
Kisspeptin-10 Rescues Cholinergic Differentiated SHSY-5Y Cells from α-Synuclein-Induced Toxicity In Vitro

 ,
,  , and
, and 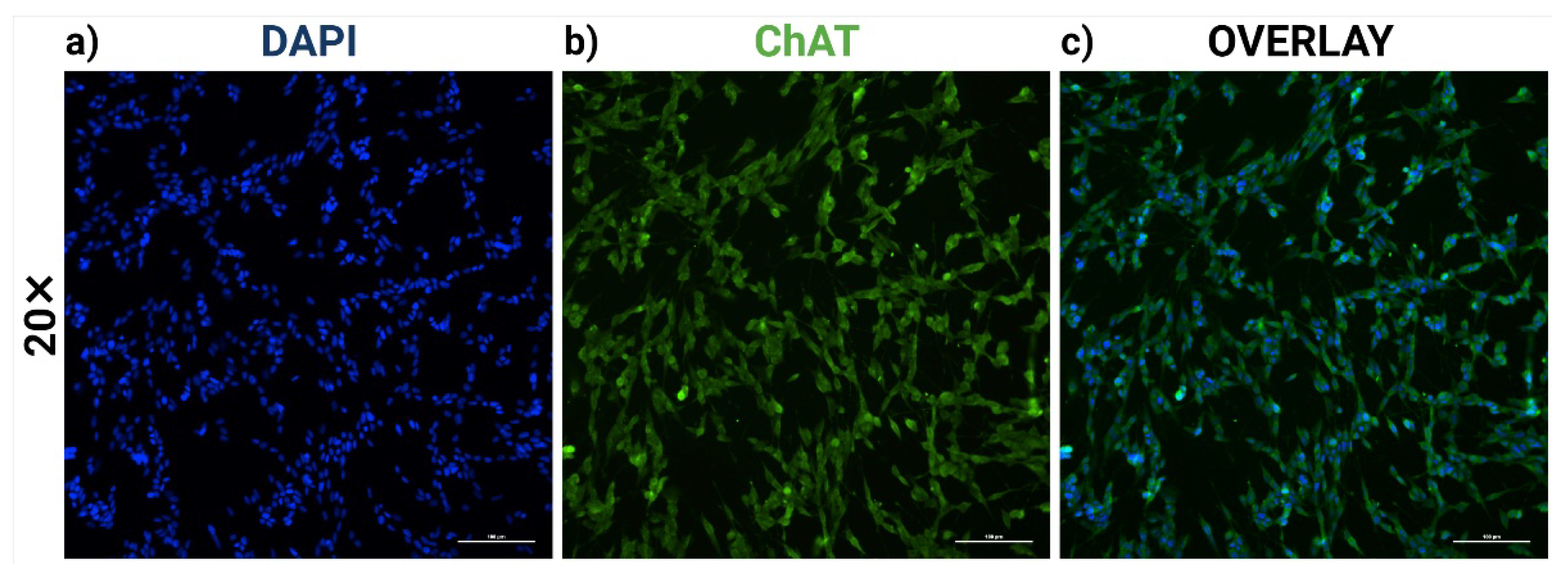{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. RA-Differentiated SH-SY5Y Cells Establish a Cholinergic-like Phenotype
