, J.; Nonaka, R.; Yamaguchi, A.; Ishikawa, K.; Kobayashi, K.; Murayama, S.; Hwang, S.H.; Saiki, S.; et al. Soluble epoxide hydrolase plays a key role in the pathogenesis of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E5815–E5823. [Google Scholar] [CrossRef] [Green Version]
- Marsden, C.D. Parkinson’s disease. Lancet 1990, 335, 948–949. [Google Scholar] [CrossRef]
- Qin, X.; Wu, Q.; Lin, L.; Sun, A.; Liu, S.; Li, X.; Cao, X.; Gao, T.; Luo, P.; Zhu, X.; et al. Soluble epoxide hydrolase deficiency or inhibition attenuates MPTP-induced Parkinsonism. Mol. Neurobiol. 2015, 52, 187–195. [Google Scholar] [CrossRef]
- Huang, H.J.; Wang, Y.T.; Lin, H.C.; Lee, Y.H.; Lin, A.M.Y. Soluble epoxide hydrolase inhibition attenuates MPTP-induced neurotoxicity in the nigrostriatal dopaminergic system: Involvement of α-synuclein aggregation and ER stress. Mol. Neurobiol. 2018, 55, 138–144. [Google Scholar] [CrossRef]
- Sun, C.P.; Zhou, J.J.; Yu, Z.L.; Huo, X.K.; Zhang, J.; Morisseau, C.; Hammock, B.D.; Ma, X.C. Kurarinone alleviated Parkinson’s disease via stabilization of epoxyeicosatrienoic acids in animal model. Proc. Natl. Acad. Sci. USA 2022, 119, e2118818119. [Google Scholar] [CrossRef]
- Lakkappa, N.; Krishnamurthy, P.T.; Pandareesh, M.D.; Hammock, B.D.; Hwang, S.H. Soluble epoxide hydrolase inhibitor, APAU, protects dopaminergic neurons against rotenone induced neurotoxicity: Implications for Parkinson’s disease. Neurotoxicology 2019, 70, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Lakkappa, N.; Krishnamurthy, P.T.; Yamjala, K.; Hwang, S.H.; Hammock, B.D.; Babu, B. Evaluation of antiparkinson activity of PTUPB by measuring dopamine and its metabolites in Drosophila melanogaster: LC–MS/MS method development. J. Pharm. Biomed. Anal. 2018, 149, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2021 update: A report from the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- Chamorro, Á.; Meisel, A.; Planas, A.M.; Urra, X.; van de Beek, D.; Veltkamp, R. The immunology of acute stroke. Nat. Rev. Neurol. 2012, 8, 401–410. [Google Scholar] [CrossRef]
- Emsley, H.C.A.; Smith, C.J.; Gavin, C.M.; Georgiou, R.F.; Vail, A.; Barberan, E.M.; Hallenbeck, J.M.; del Zoppo, G.J.; Rothwell, N.J.; Tyrrell, P.J.; et al. An early and sustained peripheral inflammatory response in acute ischaemic stroke: Relationships with infection and atherosclerosis. J. Neuroimmunol. 2003, 139, 93–101. [Google Scholar] [CrossRef]
