1. Introduction
Hepatocellular carcinoma (HCC), the most common primary malignant tumor in liver cancer cases, is a complex disease caused by a variety of risk factors. Conventional types of liver cancer treatment, including surgical resection, radiotherapy, and chemotherapy, have been either limited in application or ineffective [
1]. Transplantation of the liver is believed to be the only viable treatment; however, it is not easy to find the proper donor. Although scientists have generated intense research efforts to explore cellular, molecular, and physiological mechanisms of the disease for developing prevention and therapy strategies [
2], the mortality rate of HCC remains high.
The transcription factor p53 is activated in response to various stresses including nutrient deprivation, DNA damage, oncogene activation, and hypoxia. p53 is a well-established tumor suppressor and guardian of the genome that induces apoptosis and cell cycle arrest by activating downstream target genes [
3]. However,
p53 is mutated in around half of all human cancers. It is generally believed that p53 loses its tumor suppressor function because of a mutation in
p53. Certain types of
p53 mutations are gain-of-function mutations, which have been shown to have oncogenic functions [
4]. HCC is a lethal malignancy associated with poor prognosis and a high recurrence. Effective HCC therapeutics still await a molecular understanding of the mechanisms promoting the development of selective and precise agents. HCC has a high rate of mutation in tumor suppressor protein p53, leading to the loss of its tumor suppressor activity and, in certain cases, gain-of-function activities that promote cell proliferation, tumor progression, and drug resistance [
5]. Thus, mutant
p53 has become an important target for the development of anticancer agents in HCC.
The signal transducer and activator of transcription 3 (STAT3) is a pivotal transcriptional factor of multiple promoting genes in cancer development and immune evasion [
6]. Phosphorylated STAT3s dimerize each other and translocate into the nucleus before activating the downstream genes. Under a normal physiological state, STAT3 activation is usually transient in the continuous stimulation of cytokines and contributes to protecting normal hepatocytes from inflammatory insults. It has been reported that constitutive phosphorylation of STAT3 in tumor tissue is correlated with poor prognosis in HCC patients [
7]. Thereafter, the inactivation of the STAT3 signal pathway is a promising strategy in anti-HCC treatment.
Ailanthoidol (ATD), a neolignan, has been isolated from the bark of
