
Sulfated Hyaluronan Binds to Heparanase and Blocks Its Enzymatic and Cellular Actions in Carcinoma Cells

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Molecular Nature of the Sulfated HA Samples
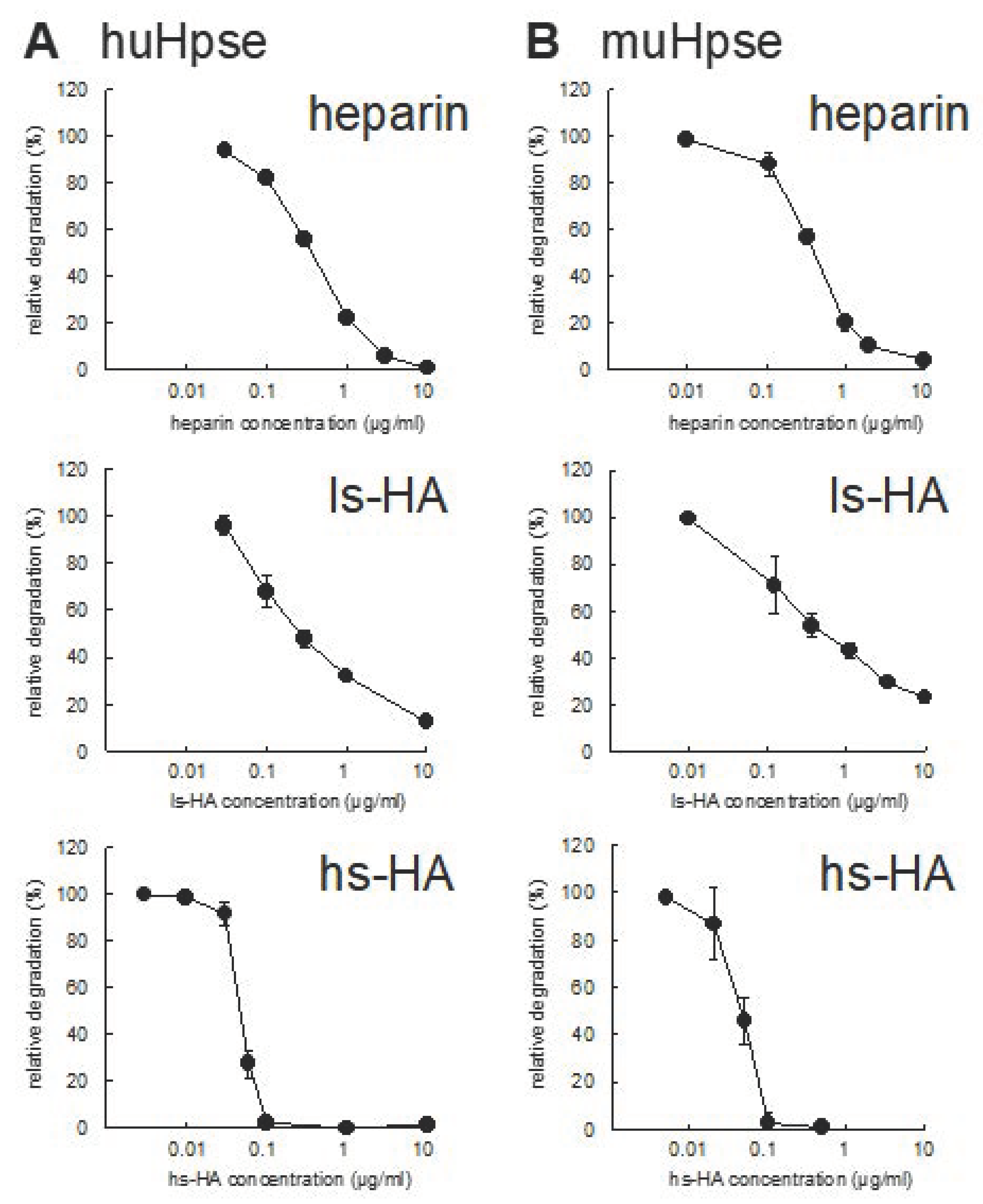
2.2. Sulfated HA Inhibited Human and Mouse Hpse-Mediated Degradation of HS
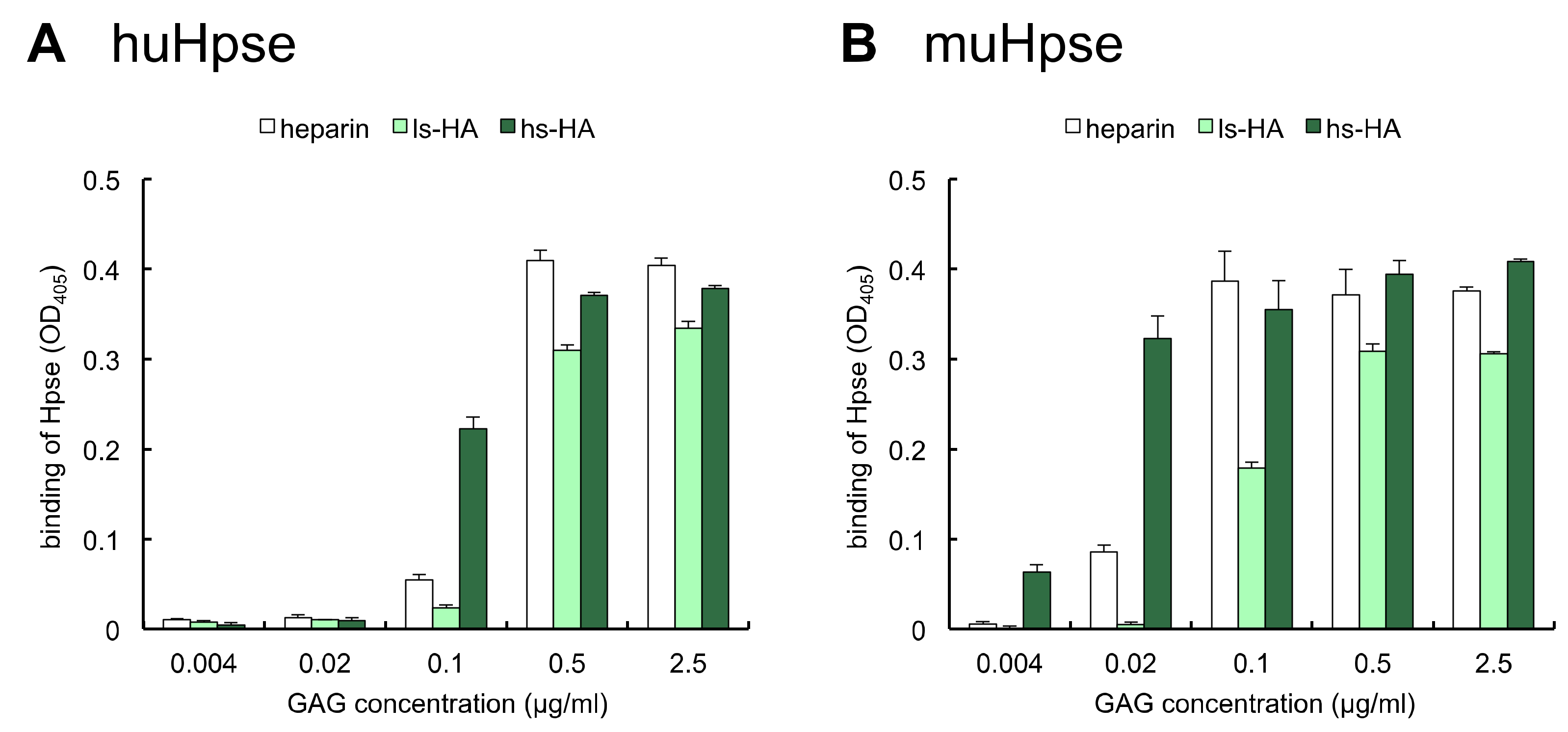
2.3. Immobilized Sulfated HA Bound to Human and Mouse Hpse Proteins
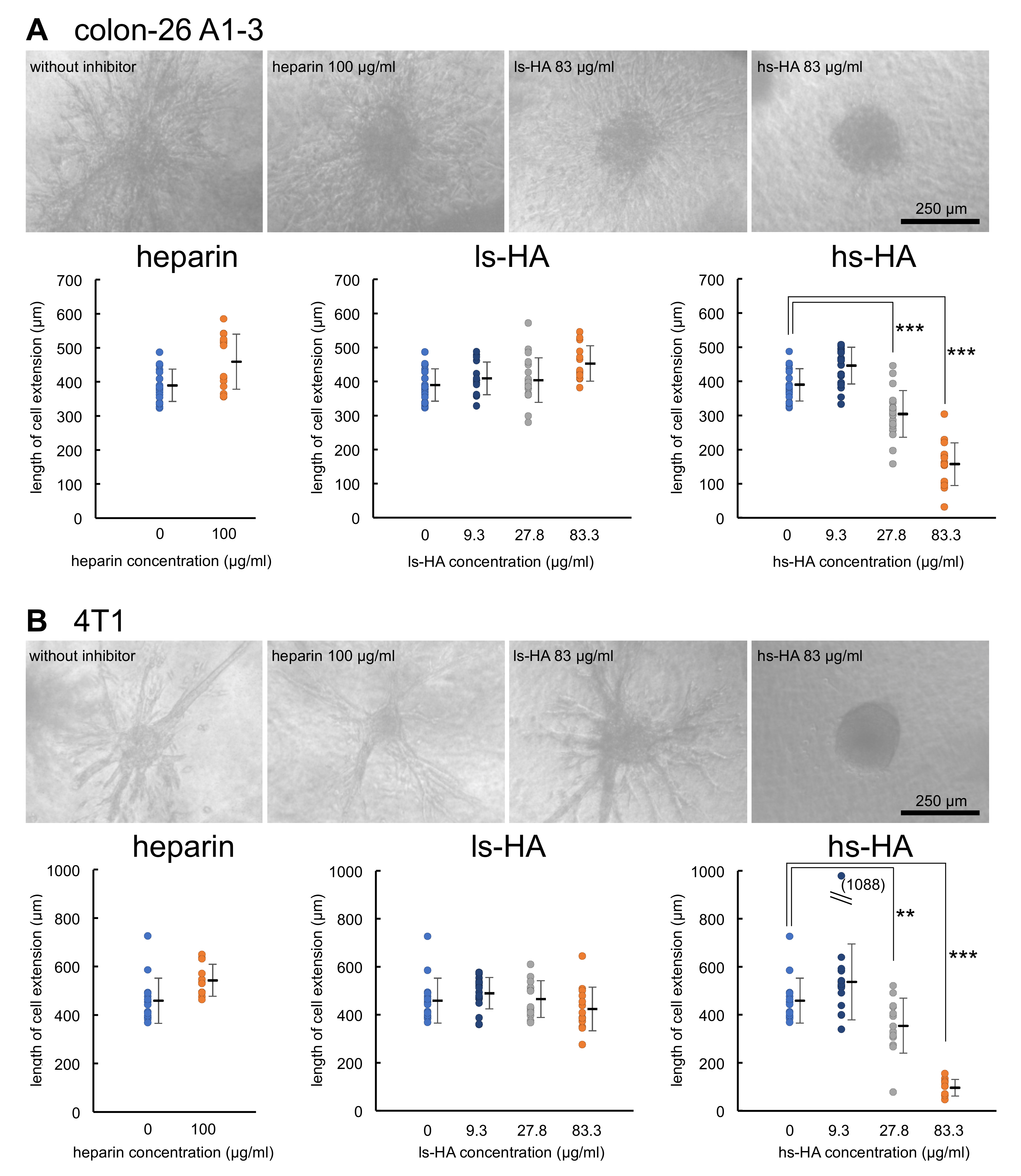
2.4. Sulfated HA Inhibited Hpse-Mediated Invasion of Colon-26 and 4T1 Cells into a Collagen Gel
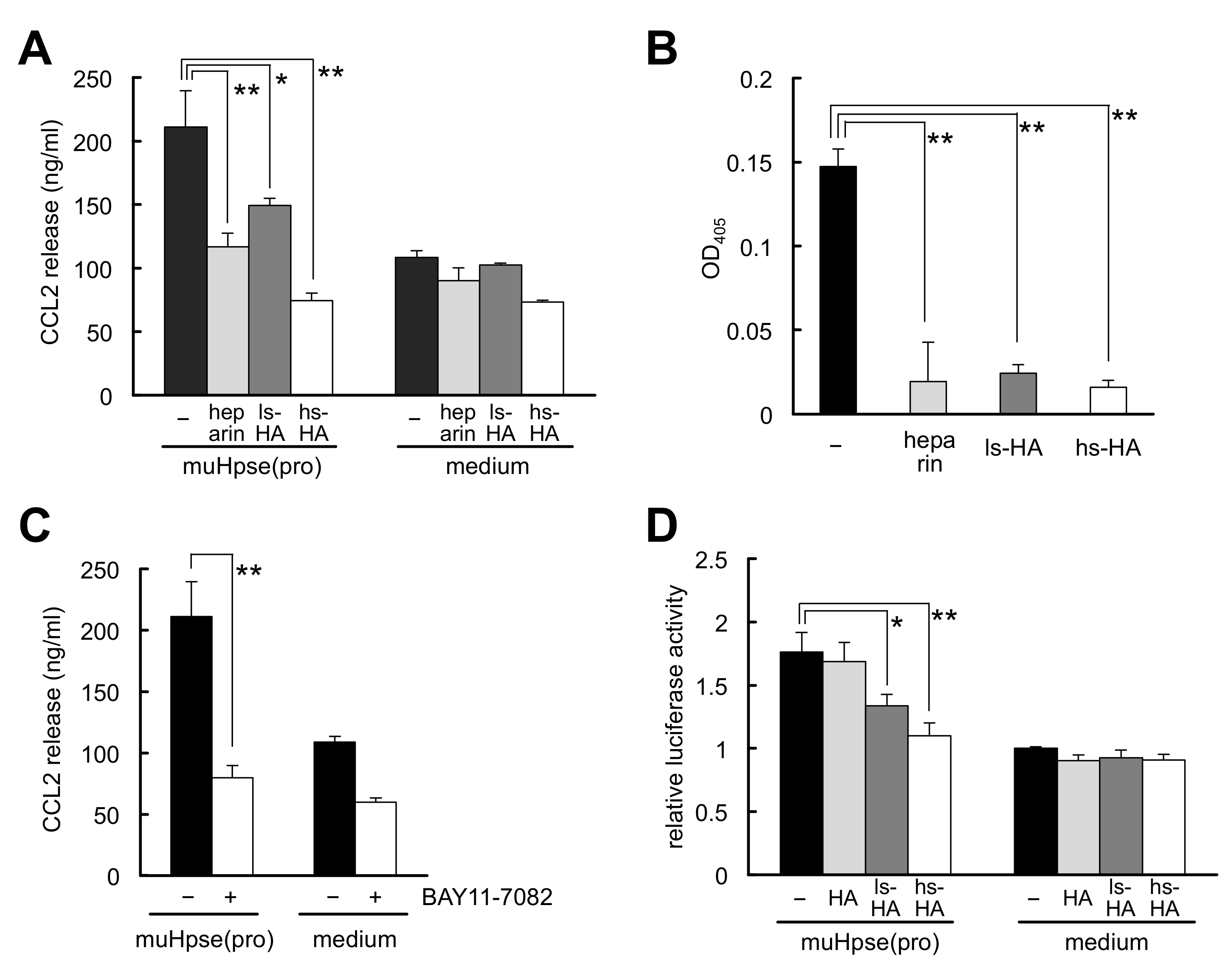
2.5. Sulfated HA Inhibited Hpse-Mediated CCL2 Release from Colon-26 Cells
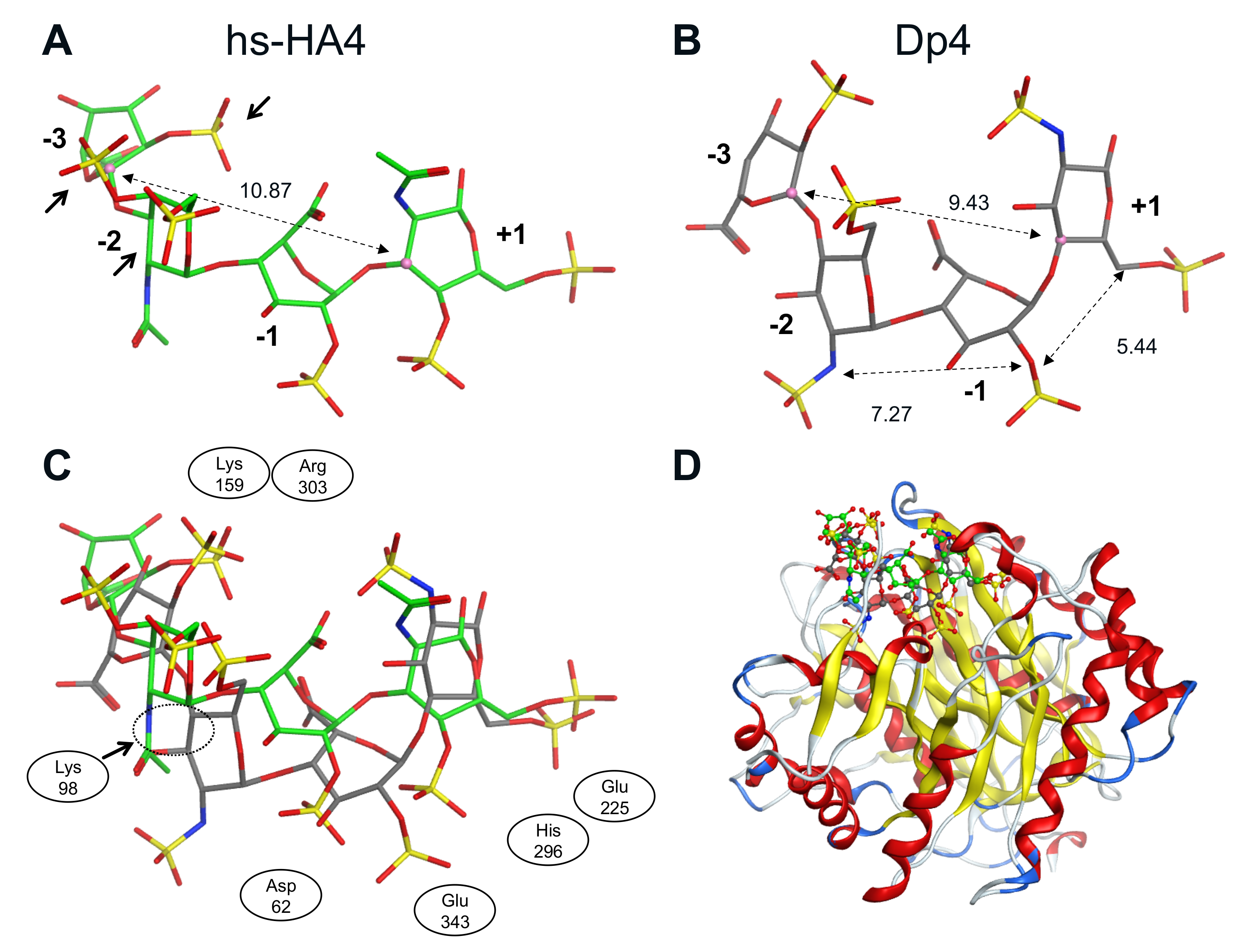
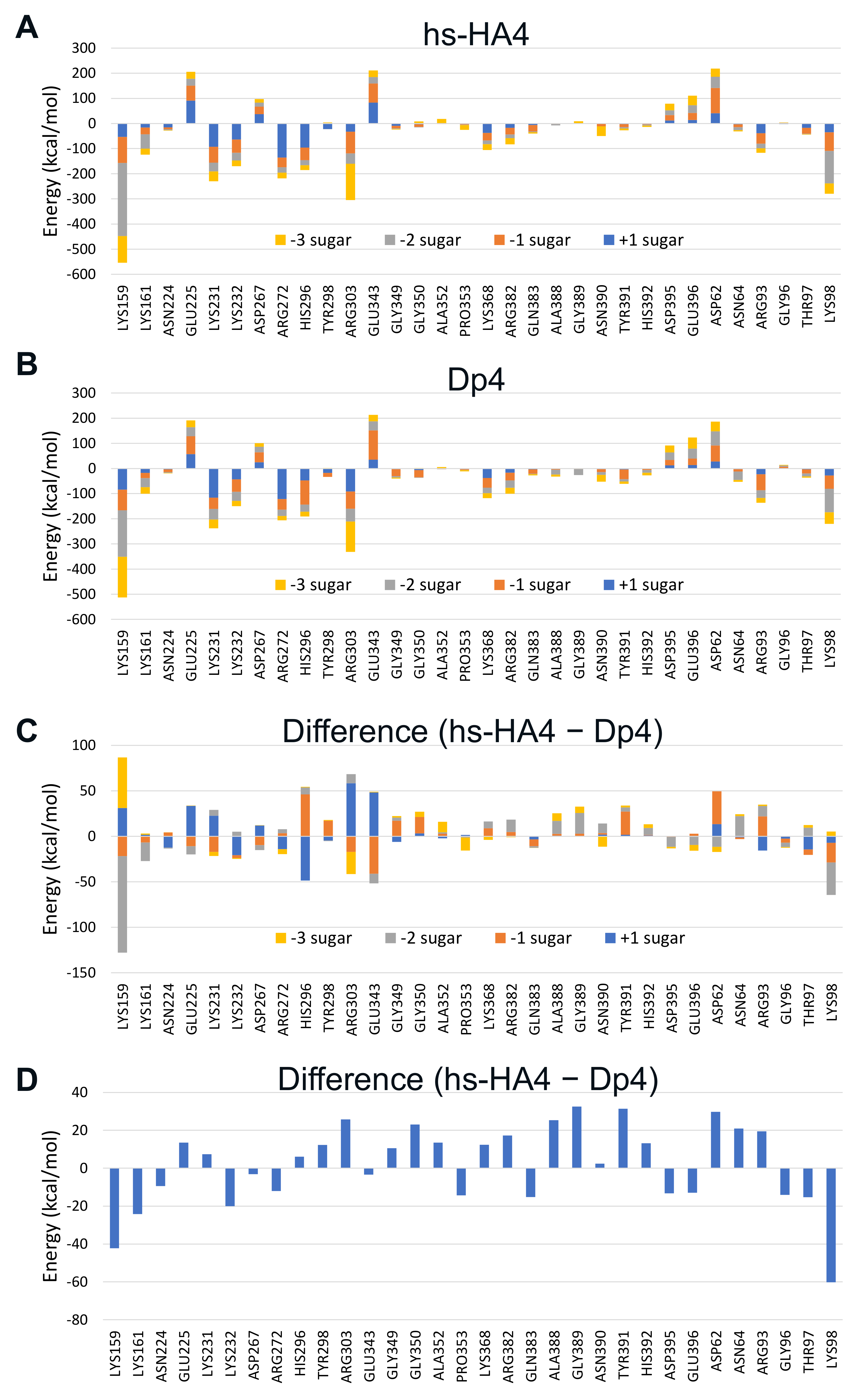
2.6. Fragmented Molecular Orbital Calculation of the Interaction between Sulfated GAG Tetrasaccharides and huHpse
3. Discussion
4. Materials and Methods
4.1. Reagents and Cells
4.2. Hpse-Mediated HS Degradation
4.3. Binding of Human and Murine Hpse to Immobilized GAGs
4.4. Surface Plasmon Resonance
4.5. Invasion Assay of 3D-Cultured Cancer Cell Clusters in a Collagen Gel
4.6. Hpse-Dependent CCL2 Release from Colon-26 Cells as Well as Cellular Binding of Colon-26 Cells to Hpse
4.7. NF-κB-Dependent Luciferase Assay in Colon-26 Cells
4.8. Statistical Analysis
4.9. Fragmented Molecular Orbital Calculation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Hpse | Heparanase |
| HS | heparan sulfate |
| CS | chondroitin sulfate |
| GAG | Glycosaminoglycan |
| HA | Hyaluronan |
| FMO | fragment molecular orbital |
| Dp4 | heparin-derived tetrasaccharide with six sulfation groups |
| ls-HA | low-sulfated HA |
| hs-HA | high-sulfated HA |
| huHpse | human heparanase |
| muHpse(mature) | recombinant protein mimicking mouse mature heparanase |
| muHpse(pro) | recombinant protein mimicking mouse proheparanase |
| ELISA | enzyme-linked immunosorbent assay |
| ABTS | 2,2′-azino-bis [3-ethylbenzothiazoline-6-sulfonic acid] |
| hs-HA4 | HA tetrasaccharide with six sulfation groups |
| SCIFIE | statistically corrected inter-fragment interaction energy |
| IFIE | inter-fragment interaction energy |
| ES | Electrostatic |
| EX | exchange repulsion |
| CT + mix | charge transfer and higher order mixed term |
| DI | Dispersion |
| HBD | heparin binding domain |
References
- Nakajima, M.; Irimura, T.; Di Ferrante, D.; Di Ferrante, N.; Nicolson, G.L. Heparan Sulfate Degradation: Relation to Tumor Invasive and Metastatic Properties of Mouse B16 Melanoma Sublines. Science 1983, 220, 611–613. [Google Scholar] [CrossRef] [PubMed]
- Parish, C.R.; Freeman, C.; Brown, K.J.; Francis, D.J.; Cowden, W.B. Identification of sulfated oligosaccharide-based inhibitors of tumor growth and metastasis using novel in vitro assays for angiogenesis and heparanase activity. Cancer Res. 1999, 59, 3433–3441. [Google Scholar] [PubMed]