- Adeoye, O.; Hornung, R.; Khatri, P.; Kleindorfer, D. Recombinant tissue-type plasminogen activator use for ischemic stroke in the United States. Stroke 2011, 42, 1952–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nor, A.M.; Ford, G.A. Misdiagnosis of stroke. Expert Rev. Neurother. 2007, 7, 989–1001. [Google Scholar] [CrossRef]
- Iliff, J.J.; Alkayed, N.J. Soluble epoxide hydrolase inhibition: Targeting multiple mechanisms of ischemic brain injury with a single agent. Future Neurol. 2009, 4, 179–199. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Iliff, J.J.; Campbell, C.J.; Wang, R.K.; Hurn, P.D.; Alkayed, N.J. Role of soluble epoxide hydrolase in the sex-specific vascular response to cerebral ischemia. J. Cereb. Blood Flow Metab. 2009, 29, 1475–1481. [Google Scholar] [CrossRef]
- Demirdöğen, B.C.; Miçooğulları, Y.; Özçelik, A.T.; Adalı, O. Missense genetic polymorphisms of microsomal (EPHX1) and soluble epoxide hydrolase (EPHX2) and their relation to the risk of large artery atherosclerotic ischemic stroke in a Turkish population. Neuropsychiatr. Dis. Treat. 2020, 16, 3251. [Google Scholar] [CrossRef]
- Yu, D.; Hennebelle, M.; Sahlas, D.J.; Ramirez, J.; Gao, F.; Masellis, M.; Cogo-Moreira, H.; Swartz, R.H.; Herrmann, N.; Chan, P.C.; et al. Soluble epoxide hydrolase-derived linoleic acid oxylipins in serum are associated with periventricular white matter hyperintensities and vascular cognitive impairment. Transl. Stroke Res. 2019, 10, 522–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matin, N.; Fisher, C.; Lansdell, T.A.; Hammock, B.D.; Yang, J.; Jackson, W.F.; Dorrance, A.M. Soluble epoxide hydrolase inhibition improves cognitive function and parenchymal artery dilation in a hypertensive model of chronic cerebral hypoperfusion. Microcirculation 2021, 28, e12653. [Google Scholar] [CrossRef]
- Hao, J.; Chen, Y.; Yao, E.; Liu, X. Soluble epoxide hydrolase inhibition alleviated cognitive impairments via NRG1/ErbB4 signaling after chronic cerebral hypoperfusion induced by bilateral carotid artery stenosis in mice. Brain Res. 2018, 1699, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Tu, R.; Armstrong, J.; Lee, K.S.S.; Hammock, B.D.; Sapirstein, A.; Koehler, R.C. Soluble epoxide hydrolase inhibition decreases reperfusion injury after focal cerebral ischemia. Sci. Rep. 2018, 8, 5279. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.H.; Lin, H.C.; Huang, S.S.; Chen, I.C.; Chu, K.W.; Chih, C.L.; Liang, Y.W.; Lee, Y.C.; Chen, Y.Y.; Lee, Y.H.; et al. Blockade of soluble epoxide hydrolase attenuates post-ischemic neuronal hyperexcitation and confers resilience against stroke with TrkB activation. Sci. Rep. 2018, 8, 118. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, N.; Nakayama, S.; Tanaka, M. Single administration of soluble epoxide hydrolase inhibitor suppresses neuroinflammation and improves neuronal damage after cardiac arrest in mice. Neurosci. Res. 2016, 111, 56–63. [Google Scholar] [CrossRef]