Zanthoxylum ailanthoides (Rutaceae), of which the dried fruit is used as a spice in Taiwan. Our previous study demonstrated that ATD displays antitumor promotion effects using the multistep skin cancer model induced by 12-o-tetradecanoylphobol-13-acetate [
8]. Recently, we found that ATD suppresses TGF-β1-promoted migration and invasion in HepG2 cells [
9]. Kim and Jun reported that ATD has in vitro and in vivo anti-inflammatory effects [
10]. Park et al. found that ATD possesses antiadipogenic activities [
11]. In addition, ATD is a benzofuran derivative and indicates diverse pharmacological activities, including anticancer activities [
12]. As the anticancer properties of ATD have not been well clarified, this study investigated the antiproliferation effects and molecular mechanism of ATD in hepatoma cells.
3. Discussion
HCC, which accounts for nearly 80% of all liver cancer cases, is a heterogeneous type of cancer caused by a variety of risk factors, including exposure to the hepatitis virus, food contaminated with Aflatoxin B1, heavy alcohol intake, and obesity [
14,
15]. The incidence of HCC is rising rapidly worldwide. In addition, since HCC is diagnosed at a late stage in most cases, surgical resection and liver transplantation are not practical therapy options. Metastasis and recurrence are quite common. Therefore, the development of a promising compound with target therapy potential is an urgent task. Plants are major food and pharmaceutical sources for humans. Some phytochemicals, such as alkaloids, diterpenoids, and sesquiterpenes, display therapeutic potential for cancer treatment [
16]. However, these therapeutic phytochemicals are also associated with adverse side effects, such as cardiovascular diseases, vomiting, renal dysfunction, and myelotoxicity. Thereafter, scientists have dedicated themselves to developing phytochemicals with minimal side effects and good bioavailability. Lignans and neolignans may possess great potential for anticancer treatment and display good safety profiles [
12,
17,
18]. In the present study, ATD, a neolignan isolated from the bark of
Zanthoxylum ailanthoides [
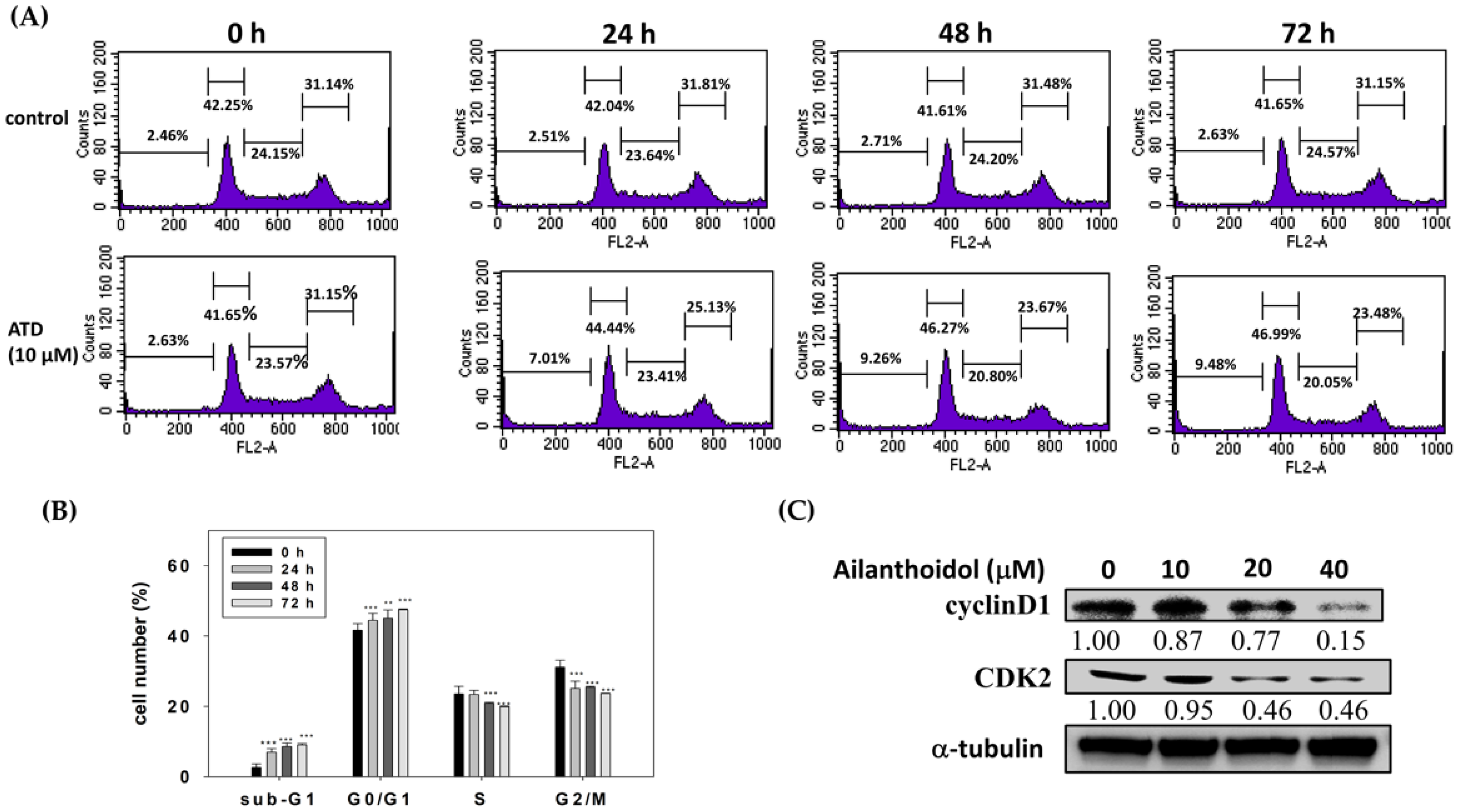
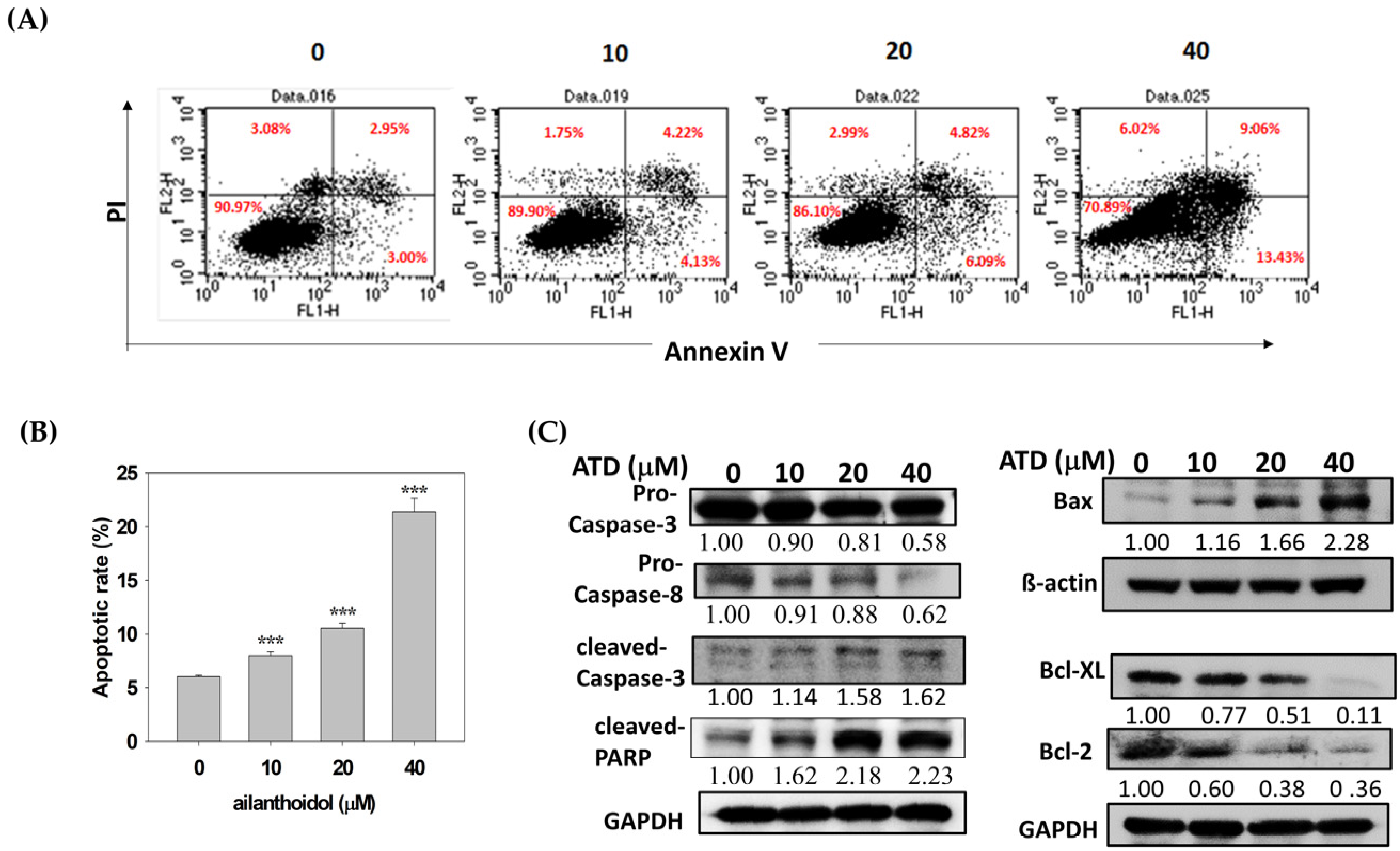
19], exhibited antiproliferation potential in Huh7 hepatoma cells, which was related to the induction of cell cycle arrest and the activation of apoptosis. Cell cycle arrest was mediated by the ATD-induced cyclin D1 and CDK2 expression, while apoptosis was activated by ATD-downregulated Bcl-xL/Bcl2 and augmented Bax, resulting in the activation of caspase 3. For a real application, animal studies of ATD are required in the future.
