
Toward the Discovery of a Novel Class of Leads for High Altitude Disorders by Virtual Screening and Molecular Dynamics Approaches Targeting Carbonic Anhydrase

 , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
2. Result and Discussion
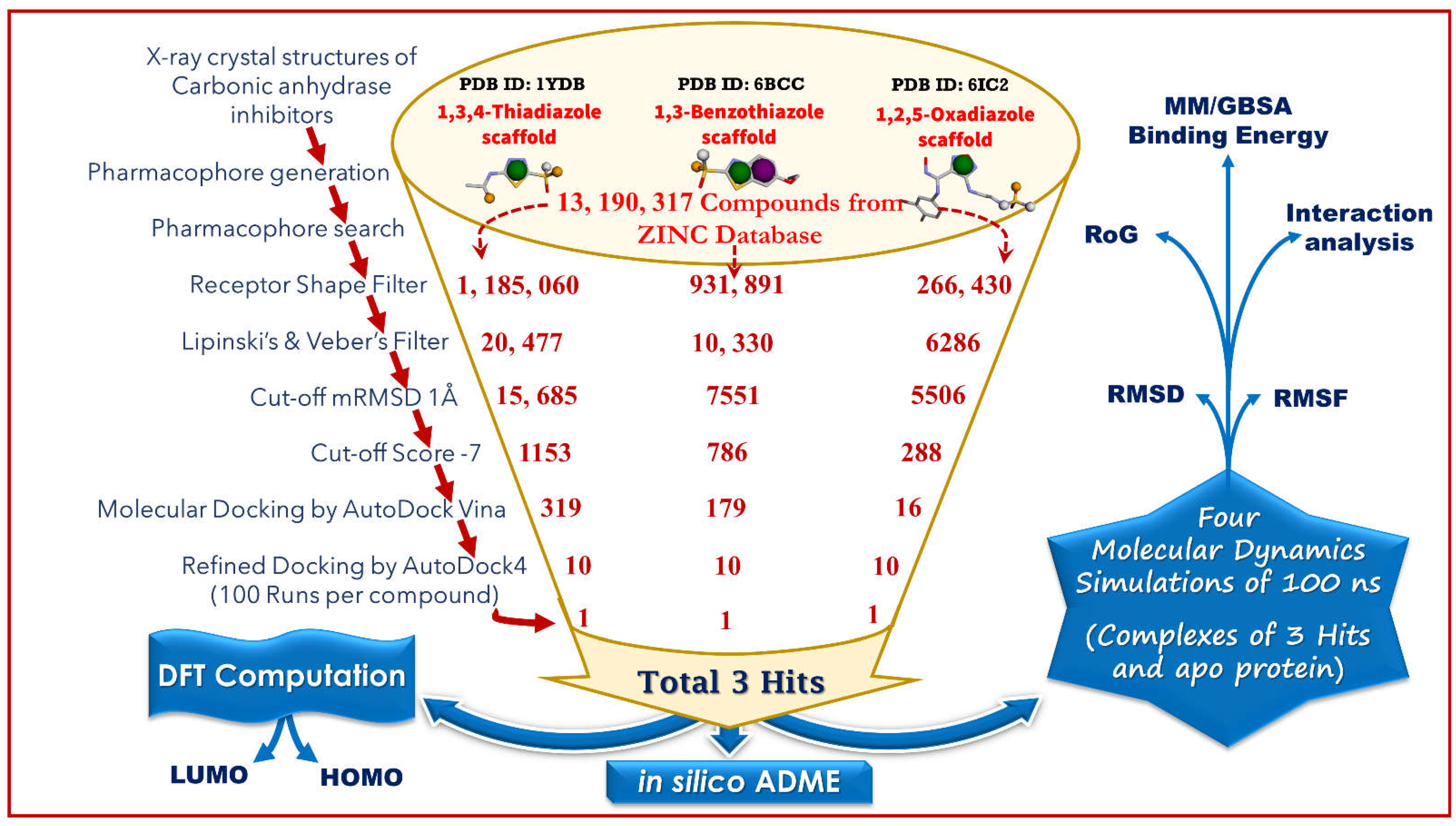
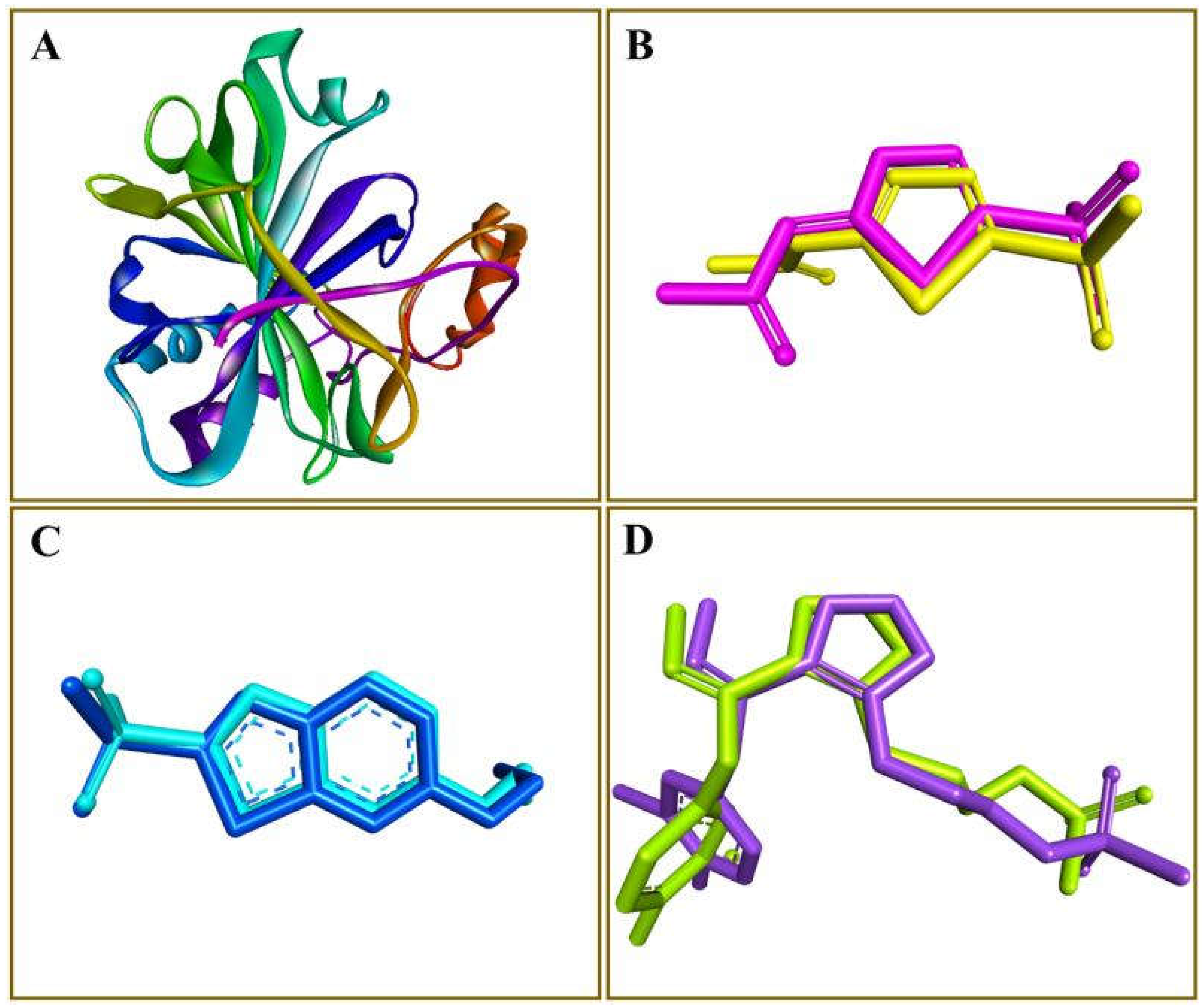
2.1. Generation of Pharmacophore Model and Virtual Screening
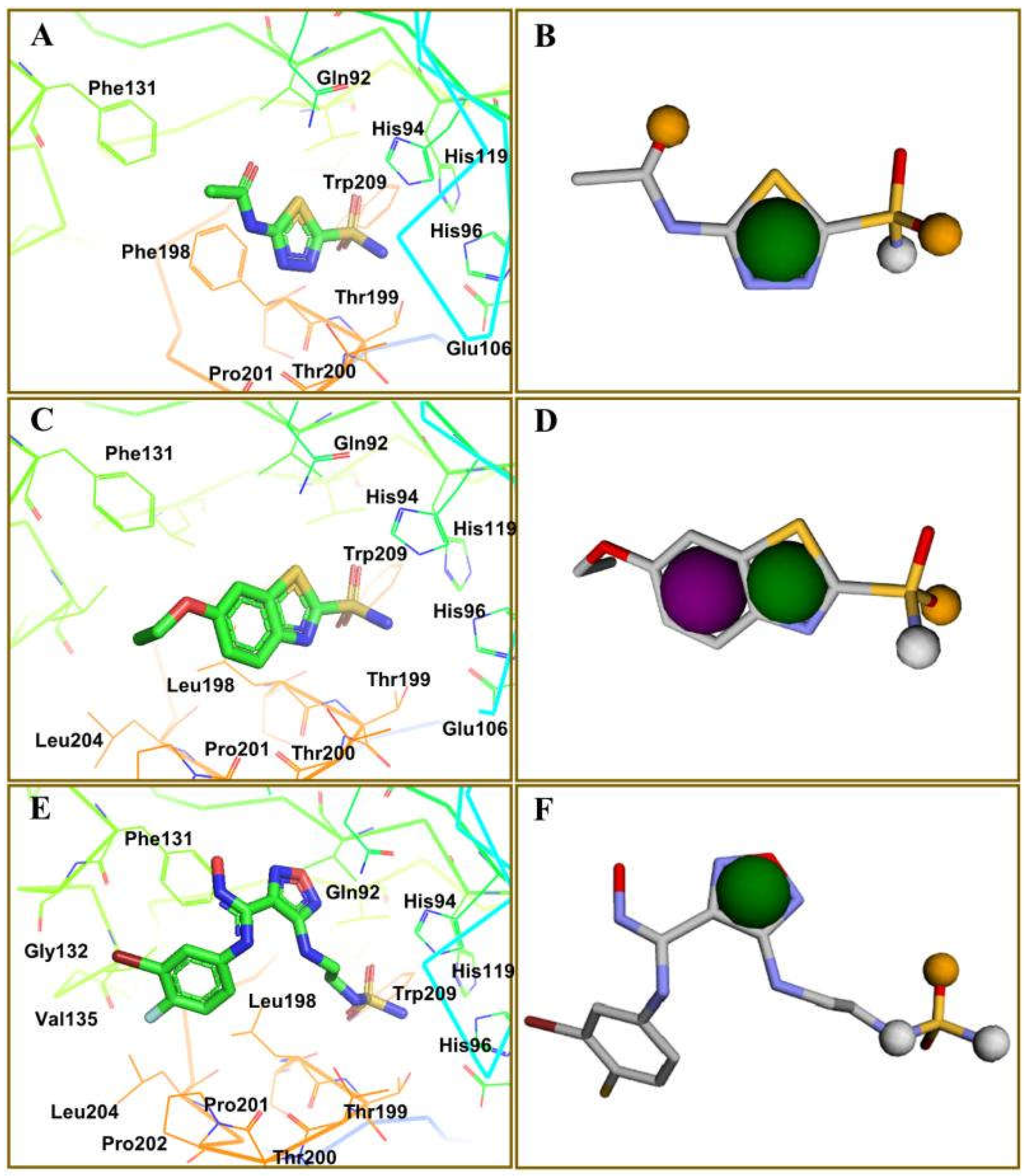
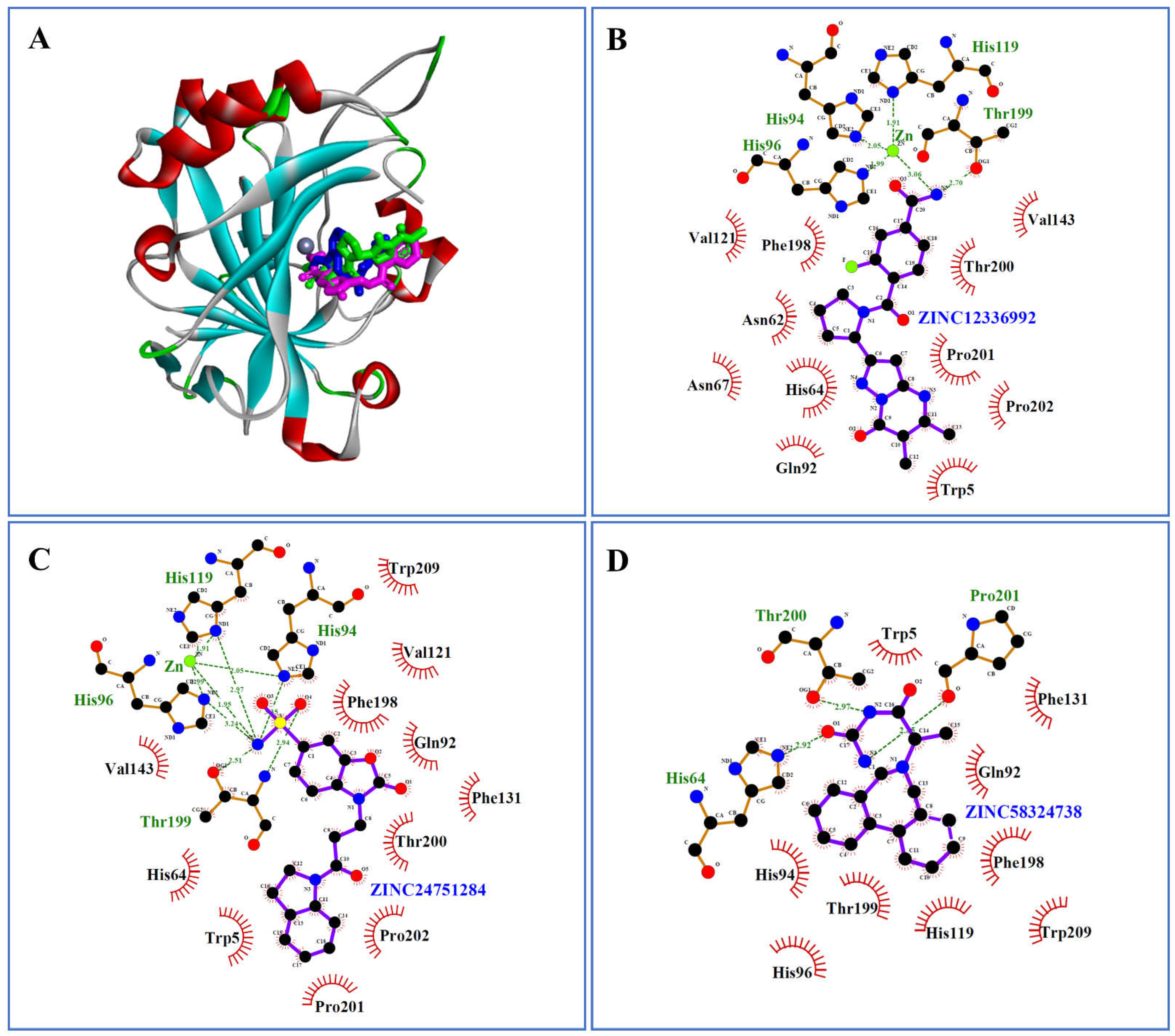
2.2. Molecular Docking by AutoDock Vina
2.3. Docking Refinement by AutoDock 4.2
2.4. Molecular Dynamics Simulation
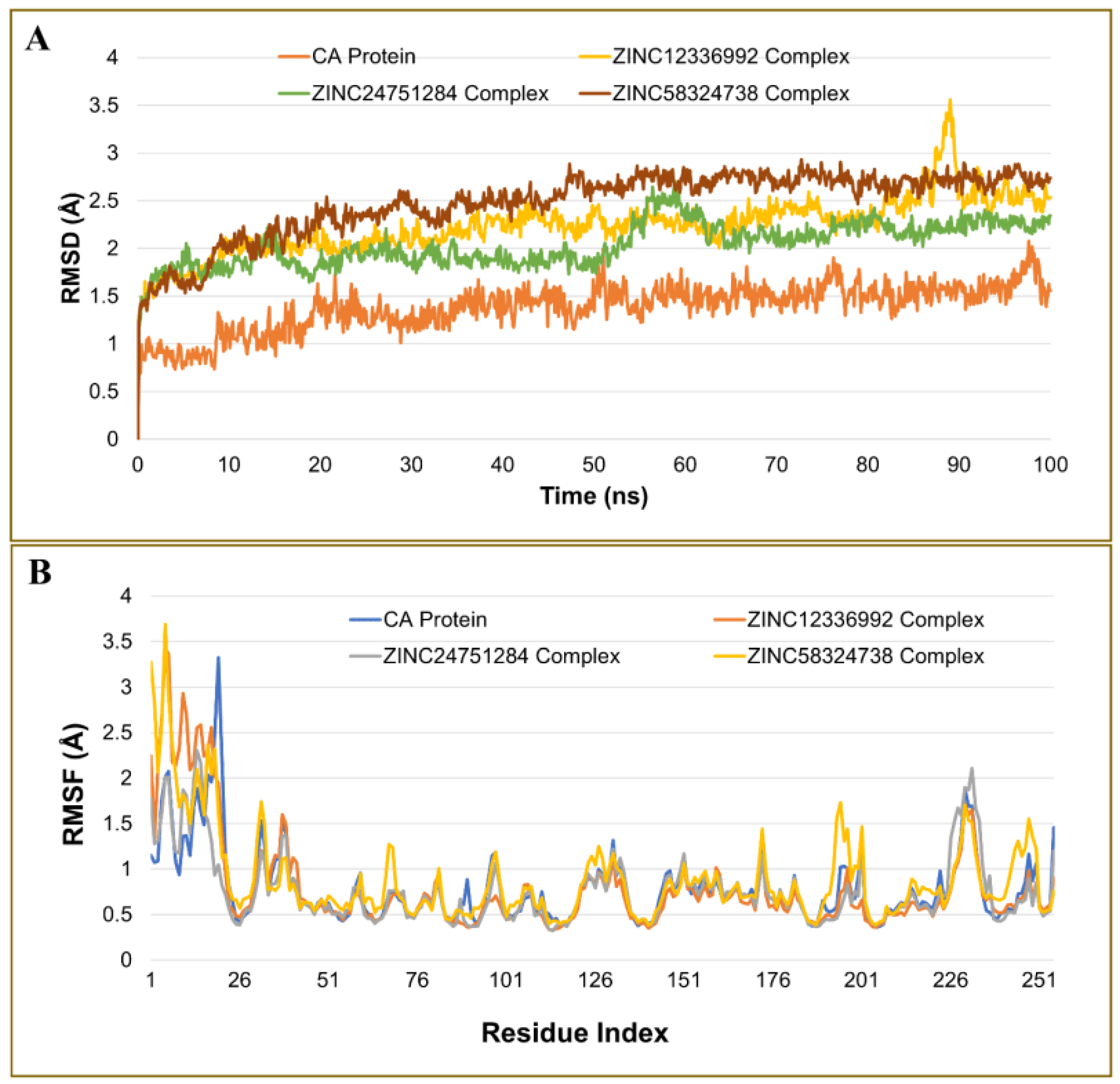
2.4.1. Analysis of the Root Mean Square Deviation
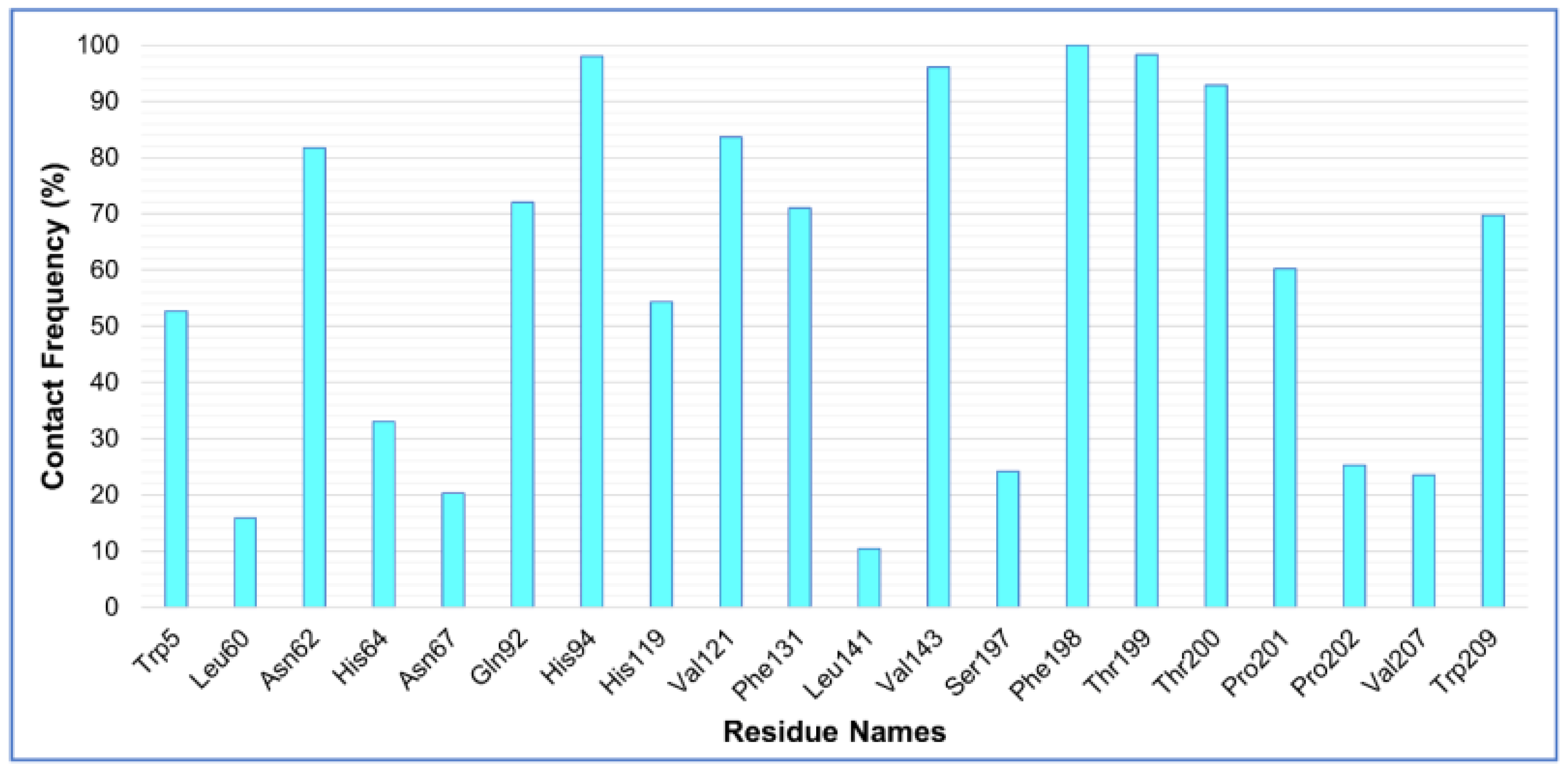
2.4.2. Analysis of Residue Mobility
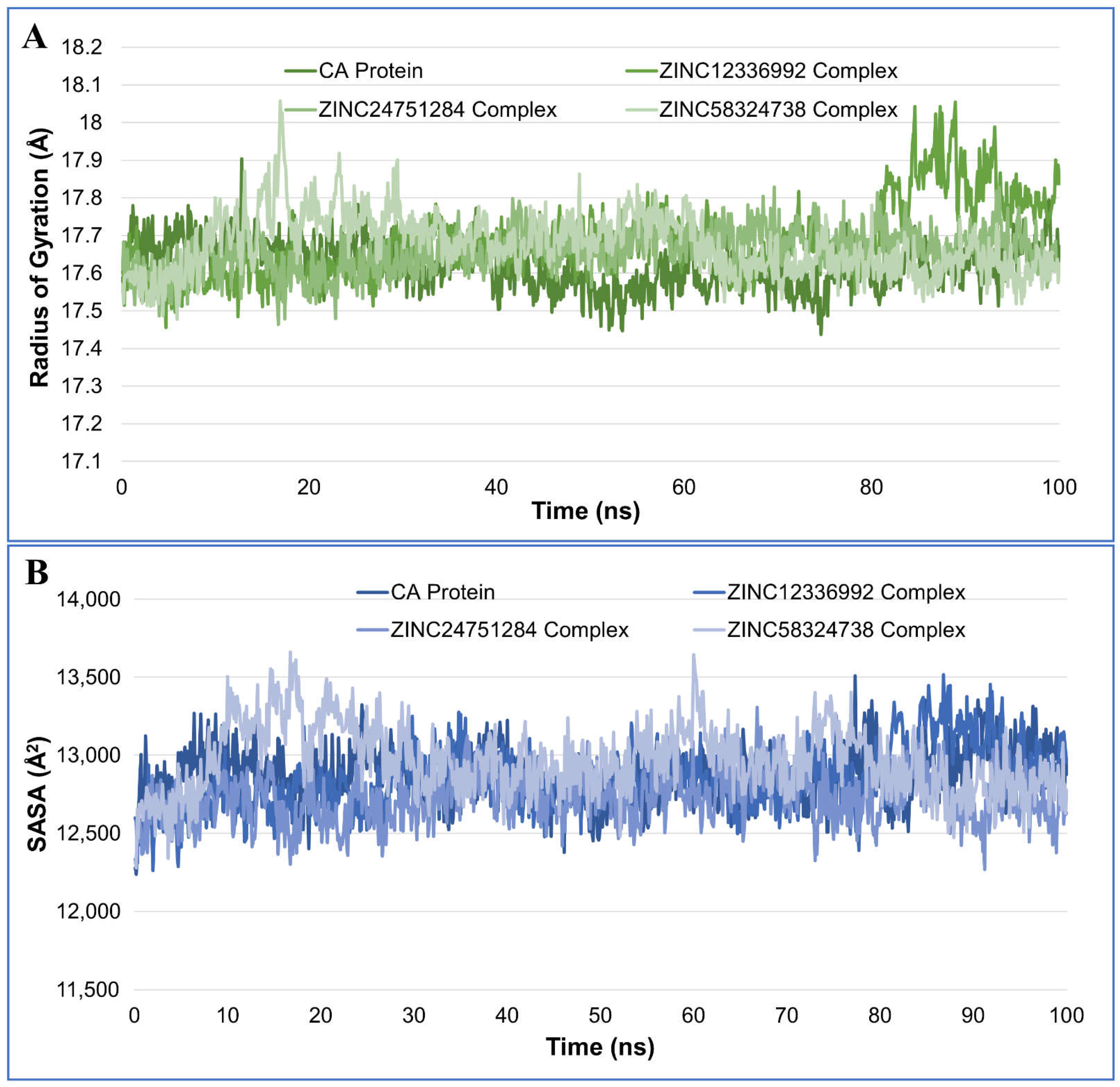
2.4.3. Radius of Gyration Analysis
2.4.4. Solvent-Accessible Surface Area Analysis
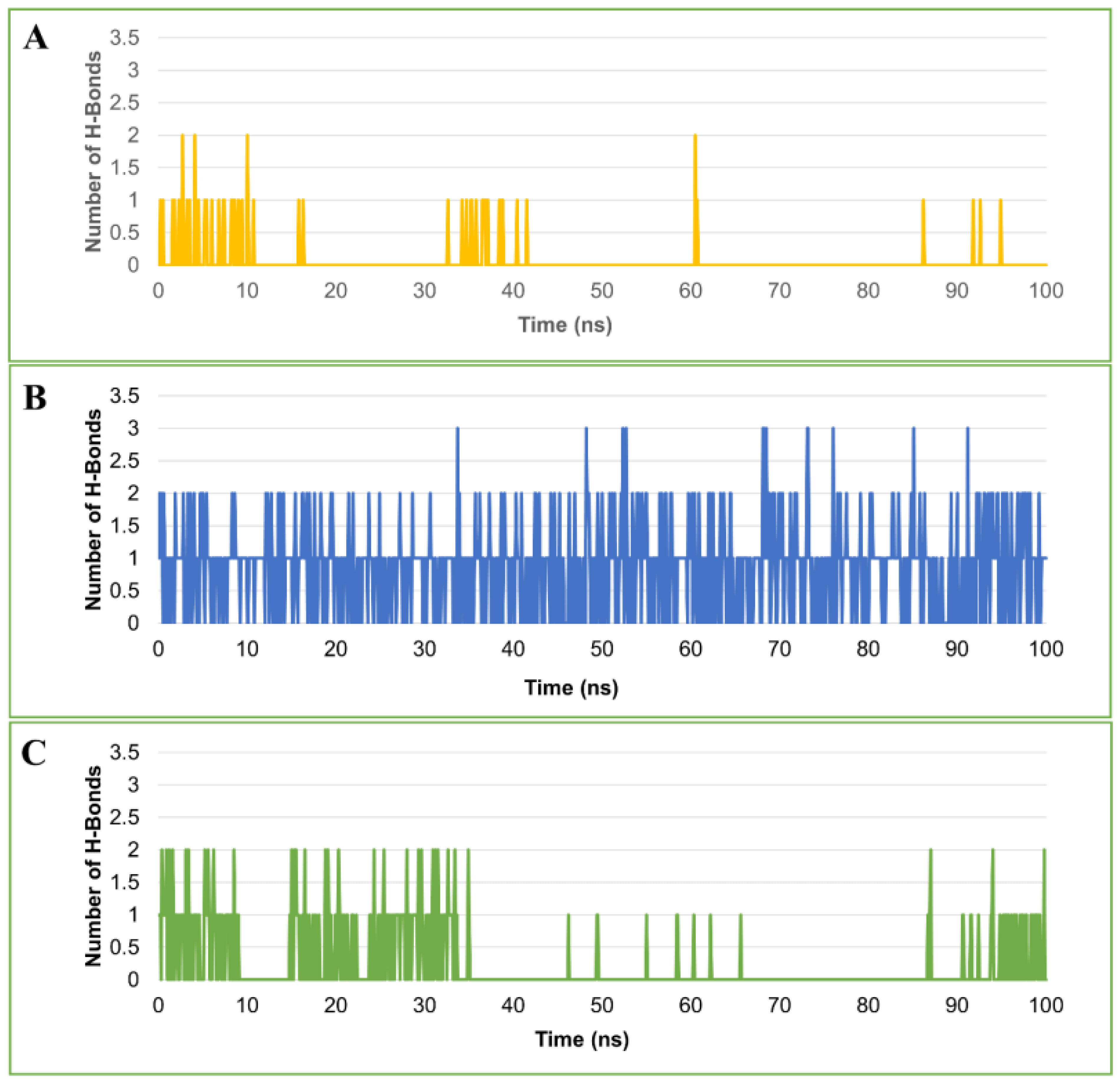
2.4.5. Hydrogen Bond Analysis
2.4.6. Binding Energy Estimation by MM/GBSA Method
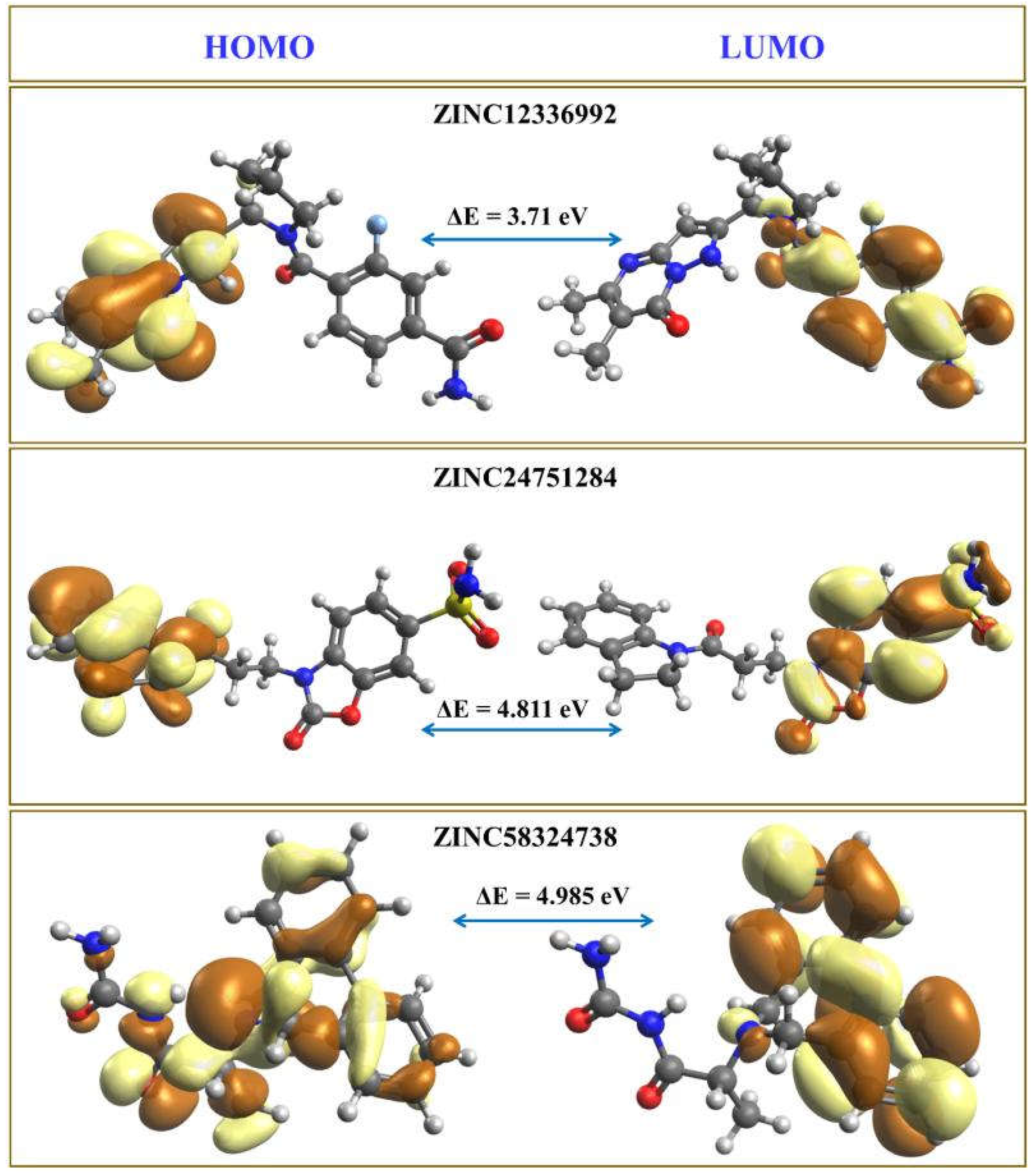
2.5. Density Functional Theory Computations
2.6. ADME Analysis
3. Materials and Methods
3.1. Virtual Screening
3.2. Molecular Docking by AutoDock Vina
3.3. Molecular Docking by AutoDock 4.2
3.4. Molecular Dynamics Simulation
3.5. Binding Energy Calculations
3.6. Density Functional Theory Computations
3.7. Pharmacokinetic Properties of the Top-Scoring Molecules
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davis, C.; Hackett, P. Advances in the Prevention and Treatment of High Altitude Illness. Emerg. Med. Clin. N. Am. 2017, 35, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Bradwell, A.R.; Wright, A.D.; Winterborn, M.; Imray, C. Acetazolamide and high altitude diseases. Int. J. Sports Med. 1992, 13, S63–S64. [Google Scholar] [CrossRef]