- Wang, J.; Fujiyoshi, T.; Kosaka, Y.; Raybuck, J.D.; Lattal, K.M.; Ikeda, M.; Herson, P.S.; Koerner, I.P. Inhibition of soluble epoxide hydrolase after cardiac arrest/cardiopulmonary resuscitation induces a neuroprotective phenotype in activated microglia and improves neuronal survival. J. Cereb. Blood Flow Metab. 2013, 33, 1574–1581. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Tian, H.; Yao, E.; Tian, Y.; Zhang, H.; Xu, L.; Yu, Z.; Fang, Y.; Wang, W.; Du, P.; et al. Soluble epoxide hydrolase inhibition promotes white matter integrity and long-term functional recovery after chronic hypoperfusion in mice. Sci. Rep. 2017, 7, 7758. [Google Scholar] [CrossRef] [Green Version]
- Yeh, C.F.; Chuang, T.Y.; Hung, Y.W.; Lan, M.Y.; Tsai, C.H.; Huang, H.X.; Lin, Y.Y. Soluble epoxide hydrolase inhibition enhances anti-inflammatory and antioxidative processes, modulates microglia polarization, and promotes recovery after ischemic stroke. Neuropsychiatr. Dis. Treat. 2019, 15, 2927. [Google Scholar] [CrossRef] [Green Version]
- Yeh, C.F.; Chuang, T.Y.; Hung, Y.W.; Lan, M.Y.; Tsai, C.H.; Huang, H.X.; Lin, Y.Y. Inhibition of soluble epoxide hydrolase regulates monocyte/macrophage polarization and improves neurological outcome in a rat model of ischemic stroke. Neuroreport 2019, 30, 567–572. [Google Scholar] [CrossRef]
- Davis, C.M.; Zhang, W.H.; Allen, E.M.; Bah, T.M.; Shangraw, R.E.; Alkayed, N.J. Soluble epoxide hydrolase blockade after stroke onset protects normal but not diabetic mice. Int. J. Mol. Sci. 2021, 22, 5419. [Google Scholar] [CrossRef] [PubMed]
- Hasumi, K.; Suzuki, E. Impact of SMTP targeting plasminogen and soluble epoxide hydrolase on thrombolysis, inflammation, and ischemic stroke. Int. J. Mol. Sci. 2021, 2, 954. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, E.; Nishimura, N.; Yoshikawa, T.; Kunikiyo, Y.; Hasegawa, K.; Hasumi, K. Efficacy of SMTP-7, a small-molecule anti-inflammatory thrombolytic, in embolic stroke in monkeys. Pharmacol. Res. Perspect. 2018, 6, e00448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Xu, S.; Wu, X.; Muse, F.M.; Chen, J.; Cao, Y.; Yan, J.; Cheng, Z.; Yi, X.; Han, Z. Protective effects of the soluble epoxide hydrolase inhibitor 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea in a rat model of permanent middle cerebral artery occlusion. Front. Pharmacol. 