- Miao, H.-Q.; Elkin, M.; Aingorn, E.; Ishai-Michaeli, R.; Stein, C.A.; Vlodavsky, I. Inhibition of heparanase activity and tumor metastasis by laminarin sulfate and synthetic phosphorothioate oligodeoxynucleotides. Int. J. Cancer 1999, 83, 424–431. [Google Scholar] [CrossRef]
- Reiland, J.; Sanderson, R.D.; Waguespack, M.; Barker, S.A.; Long, R.; Carson, D.D.; Marchetti, D. Heparanase Degrades Syndecan-1 and Perlecan Heparan Sulfate: Functional implications for tumor cell invasion. J. Biol. Chem. 2004, 279, 8047–8055. [Google Scholar] [CrossRef] [Green Version]
- Stoler-Barak, L.; Petrovich, E.; Aychek, T.; Gurevich, I.; Tal, O.; Hatzav, M.; Ilan, N.; Feigelson, S.W.; Shakhar, G.; Vlodavsky, I.; et al. Heparanase of murine effector lymphocytes and neutrophils is not required for their diapedesis into sites of inflammation. FASEB J. 2015, 29, 2010–2021. [Google Scholar] [CrossRef]
- Morris, A.; Wang, B.; Waern, I.; Venkatasamy, R.; Page, C.; Schmidt, E.P.; Wernersson, S.; Li, J.-P.; Spina, M. The Role of Heparanase in Pulmonary Cell Recruitment in Response to an Allergic but Not Non-Allergic Stimulus. PLoS ONE 2015, 10, e0127032. [Google Scholar] [CrossRef] [Green Version]
- Sue, M.; Higashi, N.; Shida, H.; Kogane, Y.; Nishimura, Y.; Adachi, H.; Kolaczkowska, E.; Kepka, M.; Nakajima, M.; Irimura, T. An iminosugar-based heparanase inhibitor heparastatin (SF4) suppresses infiltration of neutrophils and monocytes into inflamed dorsal air pouches. Int. Immunopharmacol. 2016, 35, 15–21. [Google Scholar] [CrossRef]
- Goldshmidt, O.; Zcharia, E.; Cohen, M.; Aingorn, H.; Cohen, I.; Nadav, L.; Katz, B.-Z.; Geiger, B.; Vlodavsky, I. Heparanase mediates cell adhesion independent of its enzymatic activity. FASEB J. 2003, 17, 1015–1025. [Google Scholar] [CrossRef] [Green Version]
- Gingis-Velitski, S.; Zetser, A.; Flugelman, M.Y.; Vlodavsky, I.; Ilan, N. Heparanase Induces Endothelial Cell Migration via Protein Kinase B/Akt Activation. J. Biol. Chem. 2004, 279, 23536–23541. [Google Scholar] [CrossRef] [Green Version]
- Lerner, I.; Hermano, E.; Zcharia, E.; Rodkin, D.; Bulvik, R.; Doviner, V.; Rubinstein, A.M.; Ishai-Michaeli, R.; Atzmon, R.; Sherman, Y.; et al. Heparanase powers a chronic inflammatory circuit that promotes colitis-associated tumorigenesis in mice. J. Clin. Investig. 2011, 121, 1709–1721. [Google Scholar] [CrossRef]
- Blich, M.; Golan, A.; Arvatz, G.; Sebbag, A.; Shafat, I.; Sabo, E.; Cohen-Kaplan, V.; Petcherski, S.; Avniel-Polak, S.; Eitan, A.; et al. Macrophage Activation by Heparanase Is Mediated by TLR-2 and TLR-4 and Associates with Plaque Progression. Arter. Thromb. Vasc. Biol. 2013, 33, e56–e65. [Google Scholar] [CrossRef] [Green Version]
- Quaglio, A.E.; Castilho, A.C.; Di Stasi, L.C. Experimental evidence of heparanase, Hsp70 and NF-κB gene expression on the response of anti-inflammatory drugs in TNBS-induced colonic inflammation. Life Sci. 2015, 141, 179–187. [Google Scholar] [CrossRef]
- Gutter-Kapon, L.; Alishekevitz, D.; Shaked, Y.; Li, J.-P.; Aronheim, A.; Ilan, N.; Vlodavsky, I. Heparanase is required for activation and function of macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, E7808–E7817. [Google Scholar] [CrossRef] [Green Version]