- West, J.B. The physiologic basis of high-altitude diseases. Ann. Intern. Med. 2004, 141, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Ainslie, P.N.; Lucas, S.J.E.; Burgess, K.R. Breathing and sleep at high altitude. Respir. Physiol. Neurobiol. 2013, 188, 233–256. [Google Scholar] [CrossRef] [PubMed]
- Teppema, L.J.; Balanos, G.M.; Steinback, C.D.; Brown, A.D.; Foster, G.E.; Duff, H.J.; Leigh, R.; Poulin, M.J. Effects of acetazolamide on ventilatory, cerebrovascular, and pulmonary vascular responses to hypoxia. Am. J. Respir. Crit. Care Med. 2007, 175, 277–281. [Google Scholar] [CrossRef]
- Kazemi, H.; Choma, L. H+ transport from CNS in hypercapnia and regulation of CSF [HCO3−]. J. Appl. Physiol. 1977, 42, 667–672. [Google Scholar] [CrossRef]
- Parati, G.; Revera, M.; Giuliano, A.; Faini, A.; Bilo, G.; Gregorini, F.; Lisi, E.; Salerno, S.; Lombardi, C.; Ramos Becerra, C.G. Effects of acetazolamide on central blood pressure, peripheral blood pressure, and arterial distensibility at acute high altitude exposure. Eur. Heart J. 2013, 34, 759–766. [Google Scholar] [CrossRef]
- Burtscher, M.; Gatterer, H.; Faulhaber, M.; Burtscher, J. Acetazolamide pre-treatment before ascending to high altitudes: When to start? Int. J. Clin. Exp. Med. 2014, 7, 4378–4383. [Google Scholar]
- Mishra, C.B.; Tiwari, M.; Supuran, C.T. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: Where are we today? Med. Res. Rev. 2020, 40, 2485–2565. [Google Scholar] [CrossRef]