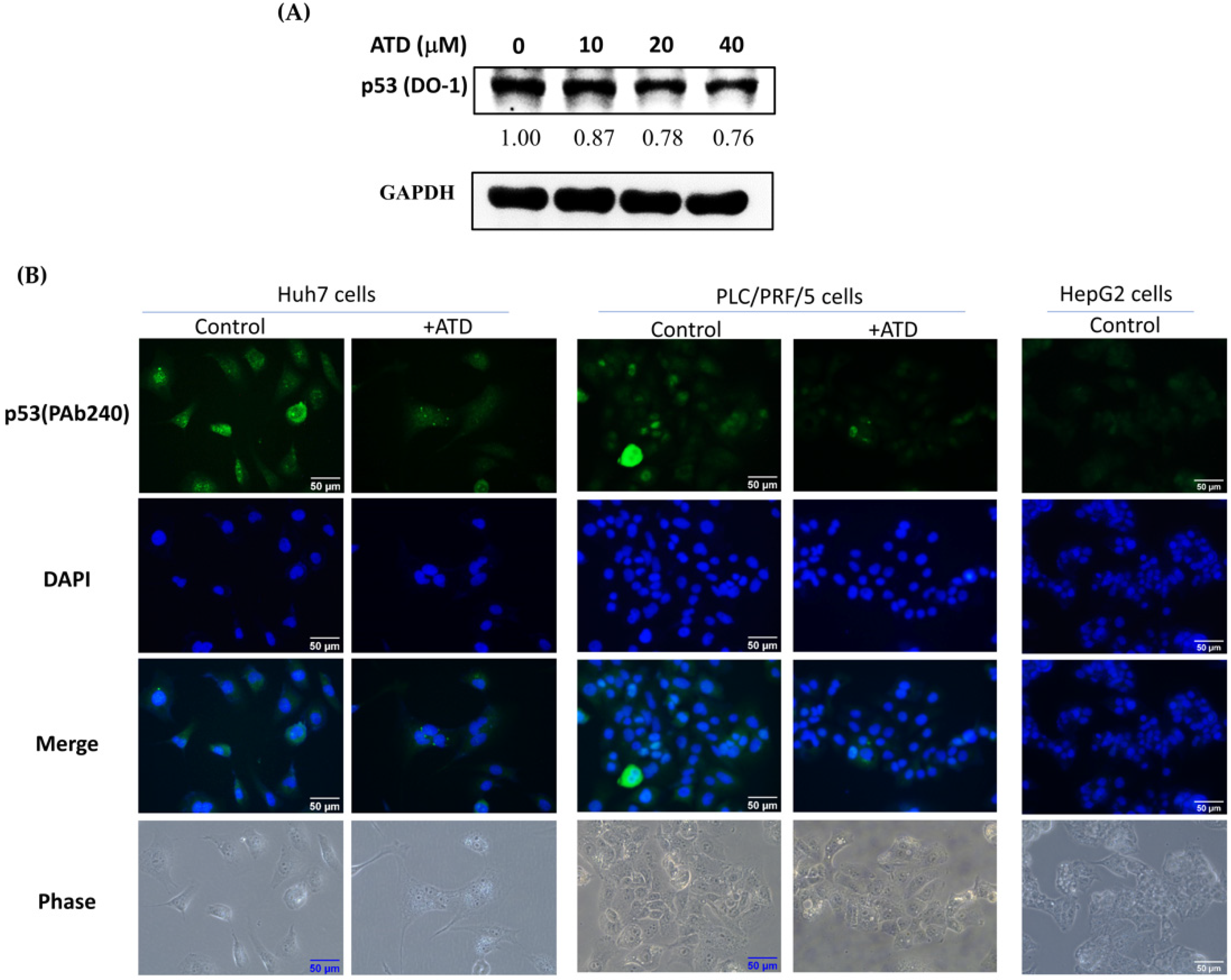
The tumor suppressor p53 regulates the transcription of numerous downstream target genes involved in cell cycle arrest, apoptosis, and metabolism. Loss of p53 activity by gene deletion or mutations in normal cells causes uncontrolled cell proliferation, leading to immortalization and, ultimately, cancer. Additionally, mutant
p53 shows oncogenic gain-of-function activities, such as enhanced tumor progression, metastasis potential, and drug resistance [
20]. As a result, obtaining efficient inhibitors against mutant
p53 cancer cells remains an urgent task for medicine development. Reactivation of the wild-type
p53 function and expression or abrogation of mutant
p53 protein may halt cancer progression [
21]. Accumulation of mutant
p53 is critical for the gain of function related to
p53 mutation, including enhanced cell growth and tumor progression; however, the manner in which mut
p53 is regulated and promotes cancer progression is not well understood [
4]. Enzymes controlling p53 proteasomal degradation or stability and some microRNA have been considered to regulate mutant
p53 levels [
13,
22]. In the present study, we found that ATD had more potent cytotoxicity in Huh7 cells (mutant
p53) than in HepG2 cells (WT
p53), which was associated with reducing the level of mut
p53. According to our results, ATD blocked the STAT3 pathway and mediated the abrogation of mut
p53. Whether ATD affects the miRNA or enzymes associated with proteasomal degradation requires further clarification. Our data implicated that ATD displayed potent anticancer potential in mut
p53-based HCC by impairing the gain of function of mutant
p53.