- Tsunekawa, N.; Higashi, N.; Kogane, Y.; Waki, M.; Shida, H.; Nishimura, Y.; Adachi, H.; Nakajima, M.; Irimura, T. Heparanase augments inflammatory chemokine production from colorectal carcinoma cell lines. Biochem. Biophys. Res. Commun. 2016, 469, 878–883. [Google Scholar] [CrossRef]
- Levy-Adam, F.; Feld, S.; Suss-Toby, E.; Vlodavsky, I.; Ilan, N. Heparanase Facilitates Cell Adhesion and Spreading by Clustering of Cell Surface Heparan Sulfate Proteoglycans. PLoS ONE 2008, 3, e2319. [Google Scholar] [CrossRef] [Green Version]
- Rivara, S.; Milazzo, F.M.; Giannini, G. Heparanase: A rainbow pharmacological target associated to multiple pathologies including rare diseases. Future Med. Chem. 2016, 8, 647–680. [Google Scholar] [CrossRef] [Green Version]
- Irimura, T.; Nakajima, M.; Nicolson, G.L. Chemically modified heparins as inhibitors of heparan sulfate specific endo-.beta.-glucuronidase (heparanase) of metastatic melanoma cells. Biochemistry 1986, 25, 5322–5328. [Google Scholar] [CrossRef]
- Naggi, A.; Casu, B.; Perez, M.; Torri, G.; Cassinelli, G.; Penco, S.; Pisano, C.; Giannini, G.; Ishai-Michaeli, R.; Vlodavsky, I. Modulation of the Heparanase-inhibiting Activity of Heparin through Selective Desulfation, Graded N-Acetylation, and Glycol Splitting. J. Biol. Chem. 2005, 280, 12103–12113. [Google Scholar] [CrossRef] [Green Version]
- Dredge, K.; Hammond, E.; Davis, K.; Li, C.P.; Liu, L.; Johnstone, K.; Handley, P.; Wimmer, N.; Gonda, T.J.; Gautam, A.; et al. The PG500 series: Novel heparan sulfate mimetics as potent angiogenesis and heparanase inhibitors for cancer therapy. Investig. New Drugs 2010, 28, 276–283. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, J.P.; Ramani, V.C.; Ren, Y.; Naggi, A.; Torri, G.; Casu, B.; Penco, S.; Pisano, C.; Carminati, P.; Tortoreto, M.; et al. SST0001, a Chemically Modified Heparin, Inhibits Myeloma Growth and Angiogenesis via Disruption of the Heparanase/Syndecan-1 Axis. Clin. Cancer Res. 2011, 17, 1382–1393. [Google Scholar] [CrossRef] [Green Version]
- Groult, H.; Cousin, R.; Chot-Plassot, C.; Maura, M.; Bridiau, N.; Piot, J.-M.; Maugard, T.; Fruitier-Arnaudin, I. λ-Carrageenan Oligosaccharides of Distinct Anti-Heparanase and Anticoagulant Activities Inhibit MDA-MB-231 Breast Cancer Cell Migration. Mar. Drugs 2019, 17, 140. [Google Scholar] [CrossRef] [Green Version]
- Higashi, N.; Maeda, R.; Sesoko, N.; Isono, M.; Ishikawa, S.; Tani, Y.; Takahashi, K.; Oku, T.; Higashi, K.; Onishi, S.; et al. Chondroitin sulfate E blocks enzymatic action of heparanase and heparanase-induced cellular responses. Biochem. Biophys. Res. Commun. 2019, 520, 152–158. [Google Scholar] [CrossRef]
- Litwiniuk, M.; Krejner, A.; Speyrer, M.S.; Gauto, A.R.; Grzela, T. Hyaluronic Acid in Inflammation and Tissue Regeneration. Wounds 2016, 28, 78–88. [Google Scholar]