- Kapetanovic, I.M. Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chem. Biol. Interact. 2008, 171, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Kaldor, S.W.; Kalish, V.J.; Davies, J.F.; Shetty, B.V.; Fritz, J.E.; Appelt, K.; Burgess, J.A.; Campanale, K.M.; Chirgadze, N.Y.; Clawson, D.K. Viracept (nelfinavir mesylate, AG1343): A potent, orally bioavailable inhibitor of HIV-1 protease. J. Med. Chem. 1997, 40, 3979–3985. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, X.; Lin, X. A Review on Applications of Computational Methods in Drug Screening and Design. Molecules 2020, 25, 1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotev, M.; Sarrat, L.; Gonzalez, C.D. User-Friendly Quantum Mechanics: Applications for Drug Discovery. Methods Mol. Biol. 2020, 2114, 231–255. [Google Scholar] [CrossRef] [PubMed]
- Dar, K.B.; Bhat, A.H.; Amin, S.; Hamid, R.; Anees, S.; Anjum, S.; Reshi, B.A.; Zargar, M.A.; Masood, A.; Ganie, S.A. Modern Computational Strategies for Designing Drugs to Curb Human Diseases: A Prospect. Curr. Top. Med. Chem. 2018, 18, 2702–2719. [Google Scholar] [CrossRef] [PubMed]
- Azam, F. Elucidation of teicoplanin interactions with drug targets related to COVID-19. Antibiotics 2021, 10, 856. [Google Scholar] [CrossRef] [PubMed]
- Sunseri, J.; Koes, D.R. Pharmit: Interactive exploration of chemical space. Nucleic Acids Res. 2016, 44, W442–W448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Nair, S.K.; Krebs, J.F.; Christianson, D.W.; Fierke, C.A. Structural basis of inhibitor affinity to variants of human carbonic anhydrase II. Biochemistry 1995, 34, 3981–3989. [Google Scholar] [CrossRef] [PubMed]