Among the diverse signaling molecules, STAT3 is considered an oncogenic factor in HCC [
7]. Under a normal physiological state, STAT3 activation is usually transient, even in the continuous stimulation of cytokines, and contributes to protecting normal hepatocytes from inflammatory and toxic insults. In HCC, the persistent activation of STAT3 changes the gene transcriptions associated with cell survival, proliferation, invasion, and angiogenesis. The pro-proliferative role of STAT3 is related to its antiapoptotic functions toward HCC via upregulating antiapoptotic proteins such as Bcl-xL. Furthermore, constitutive phosphorylation of STAT3 in tumor tissue is closely correlated with a poor prognosis in HCC patients [
6]. Recently, it has been reported that STAT3 sustains mut
p53 expression due to its interplay with the mevalonate pathway, which increases the stability of mut
p53 and prevents its degradation from proteasome [
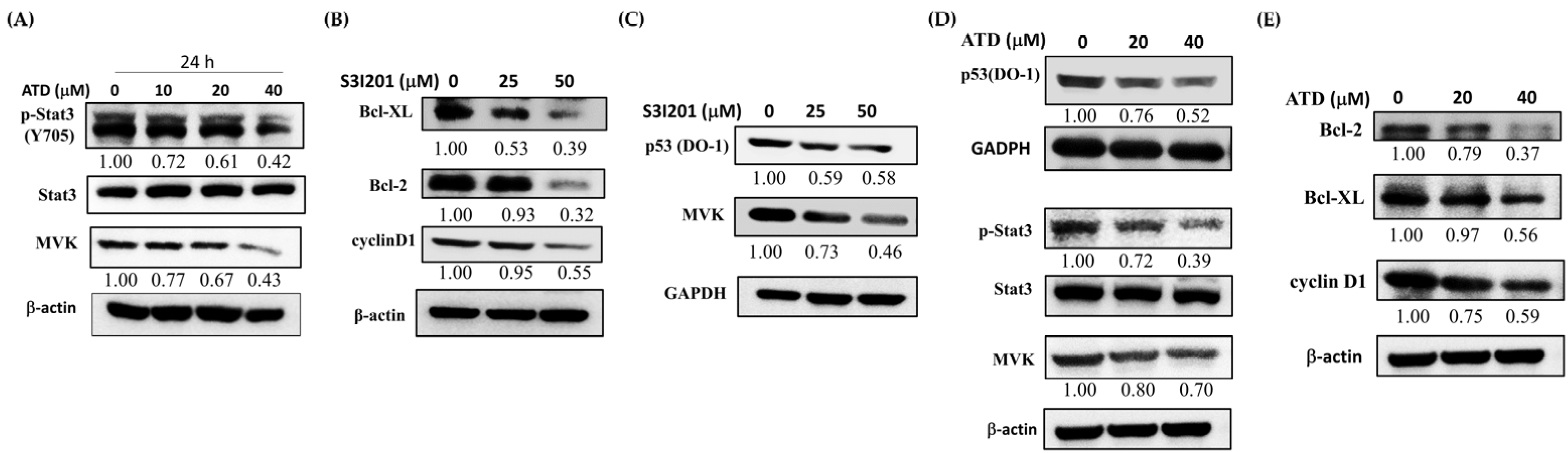