- Termeer, C.; Benedix, F.; Sleeman, J.; Fieber, C.; Voith, U.; Ahrens, T.; Miyake, K.; Freudenberg, M.; Galanos, C.; Simon, J.C. Oligosaccharides of Hyaluronan Activate Dendritic Cells via Toll-like Receptor 4. J. Exp. Med. 2002, 195, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Misra, S.; Heldin, P.; Hascall, V.C.; Karamanos, N.K.; Skandalis, S.S.; Markwald, R.R.; Ghatak, S. Hyaluronan-CD44 interactions as potential targets for cancer therapy. FEBS J. 2011, 278, 1429–1443. [Google Scholar] [CrossRef] [Green Version]
- Chang, N.-S.; Intrieri, C.; Mattison, J.; Armand, G. Synthetic polysulfated hyaluronic acid is a potent inhibitor for tumor necrosis factor production. J. Leukoc. Biol. 1994, 55, 778–784. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, X.; Rao, N.V.; Argyle, B.; McCoard, L.; Rusho, W.J.; Kennedy, T.P.; Prestwich, G.D.; Krueger, G. Novel Sulfated Polysaccharides Disrupt Cathelicidins, Inhibit RAGE and Reduce Cutaneous Inflammation in a Mouse Model of Rosacea. PLoS ONE 2011, 6, e16658. [Google Scholar] [CrossRef] [Green Version]
- Hempel, U.; Matthäus, C.; Preissler, C.; Möller, S.; Hintze, V.; Dieter, P. Artificial Matrices With High-Sulfated Glycosaminoglycans and Collagen Are Anti-Inflammatory and Pro-Osteogenic for Human Mesenchymal Stromal Cells. J. Cell. Biochem. 2014, 115, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Savage, J.R.; Pulsipher, A.; Rao, N.V.; Kennedy, T.P.; Prestwich, G.D.; Ryan, M.E.; Lee, W.Y. A Modified Glycosaminoglycan, GM-0111, Inhibits Molecular Signaling Involved in Periodontitis. PLoS ONE 2016, 11, e0157310. [Google Scholar] [CrossRef] [PubMed]
- Jouy, F.; Lohmann, N.; Wandel, E.; Ruiz-Gómez, G.; Pisabarro, M.T.; Beck-Sickinger, A.G.; Schnabelrauch, M.; Möller, S.; Simon, J.C.; Kalkhof, S.; et al. Sulfated hyaluronan attenuates inflammatory signaling pathways in macrophages involving induction of antioxidants. Proteomics 2017, 17, 1700082. [Google Scholar] [CrossRef] [PubMed]
- Thönes, S.; Rother, S.; Wippold, T.; Blaszkiewicz, J.; Balamurugan, K.; Moeller, S.; Ruiz-Gómez, G.; Schnabelrauch, M.; Scharnweber, D.; Saalbach, A.; et al. Hyaluronan/collagen hydrogels containing sulfated hyaluronan improve wound healing by sustained release of heparin-binding EGF-like growth factor. Acta Biomater. 2019, 86, 135–147. [Google Scholar] [CrossRef]
- Hauck, S.; Zager, P.; Halfter, N.; Wandel, E.; Torregrossa, M.; Kakpenova, A.; Rother, S.; Ordieres, M.; Räthel, S.; Berg, A.; et al. Collagen/hyaluronan based hydrogels releasing sulfated hyaluronan improve dermal wound healing in diabetic mice via reducing inflammatory macrophage activity. Bioact. Mater. 2021, 6, 4342–4359. [Google Scholar] [CrossRef]
- Pulsipher, A.; Savage, J.R.; Kennedy, T.P.; Gupta, K.; Cuiffo, B.G.; Sonis, S.T.; Lee, W.Y. GM-1111 reduces radiation-induced oral mucositis in mice by targeting pattern recognition receptor-mediated inflammatory signaling. PLoS ONE 2021, 16, e0249343. [Google Scholar] [CrossRef]