- Kovalevsky, A.; Aggarwal, M.; Velazquez, H.; Cuneo, M.J.; Blakeley, M.P.; Weiss, K.L.; Smith, J.C.; Fisher, S.Z.; McKenna, R. “To Be or Not to Be” Protonated: Atomic Details of Human Carbonic Anhydrase-Clinical Drug Complexes by Neutron Crystallography and Simulation. Structure 2018, 26, 383–390.e3. [Google Scholar] [CrossRef] [Green Version]
- Angeli, A.; Ferraroni, M.; Nocentini, A.; Selleri, S.; Gratteri, P.; Supuran, C.T.; Carta, F. Polypharmacology of epacadostat: A potent and selective inhibitor of the tumor associated carbonic anhydrases IX and XII. Chem. Commun. 2019, 55, 5720–5723. [Google Scholar] [CrossRef]
- Winum, J.-Y.; Montero, J.-L.; Scozzafava, A.; Supuran, C.T. Zinc Binding Functions in the Design of Carbonic Anhydrase Inhibitors. In Drug Design of Zinc-Enzyme; Wiley: Hoboken, NJ, USA, 2009; pp. 39–72. [Google Scholar]
- Azam, F.; Eid, E.E.M.; Almutairi, A. Targeting SARS-CoV-2 main protease by teicoplanin: A mechanistic insight by docking, MM/GBSA and molecular dynamics simulation. J. Mol. Struct. 2021, 1246, 131124. [Google Scholar] [CrossRef] [PubMed]
- Azam, F.; Taban, I.M.; Eid, E.E.M.; Iqbal, M.; Alam, O.; Khan, S.; Mahmood, D.; Anwar, M.J.; Khalilullah, H.; Khan, M.U. An in-silico analysis of ivermectin interaction with potential SARS-CoV-2 targets and host nuclear importin α. J. Biomol. Struct. Dyn. 2022, 40, 2851–2864. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Azam, F.; El-Gnidi, B.A.; Alkskas, I.A. Combating oxidative stress in epilepsy: Design, synthesis, quantum chemical studies and anticonvulsant evaluation of 1-(substituted benzylidene/ethylidene) -4-(naphthalen-1-yl)semicarbazides. Eur. J. Med. Chem. 2010, 45, 2817–2826. [Google Scholar] [CrossRef] [PubMed]