13]. In the present study, ATD inhibited the p-STAT3, MVK, and mut
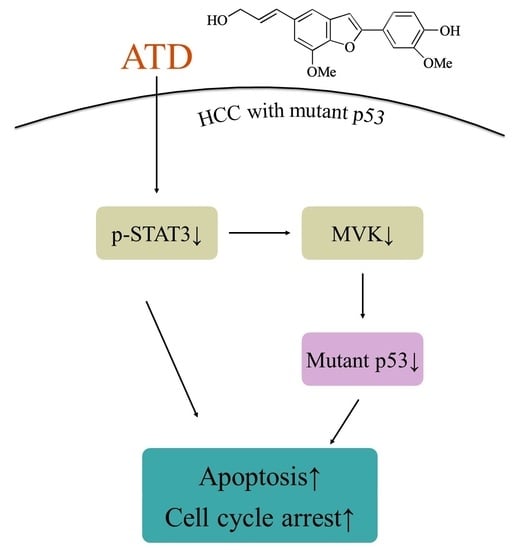
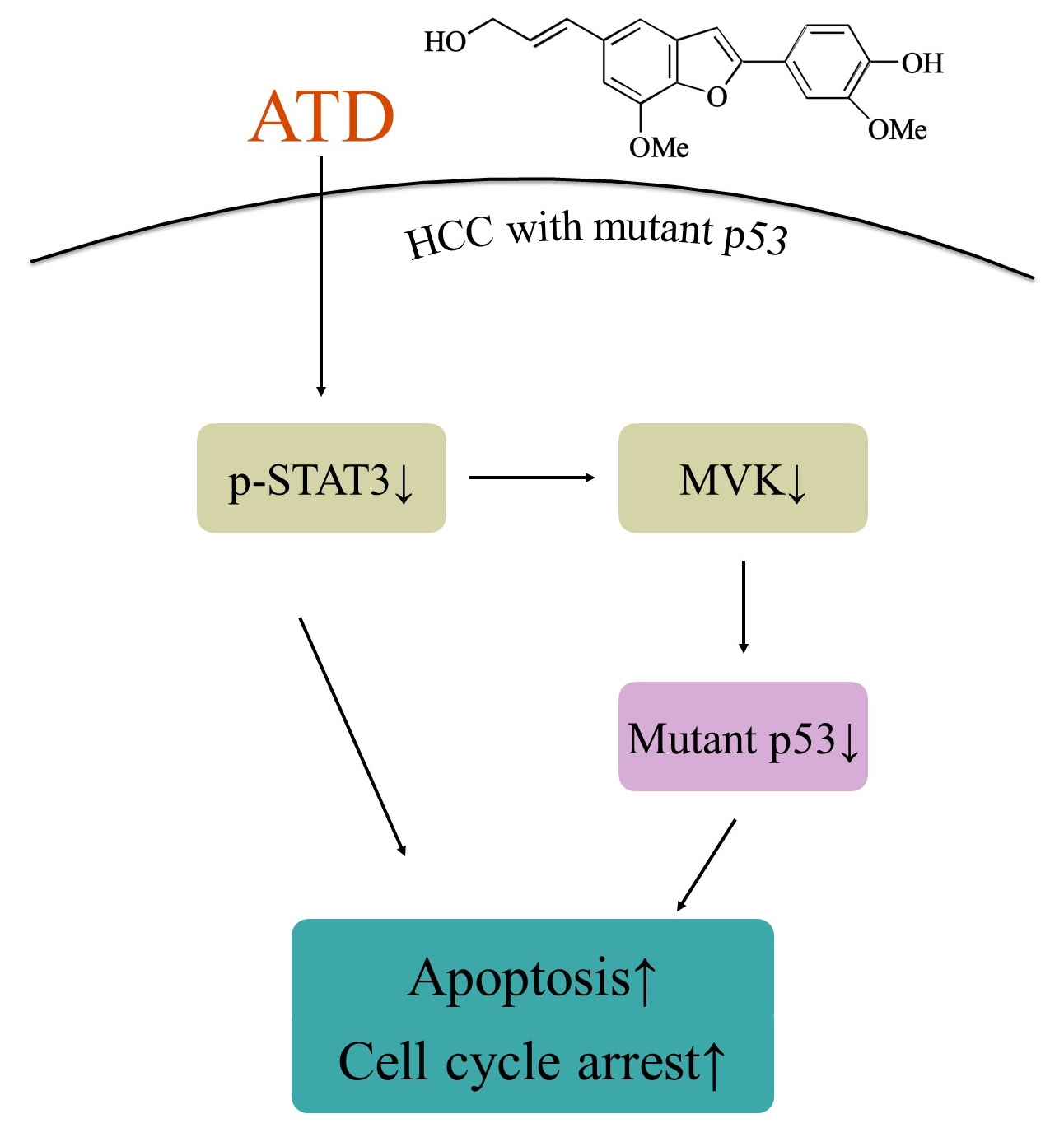
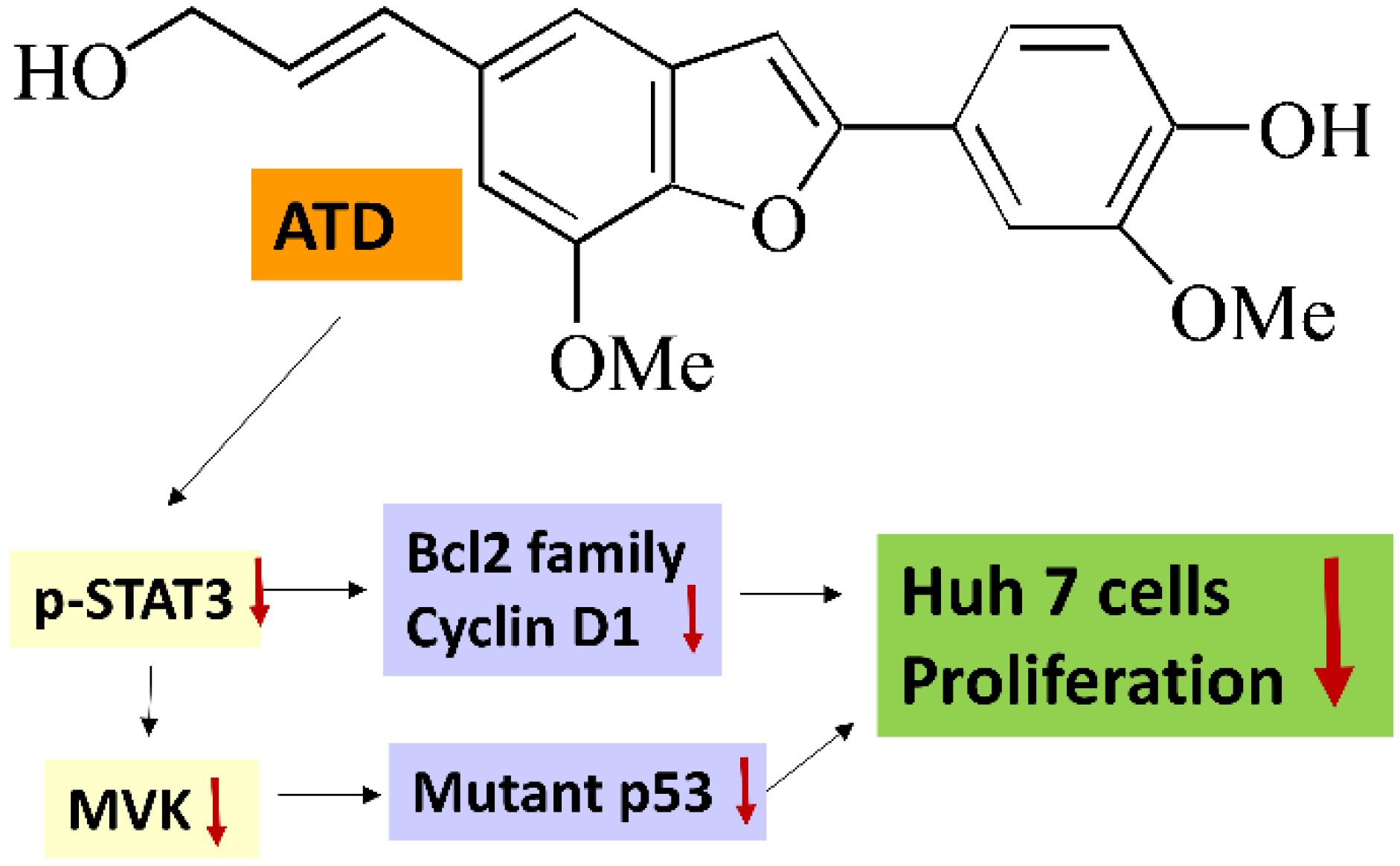
p53 levels in Huh7 cells. According to
Figure 7, we supposed that ATD blocked the STAT3 pathway mediating a reduction in mut
p53 protein in Huh7 cells. Although we found that ATD reduced the level of MVK (the downstream target gene product of STAT3, which might affect the stability of mut
p53), the real interplay between STAT3 and mut
p53 needs further elucidation. In addition to reducing the gain-of-function activity of mut
p53, ATD also triggered apoptosis by decreasing the expression of Bcl-xL and Bcl-2, which is associated with the inactivation of the STAT3 pathway. Additional studies are still needed to elucidate the action mechanisms of the ailanthoidol (ATD) as a chemopreventive and therapeutic agent in in vivo xenograft mouse models.