- Picke, A.-K.; Salbach-Hirsch, J.; Hintze, V.; Rother, S.; Rauner, M.; Kascholke, C.; Möller, S.; Bernhardt, R.; Rammelt, S.; Pisabarro, M.T.; et al. Sulfated hyaluronan improves bone regeneration of diabetic rats by binding sclerostin and enhancing osteoblast function. Biomaterials 2016, 96, 11–23. [Google Scholar] [CrossRef]
- Jordan, A.; Lokeshwar, S.D.; Lopez, L.E.; Hennig, M.; Chipollini, J.; Yates, T.; Hupe, M.C.; Merseburger, A.S.; Shiedlin, A.; Cerwinka, W.H.; et al. Antitumor activity of sulfated hyaluronic acid fragments in pre-clinical models of bladder cancer. Oncotarget 2017, 8, 24262–24274. [Google Scholar] [CrossRef] [Green Version]
- Miura, T.; Yuasa, N.; Ota, H.; Habu, M.; Kawano, M.; Nakayama, F.; Nishihara, S. Highly sulfated hyaluronic acid maintains human induced pluripotent stem cells under feeder-free and bFGF-free conditions. Biochem. Biophys. Res. Commun. 2019, 518, 506–512. [Google Scholar] [CrossRef]
- Koehler, L.; Ruiz-Gómez, G.; Balamurugan, K.; Rother, S.; Freyse, J.; Möller, S.; Schnabelrauch, M.; Köhling, S.; Djordjevic, S.; Scharnweber, D.; et al. Dual Action of Sulfated Hyaluronan on Angiogenic Processes in Relation to Vascular Endothelial Growth Factor-A. Sci. Rep. 2019, 9, 18143. [Google Scholar] [CrossRef]
- Tao, L.; Tian, S.; Zhang, J.; Liu, Z.; Robinson-McCarthy, L.; Miyashita, S.-I.; Breault, D.T.; Gerhard, R.; Oottamasathien, S.; Whelan, S.; et al. Sulfated glycosaminoglycans and low-density lipoprotein receptor contribute to Clostridium difficile toxin A entry into cells. Nat. Microbiol. 2019, 4, 1760–1769. [Google Scholar] [CrossRef]
- Kitaura, K.; Ikeo, E.; Asada, T.; Nakano, T.; Uebayasi, M. Fragment molecular orbital method: An approximate computational method for large molecules. Chem. Phys. Lett. 1999, 313, 701–706. [Google Scholar] [CrossRef]
- Mochizuki, Y.; Tanaka, S.; Fukuzawa, K. (Eds.) Recent Advances of the Fragment Molecular Orbital Method; Springer: Singapore, 2021. [Google Scholar] [CrossRef]
- Fukuzawa, K.; Tanaka, S. Fragment molecular orbital calculations for biomolecules. Curr. Opin. Struct. Biol. 2021, 72, 127–134. [Google Scholar] [CrossRef]
- Wu, L.; Viola, C.M.; Brzozowski, A.M.; Davies, G.J. Structural characterization of human heparanase reveals insights into substrate recognition. Nat. Struct. Mol. Biol. 2015, 22, 1016–1022. [Google Scholar] [CrossRef] [Green Version]
- Casu, B.; Gennaro, U. A conductimetric method for the determination of sulphate and carboxyl groups in heparin and other mucopolysaccharides. Carbohydr. Res. 1975, 39, 168–176. [Google Scholar] [CrossRef]
- Vinci, M.; Box, C.; Eccles, S.A. Three-Dimensional (3D) Tumor Spheroid Invasion Assay. J. Vis. Exp. 2015, 99, e52686. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Watanabe, C.; Okiyama, Y. Statistical correction to effective interactions in the fragment molecular orbital method. Chem. Phys. Lett. 2013, 556, 272–277. [Google Scholar] [CrossRef] [Green Version]