- Azam, F.; Abodabos, H.S.; Taban, I.M.; Rfieda, A.R.; Mahmood, D.; Anwar, M.J.; Khan, S.; Sizochenko, N.; Poli, G.; Tuccinardi, T.; et al. Rutin as promising drug for the treatment of Parkinson’s disease: An assessment of MAO-B inhibitory potential by docking, molecular dynamics and DFT studies. Mol. Simul. 2019, 45, 1563–1571. [Google Scholar] [CrossRef]
- Ovung, A.; Bhattacharyya, J. Sulfonamide drugs: Structure, antibacterial property, toxicity, and biophysical interactions. Biophys. Rev. 2021, 13, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.H.; Abraham, M.H.; Le, J.; Hersey, A.; Luscombe, C.N.; Beck, G.; Sherborne, B.; Cooper, I. Rate-Limited Steps of Human Oral Absorption and QSAR Studies. Pharm. Res. 2002, 19, 1446–1457. [Google Scholar] [CrossRef]
- Azam, F.; Madi, A.M.; Ali, H.I. Molecular docking and prediction of pharmacokinetic properties of dual mechanism drugs that block MAO-B and adenosine A 2A receptors for the treatment of Parkinson′s disease. J. Young Pharm. 2012, 4, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Refsgaard, H.H.F.; Jensen, B.F.; Brockhoff, P.B.; Padkjær, S.B.; Guldbrandt, M.; Christensen, M.S. In Silico Prediction of Membrane Permeability from Calculated Molecular Parameters. J. Med. Chem. 2005, 48, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 44130. [Google Scholar] [CrossRef] [PubMed]
- Nosé, S.; Klein, M.L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Grest, G.S.; Kremer, K. Molecular dynamics simulation for polymers in the presence of a heat bath. Phys. Rev. A 1986, 33, 3628. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Bai, Q.; Tan, S.; Xu, T.; Liu, H.; Huang, J.; Yao, X. MolAICal: A soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Brief. Bioinform. 2021, 22, bbaa161. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeerschd, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avogadro: An Open-Source Molecular Builder and Visualization Tool, Version 1.2.0. Available online: http://avogadro.cc/ (accessed on 1 March 2022).
- Duffel, M.W.; Ing, I.S.; Segarra, T.M.; Dixson, J.A.; Barfknecht, C.F.; Schoenwald, R.D. N-Substituted sulfonamide carbonic anhydrase inhibitors with topical effects on intraocular pressure. J. Med. Chem. 1986, 29, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Carta, F.; Ferraroni, M.; Scozzafava, A.; Supuran, C.T. Fluorescent sulfonamide carbonic anhydrase inhibitors incorporating 1,2,3-triazole moieties: Kinetic and X-ray crystallographic studies. Bioorg. Med. Chem. 2016, 24, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Temperini, C.; Cecchi, A.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors. Interaction of indapamide and related diuretics with 12 mammalian isozymes and X-ray crystallographic studies for the indapamide–isozyme II adduct. Bioorg. Med. Chem. Lett. 2008, 18, 2567–2573. [Google Scholar] [CrossRef]
- Scozzafava, A.; Menabuoni, L.; Mincione, F.; Briganti, F.; Mincione, G.; Supuran, C.T. Carbonic Anhydrase Inhibitors: Perfluoroalkyl/Aryl-Substituted Derivatives of Aromatic/Heterocyclic Sulfonamides as Topical Intraocular Pressure-Lowering Agents with Prolonged Duration of Action. J. Med. Chem. 2000, 43, 4542–4551. [Google Scholar] [CrossRef]