4. Materials and Methods
4.1. Materials
Dulbecco’s modified Eagle’s medium (DMEM), phosphate-buffered saline (PBS), fetal bovine serum (FBS), penicillin–streptomycin–neomycin (PSN), and trypsin–EDTA were purchased from Gibco Ltd. (Grand Island, NY, USA). Primary antibodies against p53(DO-1), p53(Pab-240), CDK2, Bax, Bcl-2, Bcl-xL pro-caspase 3/8, STAT3, MVK, GADPH, and actin were obtained from Santa Cruz Biotechnology (St. Louis, MO, USA). Anti-cyclin D1, anti-c-PARP, and anti-p-STAT3 (Tyr750) were obtained from Cell Signaling Technology (Beverly, MA, USA). Alexa 488-labeled goat anti-mouse IgG antibody was from Thermo Fisher Scientific, Waltham, MA, USA. ATD was provided by Dr. Lee and synthesized from 5-bromo-2-hydroxy-3-methoxybenzaldehyde, as previously reported [
23]. Tris base and all other materials were purchased from Sigma Chemical Co. (St. Louis, MO, USA).
4.2. Cells and Cell Culture
The human liver cancer cell line Huh7 (p53 Y220C) was obtained from the Food Industry Research and Development Institute (Hsinchu, Taiwan) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco BRL, Grand Island, NY, USA), supplemented with 10% FBS, 1% PSN, 1% essential amino acid, and 1mM glutamine. HepG2 (p53 WT) cells were cultured in DMEM supplemented with 10% FBS, 1% PSN, 1% essential amino acid, 1% sodium pyruvate, and 1mM glutamine. PLC/PRF/5 (R249S) cells were cultured in MEM supplemented with 10% FBS and 1% PSN. The cell cultures were maintained at 37 °C in a humidified atmosphere of 5% CO2.
4.3. Cell Viability Assay
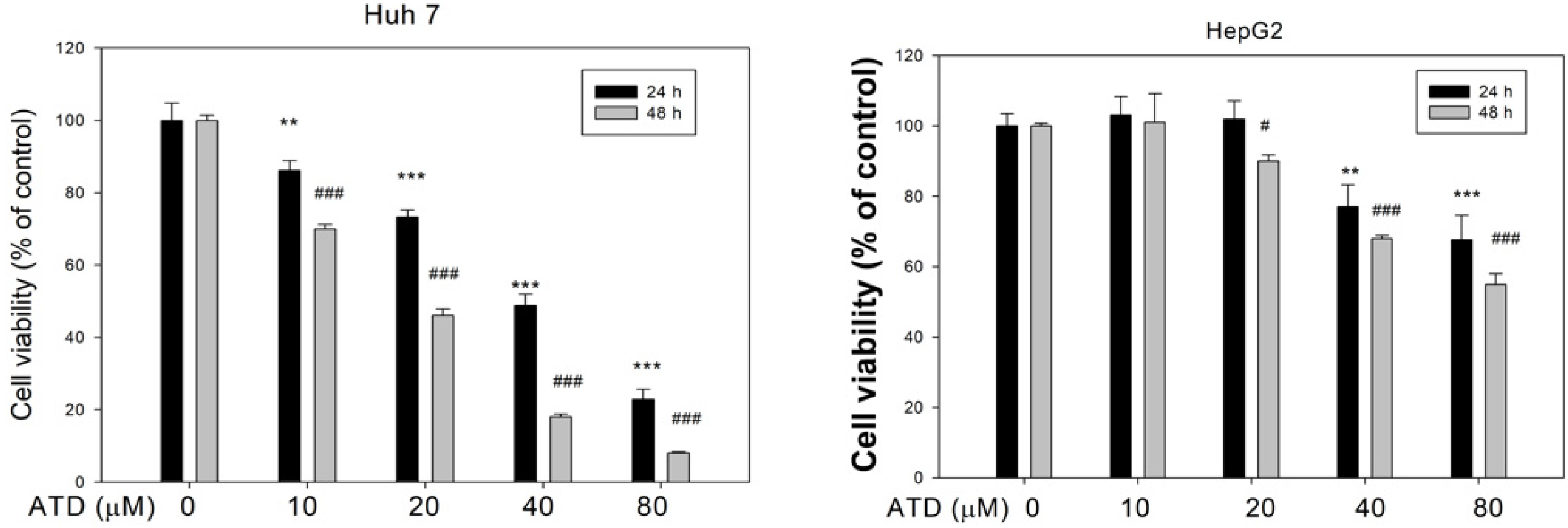
Huh7 and HepG2 cells were placed in a 24-well plate at a density of 2 × 104 cells/well, respectively, and treated with various concentrations of ATD (10–80 μM) or solvent control (0.2% DMSO) for 24 h and 48 h. Cell viability was determined in the presence of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) dye solution for 4 h. The medium was removed, and formazan was solubilized in isopropanol and measured spectrophotometrically at 560 nm using a microplate reader.