- Miura, T.; Kawano, M.; Takahashi, K.; Yuasa, N.; Habu, M.; Kimura, F.; Imamura, T.; Nakayama, F. High-Sulfated Hyaluronic Acid Ameliorates Radiation-Induced Intestinal Damage Without Blood Anticoagulation. Adv. Radiat. Oncol. 2022, 7, 100900. [Google Scholar] [CrossRef]
- Sache, E.; Maillard, M.; Malazzi, P.; Bertrand, H. Partially N-desulfated heparin as a non-anticoagulant heparin: Some physico-chemical and biological properties. Thromb. Res. 1989, 55, 247–258. [Google Scholar] [CrossRef]
- Miron, A.; Rother, S.; Huebner, L.; Hempel, U.; Käppler, I.; Moeller, S.; Schnabelrauch, M.; Scharnweber, D.; Hintze, V. Sulfated Hyaluronan Influences the Formation of Artificial Extracellular Matrices and the Adhesion of Osteogenic Cells. Macromol. Biosci. 2014, 14, 1783–1794. [Google Scholar] [CrossRef]
- Rother, S.; Salbach-Hirsch, J.; Moeller, S.; Seemann, T.; Schnabelrauch, M.; Hofbauer, L.C.; Hintze, V.; Scharnweber, D. Bioinspired Collagen/Glycosaminoglycan-Based Cellular Microenvironments for Tuning Osteoclastogenesis. ACS Appl. Mater. Interfaces 2015, 7, 23787–23797. [Google Scholar] [CrossRef]
- Metzger, W.; Rother, S.; Pohlemann, T.; Möller, S.; Schnabelrauch, M.; Hintze, V.; Scharnweber, D. Evaluation of cell-surface interaction using a 3D spheroid cell culture model on artificial extracellular matrices. Mater. Sci. Eng. C 2017, 73, 310–318. [Google Scholar] [CrossRef]
- Pączek, S.; Łukaszewicz-Zając, M.; Mroczko, B. Chemokines—What Is Their Role in Colorectal Cancer? Cancer Control 2020, 27, 1073274820903384. [Google Scholar] [CrossRef]
- Itatani, Y.; Kawada, K.; Inamoto, S.; Yamamoto, T.; Ogawa, R.; Taketo, M.M.; Sakai, Y. The Role of Chemokines in Promoting Colorectal Cancer Invasion/Metastasis. Int. J. Mol. Sci. 2016, 17, 643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Sun, L.; Guo, C.; Liu, Q.; Zhou, Z.; Peng, L.; Pan, J.; Yu, L.; Lou, J.; Yang, Z.; et al. Tumor Cell-Microenvironment Interaction Models Coupled with Clinical Validation Reveal CCL2 and SNCG as Two Predictors of Colorectal Cancer Hepatic Metastasis. Clin. Cancer Res. 2009, 15, 5485–5493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zajkowska, M.; Dulewicz, M.; Kulczyńska-Przybik, A.; Safiejko, K.; Juchimiuk, M.; Konopko, M.; Kozłowski, L.; Mroczko, B. The Significance of Selected C-C Motif Chemokine Ligands in Colorectal Cancer Patients. J. Clin. Med. 2022, 11, 1794. [Google Scholar] [CrossRef] [PubMed]
- Goodall, K.; Poon, I.; Phipps, S.; Hulett, M.D. Soluble Heparan Sulfate Fragments Generated by Heparanase Trigger the Release of Pro-Inflammatory Cytokines through TLR-4. PLoS ONE 2014, 9, e109596. [Google Scholar] [CrossRef]
- O’Callaghan, P.; Li, J.-P.; Lannfelt, L.; Lindahl, U.; Zhang, X. Microglial Heparan Sulfate Proteoglycans Facilitate the Cluster-of-Differentiation 14 (CD14)/Toll-like Receptor 4 (TLR4)-Dependent Inflammatory Response. J. Biol. Chem. 2015, 290, 14904–14914. [Google Scholar] [CrossRef] [Green Version]
- Pala, D.; Rivara, S.; Mor, M.; Milazzo, F.M.; Roscilli, G.; Pavoni, E.; Giannini, G. Kinetic analysis and molecular modeling of the inhibition mechanism of roneparstat (SST0001) on human heparanase. Glycobiology 2016, 26, 640–654. [Google Scholar] [CrossRef] [Green Version]