- Vullo, D.; Voipio, J.; Innocenti, A.; Rivera, C.; Ranki, H.; Scozzafava, A.; Kaila, K.; Supuran, C.T. Carbonic anhydrase inhibitors. Inhibition of the human cytosolic isozyme VII with aromatic and heterocyclic sulfonamides. Bioorg. Med. Chem. Lett. 2005, 15, 971–976. [Google Scholar] [CrossRef]
- Güzel-Akdemir, Ö.; Akdemir, A.; Isik, S.; Vullo, D.; Supuran, C.T. o-Benzenedisulfonimido–sulfonamides are potent inhibitors of the tumor-associated carbonic anhydrase isoforms CA IX and CA XII. Bioorg. Med. Chem. 2013, 21, 1386–1391. [Google Scholar] [CrossRef]
- Temperini, C.; Innocenti, A.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors. Interaction of the antitumor sulfamate EMD 486019 with twelve mammalian carbonic anhydrase isoforms: Kinetic and X-ray crystallographic studies. Bioorg. Med. Chem. Lett. 2008, 18, 4282–4286. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SN | Pharmacophore | Top Hits | Chemical Structure | Docking Score (kcal/mol) |
|---|---|---|---|---|
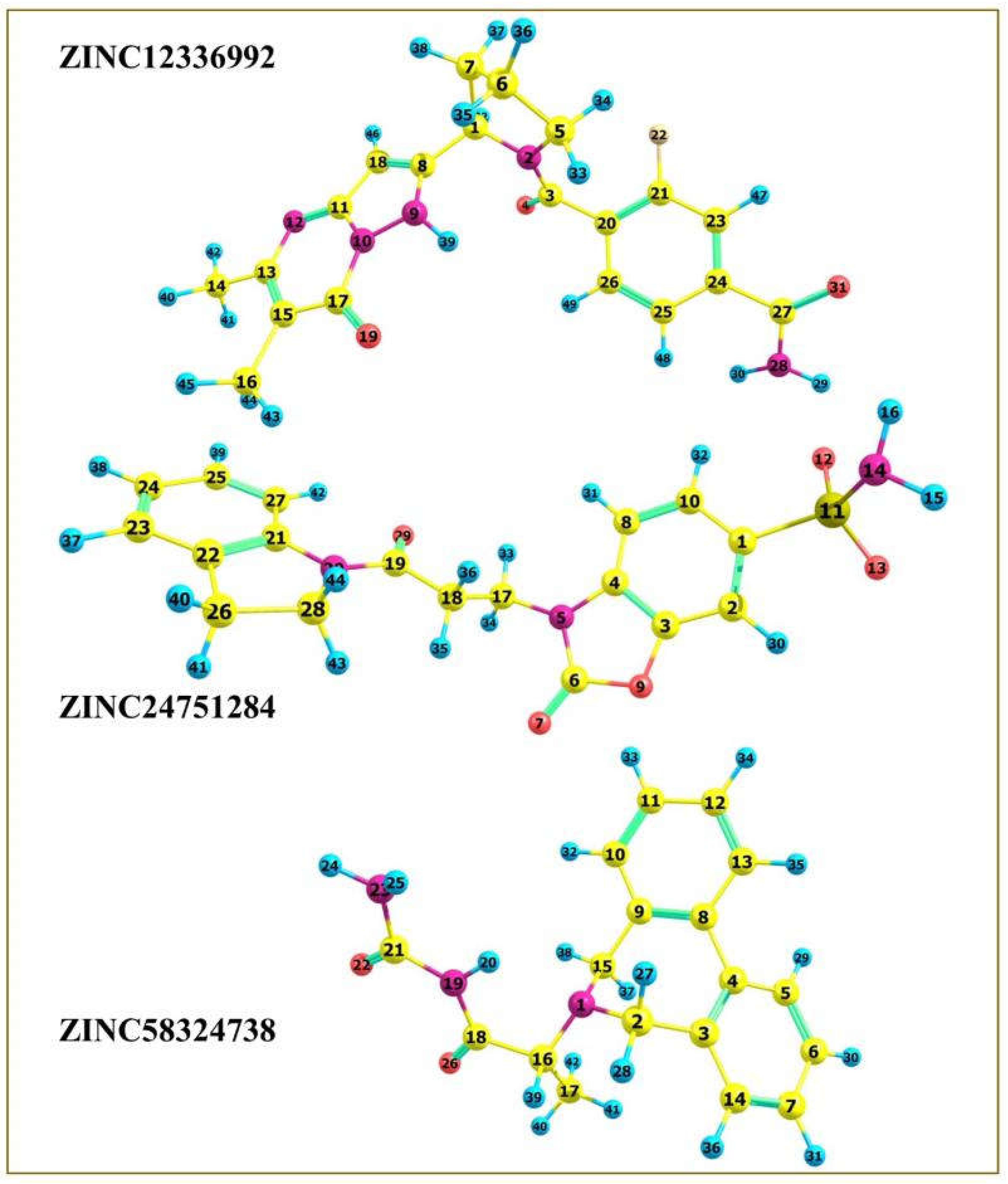
| 1. |  | ZINC12336992 |  | −9.0 |
| 2. |  | ZINC24751284 |  | −9.0 |
| 3. |  | ZINC58324738 |  | −8.9 |
| Complex | ΔG | ΔE(electrostat.) + ΔE(sol.) | ΔE(VDW) |
|---|---|---|---|
| ZINC12336992 | −16.00 ± 0.19 | 8.3899 | −24.3925 |
| ZINC24751284 | −21.04 ± 0.17 | 10.9063 | −31.9482 |
| ZINC58324738 | −19.70 ± 0.18 | 9.5605 | −29.2571 |
| Parameters | Compounds | ||
|---|---|---|---|
 ZINC12336992 |  ZINC24751284 |  ZINC58324738 | |
| Molecular weight | 397.40 | 387.41 | 309.36 |
| No. H-bond acceptor | 5 | 6 | 3 |
| No. H-bond donor | 2 | 1 | 2 |
| Log PO/W (iLOGP) | 2.52 | 1.62 | 1.65 |
| No. rotatable bonds | 4 | 5 | 4 |
| TPSA | 113.56 | 133.99 | 75.43 |