4.4. Trypan Blue Dye Exclusion Assay
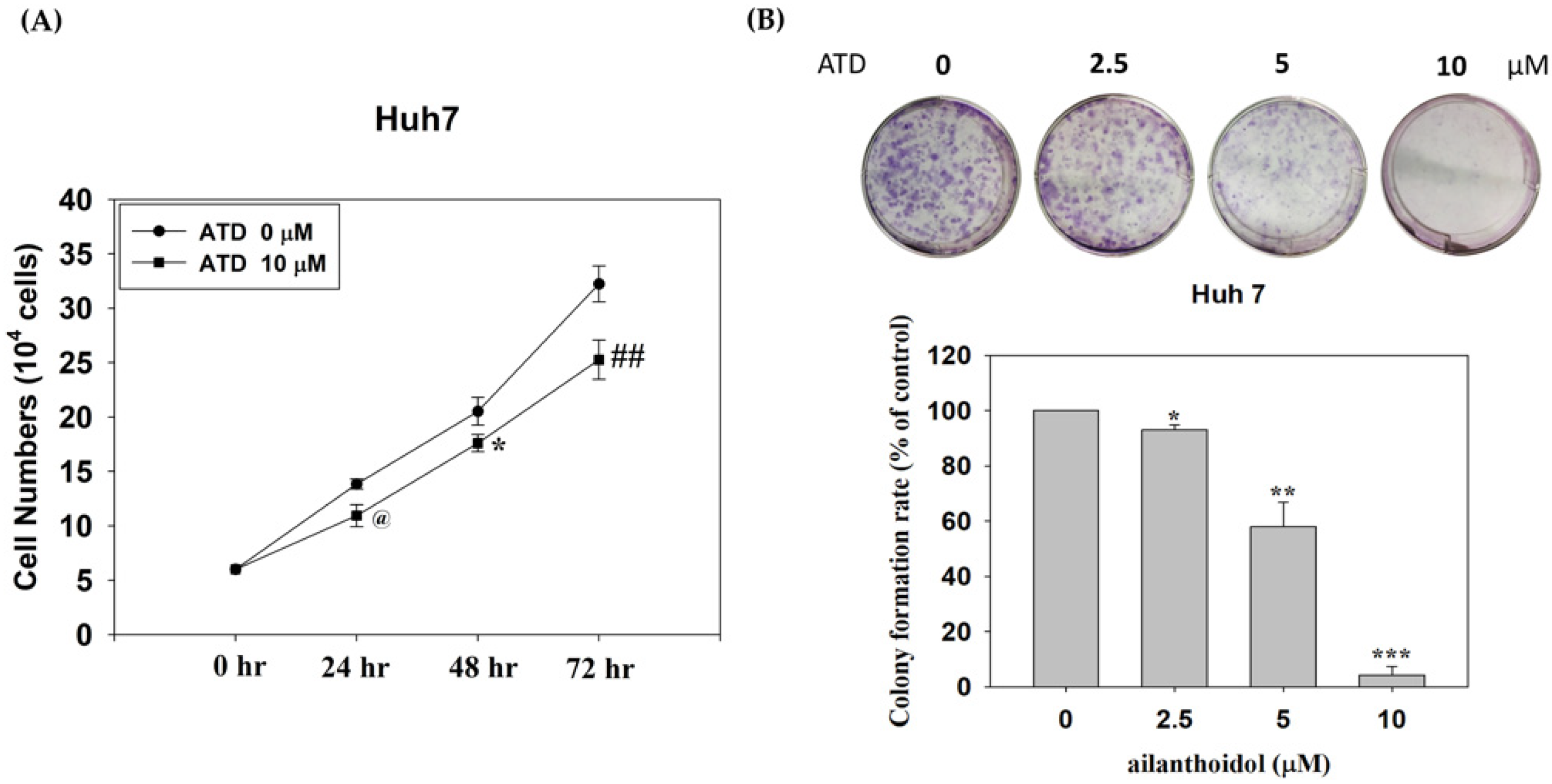
Huh7 cells were placed in a 10 cm dish at a density of 4 × 104 cells/dish and treated with ATD (10 μM) or solvent control (0.2% DMSO) for 24, 48, and 72 h. After treatment, trypan blue was added to the cell suspension, and viable cells that excluded the dye were counted on a hemacytometer.
4.5. Colony Formation Assay
Cells were plated in 6-well plates, at a density of 500 cells/well. On the next day, cells were treated with 0.2% DMSO (control) or ATD at the indicated concentration for 48 h, then cultured for 7 days. The colony was fixed with methanol for 15 min and stained with Giemsa. Cell colonies were photographed and counted.
4.6. Cell Cycle Analysis
Cell cycle distribution was determined using a flow cytometer with propidium iodide (PI) staining. Briefly, 6 × 105 cells/dish were treated with 0.2% dimethyl sulfoxide (DMSO; control) or 10 μM ATD for indicated time. Then, cells were harvested, fixed with cold 75% alcohol, and stained with 50 μg/mL PI solution in darkness for 30 min on ice. The distribution of cells in different cell cycle phases was determined using flow cytometry (FACSCalibur, BD Biosciences, San Jose, CA, USA). In total, 10,000 cells per sample were counted, and DNA histograms were analyzed using Cell Quest software (BD Biosciences, San Jose, CA, USA) to calculate the percentage of cells in each peak.
4.7. Annexin V/PI Double Staining Assay
For this assay, 6 × 105 cells were plated in a 10 cm culture dish. After attachment, cells were treated with DMSO or ATD at the indicated concentration for 48 h and then harvested and resuspended in PBS. Apoptotic cells were measured with a FITC-Annexin V Apoptosis Detection Kit (BD Biosciences, San Jose, CA, USA) according to the manufacturer’s protocol. Briefly, cells were stained with FITC annexin V and propidium iodide (PI) solution for 15 min at room temperature in darkness. In total, 10,000 cells were analyzed for each histogram. Flow cytometry demonstrated that the early apoptotic cells were in the lower-right quadrant, and the advanced apoptotic cells were in the upper-right quadrant. The apoptotic rate was the sum of the early and advanced apoptotic rates.
4.8. Western Immunoblotting
Equal amounts of protein from total cell lysates were separated in 8–12% polyacrylamide gel and transferred onto the PVDF membrane. The blot was subsequently incubated in blocking buffer (5% nonfat milk in PBS) for 1 h and then probed with a corresponding antibody against a specific protein overnight at 4 °C and washed with tris-buffered saline; the membrane was then incubated with an appropriate peroxidase-conjugated secondary antibody for 1 h. Finally, antigen–antibody complex was developed by ECL detection system. The relative image density was quantitated with densitometry.