- Chhabra, M.; Wilson, J.C.; Wu, L.; Davies, G.J.; Gandhi, N.S.; Ferro, V. Structural Insights into Pixatimod (PG545) Inhibition of Heparanase, a Key Enzyme in Cancer and Viral Infections. Chem. Eur. J. 2022, 28, e202104222. [Google Scholar] [CrossRef]
- Loka, R.S.; Yu, F.; Sletten, E.T.; Nguyen, H.M. Design, synthesis, and evaluation of heparan sulfate mimicking glycopolymers for inhibiting heparanase activity. Chem. Commun. 2017, 53, 9163–9166. [Google Scholar] [CrossRef]
- Yang, W.; Eken, Y.; Zhang, J.; Cole, L.E.; Ramadan, S.; Xu, Y.; Zhang, Z.; Liu, J.; Wilson, A.K.; Huang, X. Chemical synthesis of human syndecan-4 glycopeptide bearing O-, N-sulfation and multiple aspartic acids for probing impacts of the glycan chain and the core peptide on biological functions. Chem. Sci. 2020, 11, 6393–6404. [Google Scholar] [CrossRef]
- Fedorov, D.G.; Kitaura, K. Pair interaction energy decomposition analysis. J. Comput. Chem. 2006, 28, 222–237. [Google Scholar] [CrossRef]
- Levy-Adam, F.; Abboud-Jarrous, G.; Guerrini, M.; Beccati, D.; Vlodavsky, I.; Ilan, N. Identification and Characterization of Heparin/Heparan Sulfate Binding Domains of the Endoglycosidase Heparanase. J. Biol. Chem. 2005, 280, 20457–20466. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, N.S.; Freeman, C.; Parish, C.R.; Mancera, R.L. Computational analyses of the catalytic and heparin-binding sites and their interactions with glycosaminoglycans in glycoside hydrolase family 79 endo-β-d-glucuronidase (heparanase). Glycobiology 2011, 22, 35–55. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, N.; Waki, M.; Sue, M.; Tokuda, C.; Kasaoka, T.; Nakajima, M.; Higashi, N.; Irimura, T. Heparanase expression in B16 melanoma cells and peripheral blood neutrophils before and after extravasation detected by novel anti-mouse heparanase monoclonal antibodies. J. Immunol. Methods 2008, 331, 82–93. [Google Scholar] [CrossRef]
- Sasaki, N.; Higashi, N.; Taka, T.; Nakajima, M.; Irimura, T. Cell Surface Localization of Heparanase on Macrophages Regulates Degradation of Extracellular Matrix Heparan Sulfate. J. Immunol. 2004, 172, 3830–3835. [Google Scholar] [CrossRef] [Green Version]
- Oku, T.; Ando, Y.; Ogura, M.; Tsuji, T. Development of Splice Variant-Specific Monoclonal Antibodies Against Human α3 Integrin. Monoclon. Antib. Immunodiagn. Immunother. 2016, 35, 12–17. [Google Scholar] [CrossRef]
- Oku, T.; Shimada, K.; Kenmotsu, H.; Ando, Y.; Kurisaka, C.; Sano, R.; Tsuiji, M.; Hasegawa, S.; Fukui, T.; Tsuji, T. Stimulation of Peritoneal Mesothelial Cells to Secrete Matrix Metalloproteinase-9 (MMP-9) by TNF-α: A Role in the Invasion of Gastric Carcinoma Cells. Int. J. Mol. Sci. 2018, 19, 3961. [Google Scholar] [CrossRef] [Green Version]
- Chemical Computing Group Inc. Molecular Operating Environment (MOE). Available online: https://www.chemcomp.com/index.htm (accessed on 31 December 2021).