| Log KP (skin permeation) | −8.11 | −8.02 | −6.44 |
| Lipinski’s rule violation | No | No | No |
| Bioavailability score | 0.55 | 0.55 | 0.55 |
| GI absorption | High | High | High |
| PAINS alerts | 0 | 0 | 0 |
| P-pg substrate | Yes | No | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, A.; Ali, A.; Warsi, M.H.; Rahman, M.A.; Ahsan, M.J.; Azam, F. Toward the Discovery of a Novel Class of Leads for High Altitude Disorders by Virtual Screening and Molecular Dynamics Approaches Targeting Carbonic Anhydrase. Int. J. Mol. Sci. 2022, 23, 5054. https://doi.org/10.3390/ijms23095054
Ali A, Ali A, Warsi MH, Rahman MA, Ahsan MJ, Azam F. Toward the Discovery of a Novel Class of Leads for High Altitude Disorders by Virtual Screening and Molecular Dynamics Approaches Targeting Carbonic Anhydrase. International Journal of Molecular Sciences. 2022; 23(9):5054. https://doi.org/10.3390/ijms23095054
Chicago/Turabian StyleAli, Amena, Abuzer Ali, Musarrat Husain Warsi, Mohammad Akhlaquer Rahman, Mohamed Jawed Ahsan, and Faizul Azam. 2022. "Toward the Discovery of a Novel Class of Leads for High Altitude Disorders by Virtual Screening and Molecular Dynamics Approaches Targeting Carbonic Anhydrase" International Journal of Molecular Sciences 23, no. 9: 5054. https://doi.org/10.3390/ijms23095054
APA StyleAli, A., Ali, A., Warsi, M. H., Rahman, M. A., Ahsan, M. J., & Azam, F. (2022). Toward the Discovery of a Novel Class of Leads for High Altitude Disorders by Virtual Screening and Molecular Dynamics Approaches Targeting Carbonic Anhydrase. International Journal of Molecular Sciences, 23(9), 5054. https://doi.org/10.3390/ijms23095054