4.9. Immunofluorescence
After ATD or DMSO treatment, Huh7, PLC/PRF/5, and HepG2 cells were washed with PBS and fixed with 4% paraformaldehyde for 10 min. The cells were permeated with 0.1% Triton X-100, then incubated at 4 °C overnight with a monoclonal anti-p53 (Pab-240) antibody, followed by a 1 h incubation with an Alexa 488-labeled goat anti-mouse IgG antibody (Thermo Fisher Scientific, Waltham, MA, USA). After washing with PBS containing 0.1% tween 20, the DAPI was added for 10 min. The cells were observed under a fluorescence microscope at 400× magnification.
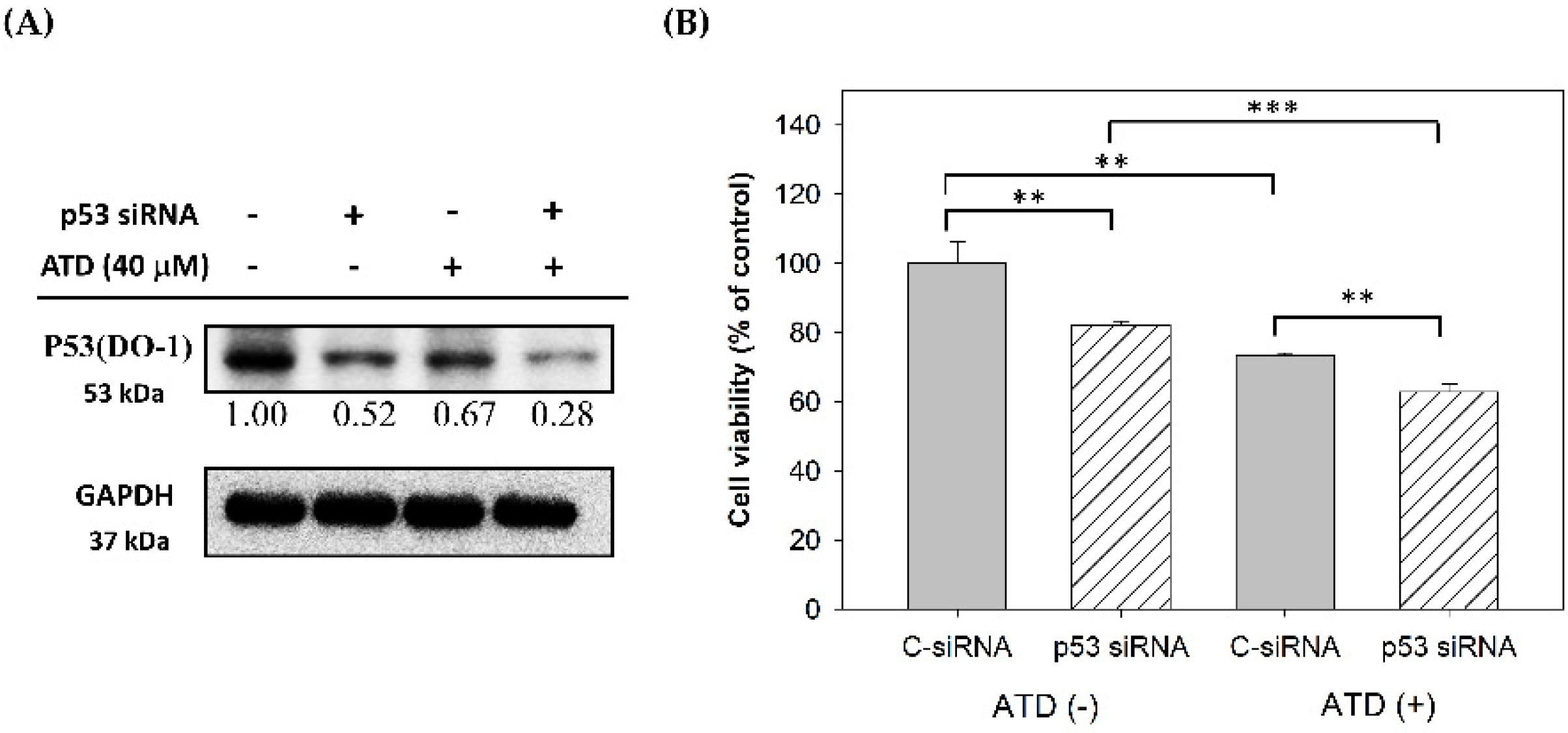
4.10. Transfection with Small Interfering RNA (siRNA)
p53 siRNAs (sense: 5′-AGA-CCU-AUG-GAA-ACU-ACU-Utt-3′) were purchased from GeneDireX, (QUANTUM BIOTECHNOLOGY, INC., Durham, NC, USA) [
24]. For transfection, 3 × 10
3 Huh7 cells were seeded on 96-well dishes or 4 × 10
5 on 10 cm dishes. After overnight incubation,
p53 siRNA or control siRNA (40 nM) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were transfected using a T-Pro NTR II transfection reagent, according to the manufacturer’s instructions. Following incubation for 48 h, the cells were treated with or without ATD for 24 h. After ATD treatment, viable-cell counting was performed using Cell Counting Kit-8 (CCK-8 kit), or the total cell lysate was prepared for immunoblotting analysis.
4.11. Cell Proliferation Assay
Following the transfection, cell proliferation was assayed using a CCK-8, according to the manufacturer’s protocols. Briefly, after transfection and ATD treatment, the CCK-8 solution was added and incubated for 3 h. The optical density was measured at 450 nm using a microplate reader.
4.12. Statistical Analysis
Data are expressed as means ± SD from three independent experiments. The statistical significance of differences throughout the study was analyzed by a one-way ANOVA test. A p value < 0.05 was considered to be statistically significant.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