- Mochizuki, Y.; Nakano, T.; Koikegami, S.; Tanimori, S.; Abe, Y.; Nagashima, U.; Kitaura, K. A parallelized integral-direct second-order Møller–Plesset perturbation theory method with a fragment molecular orbital scheme. Theor. Chim. Acta 2004, 112, 442–452. [Google Scholar] [CrossRef]
- Fukuzawa, K.; Omagari, K.; Nakajima, K.; Nobusawa, E.; Tanaka, S. Sialic acid recognition of the pandemic influenza 2009 H1N1 virus: Binding mechanism between human receptor and influenza hemagglutinin. Protein Pept. Lett. 2011, 18, 530–539. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sulfated GAGs | IC50 for HuHpse (µg/mL) | IC50 for MuHpse (µg/mL) |
|---|---|---|
| Heparin | 0.363 ± 0.006 | 0.400 ± 0.028 |
| ls-HA | 0.267 ± 0.061 | 0.542 ± 0.236 |
| hs-HA | 0.047 ± 0.003 *** | 0.046 ± 0.007 *** |
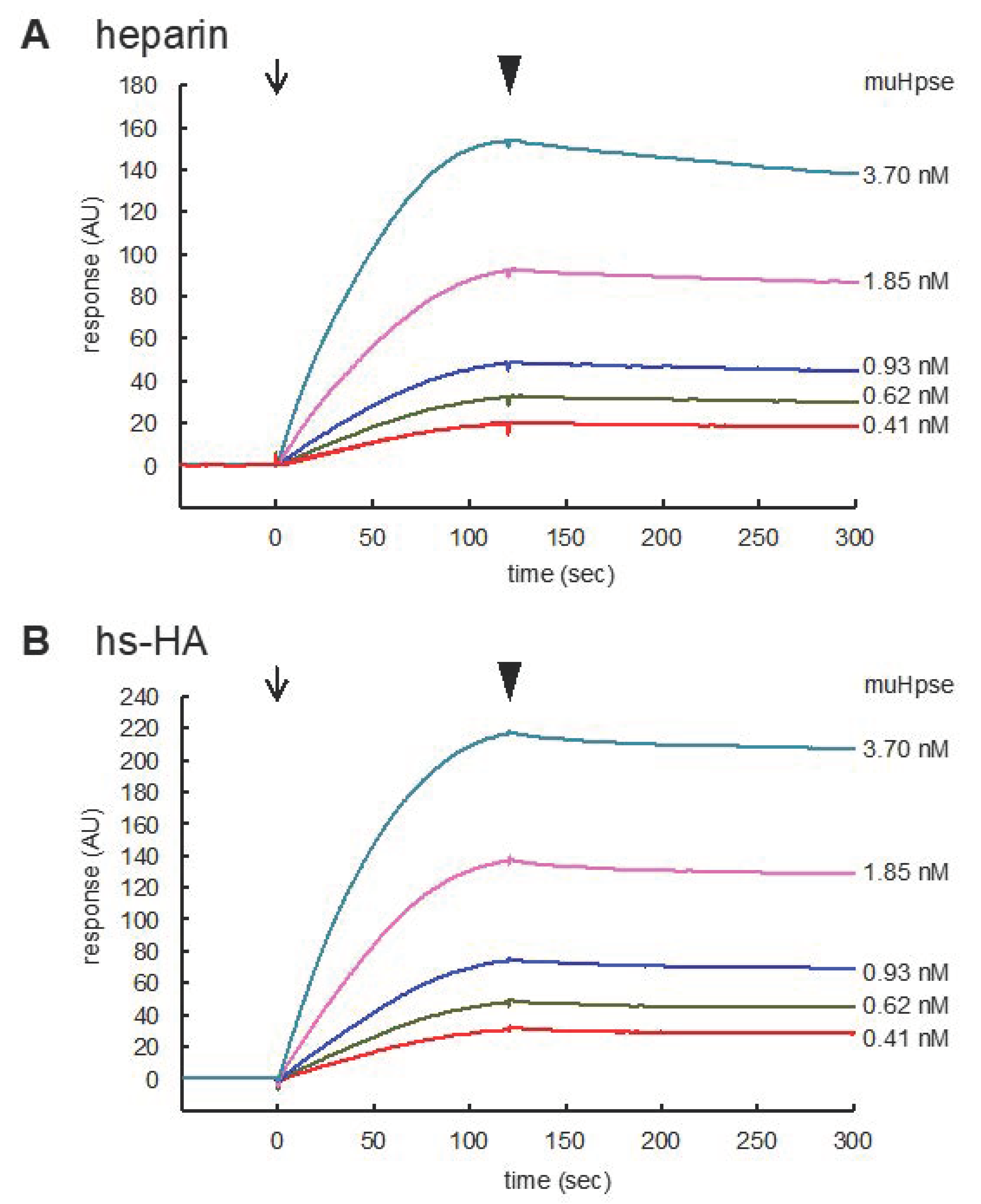
| Sulfated GAGs | ka (M−1s−1) | kd (1/s−1) | KD (M) |
|---|---|---|---|
| heparin | (1.18 ± 0.42) × 107 | (9.58 ± 0.58) × 10−4 | (8.16 ± 0.72) × 10−11 |
| hs-HA | (1.20 ± 0.18) × 107 | (3.08 ± 0.52) × 10−4 *** | (2.67 ± 0.85) × 10−11 *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, J.; Kanoya, R.; Tani, Y.; Ishikawa, S.; Maeda, R.; Suzuki, S.; Kawanami, F.; Miyagawa, N.; Takahashi, K.; Oku, T.; et al. Sulfated Hyaluronan Binds to Heparanase and Blocks Its Enzymatic and Cellular Actions in Carcinoma Cells. Int. J. Mol. Sci. 2022, 23, 5055. https://doi.org/10.3390/ijms23095055
Shi J, Kanoya R, Tani Y, Ishikawa S, Maeda R, Suzuki S, Kawanami F, Miyagawa N, Takahashi K, Oku T, et al. Sulfated Hyaluronan Binds to Heparanase and Blocks Its Enzymatic and Cellular Actions in Carcinoma Cells. International Journal of Molecular Sciences. 2022; 23(9):5055. https://doi.org/10.3390/ijms23095055
Chicago/Turabian StyleShi, Jia, Riku Kanoya, Yurina Tani, Sodai Ishikawa, Rino Maeda, Sana Suzuki, Fumiya Kawanami, Naoko Miyagawa, Katsuhiko Takahashi, Teruaki Oku, and et al. 2022. "Sulfated Hyaluronan Binds to Heparanase and Blocks Its Enzymatic and Cellular Actions in Carcinoma Cells" International Journal of Molecular Sciences 23, no. 9: 5055. https://doi.org/10.3390/ijms23095055
APA StyleShi, J., Kanoya, R., Tani, Y., Ishikawa, S., Maeda, R., Suzuki, S., Kawanami, F., Miyagawa, N., Takahashi, K., Oku, T., Yamamoto, A., Fukuzawa, K., Nakajima, M., Irimura, T., & Higashi, N. (2022). Sulfated Hyaluronan Binds to Heparanase and Blocks Its Enzymatic and Cellular Actions in Carcinoma Cells. International Journal of Molecular Sciences, 23(9), 5055. https://doi.org/10.3390/ijms23095055