2020, 11, 182. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Xu, C.; Huang, P.; Zhang, L.; Qing, T.; Li, J.; Wang, C.; Zeng, T.; Lu, J.; Han, Z. 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea protects the blood-brain barrier against ischemic injury by upregulating tight junction protein expression, mitigating apoptosis and inflammation in vivo and in vitro model. Front. Pharmacol. 2020, 11, 1197. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Cao, Q.; Luo, S.; He, L.; Yang, C.; Chen, J.; Qi, Q.; Hashimoto, K.; Zhang, J.C. Microglial ERK-NRBP1-CREB-BDNF signaling in sustained antidepressant actions of (R)-ketamine. Mol. Psychiatry 2021. [Google Scholar] [CrossRef]
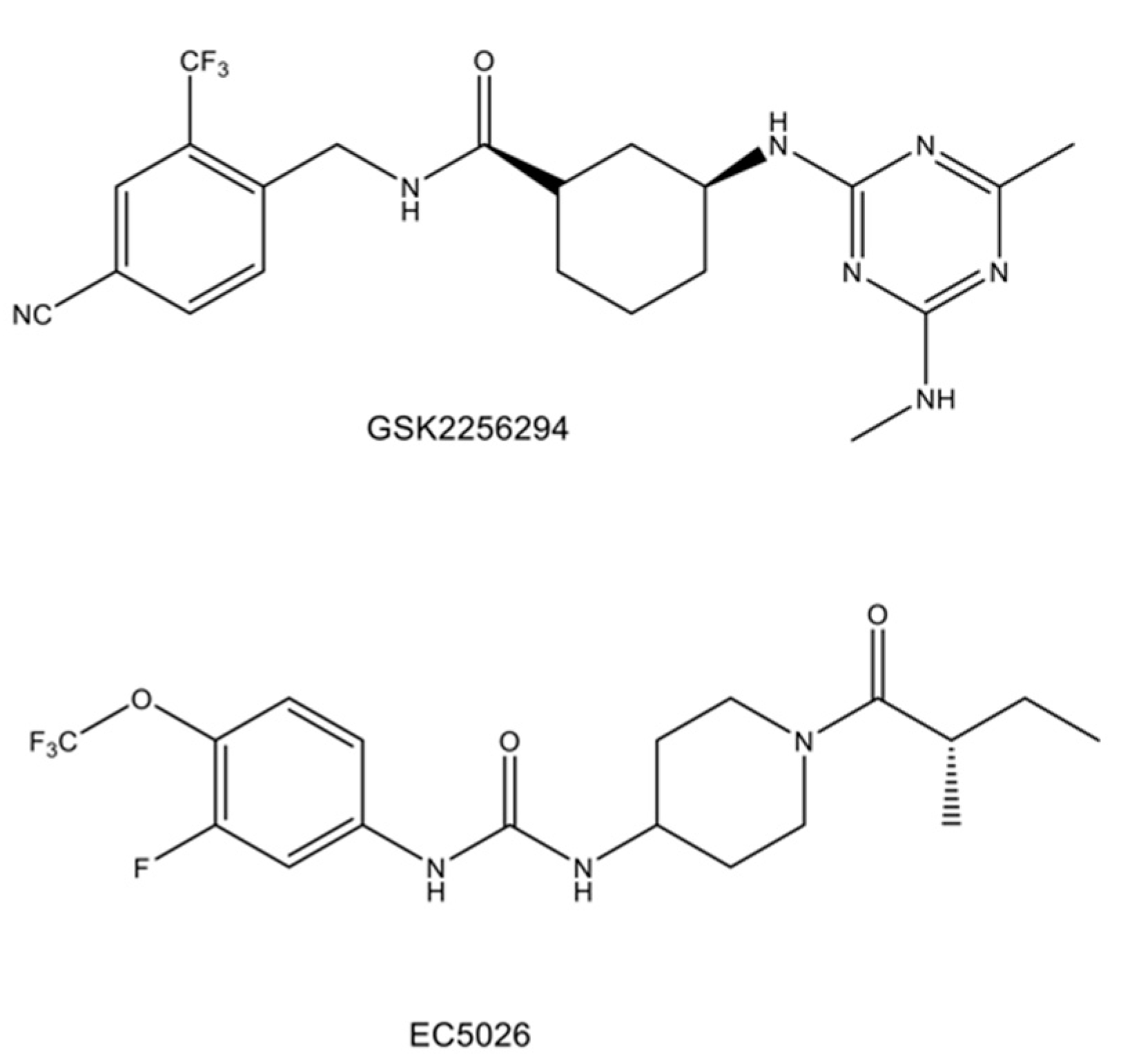
- Hammock, B.D.; McReynolds, C.B.; Wagner, K.; Buckpitt, A.; Cortes-Puch, I.; Croston, G.; Lee, K.S.S.; Yang, J.; Schmidt, W.K.; Hwang, S.H. Movement to the clinic of soluble epoxide hydrolase inhibitor EC5026 as an analgesic for neuropathic pain and for use as a nonaddictive opioid alternative. J. Med. Chem. 2021, 64, 1856–1872. [Google Scholar] [CrossRef]
- Luther, J.M.; Ray, J.; Wei, D.; Koethe, J.R.; Hannah, L.; DeMatteo, A.; Manning, R.; Terker, A.S.; Peng, D.; Nian, H.; et al. GSK2256294 decreases sEH (soluble epoxide hydrolase) activity in plasma, muscle, and adipose and reduces F2-isoprostanes but does not alter insulin sensitivity in humans. Hypertension 2021, 78, 1092–1102. [Google Scholar] [CrossRef]
- Martini, R.P.; Siler, D.; Cetas, J.; Alkayed, N.J.; Allen, E.; Treggiari, M.M. A double-blind, randomized, placebo-controlled trial of soluble epoxide hydrolase inhibition in patients with aneurysmal subarachnoid hemorrhage. Neurocrit. Care 2021. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shan, J.; Hashimoto, K. Soluble Epoxide Hydrolase as a Therapeutic Target for Neuropsychiatric Disorders. Int. J. Mol. Sci. 2022, 23, 4951. https://doi.org/10.3390/ijms23094951
Shan J, Hashimoto K. Soluble Epoxide Hydrolase as a Therapeutic Target for Neuropsychiatric Disorders. International Journal of Molecular Sciences. 2022; 23(9):4951. https://doi.org/10.3390/ijms23094951
Chicago/Turabian StyleShan, Jiajing, and Kenji Hashimoto. 2022. "Soluble Epoxide Hydrolase as a Therapeutic Target for Neuropsychiatric Disorders" International Journal of Molecular Sciences 23, no. 9: 4951. https://doi.org/10.3390/ijms23094951
APA StyleShan, J., & Hashimoto, K. (2022). Soluble Epoxide Hydrolase as a Therapeutic Target for Neuropsychiatric Disorders. International Journal of Molecular Sciences, 23(9), 4951. https://doi.org/10.3390/ijms23094951