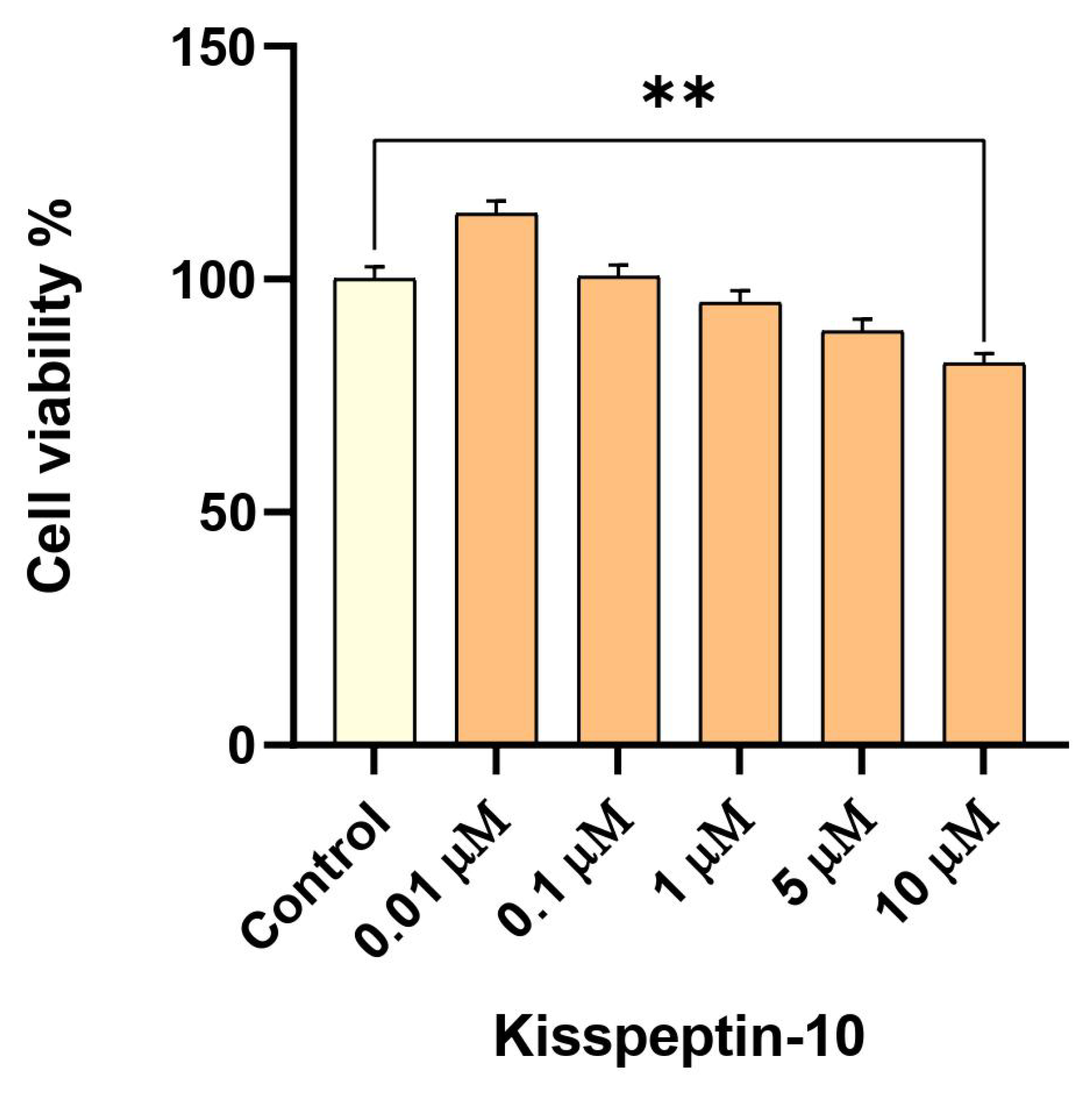
2.2. Low-Dose Exposure to KP-10 Has No Effect on RA-Differentiated Cholinergic Cell Viability
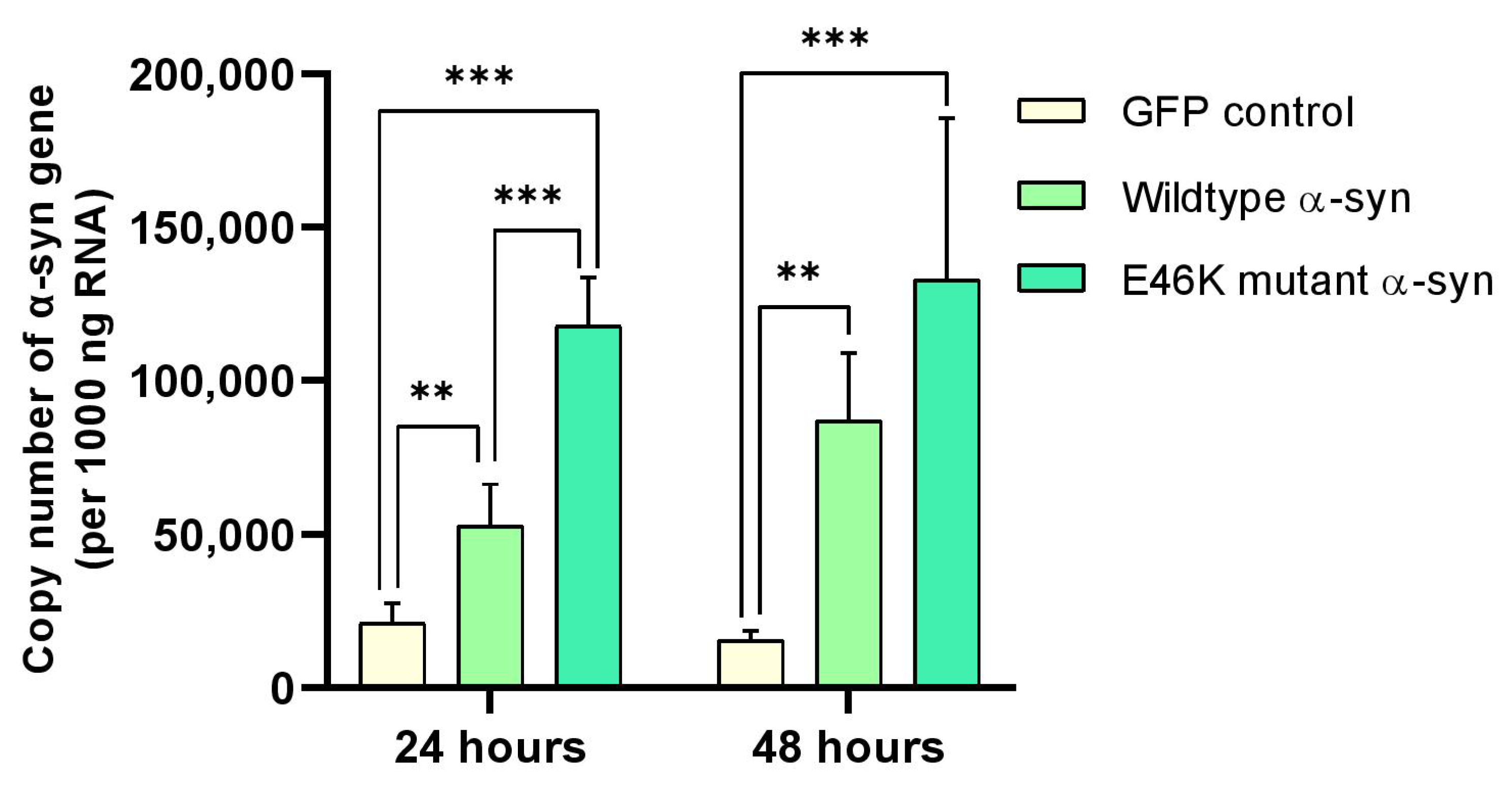
2.3. α-Syn mRNA Overexpression in RA-Differentiated Cholinergic Cells
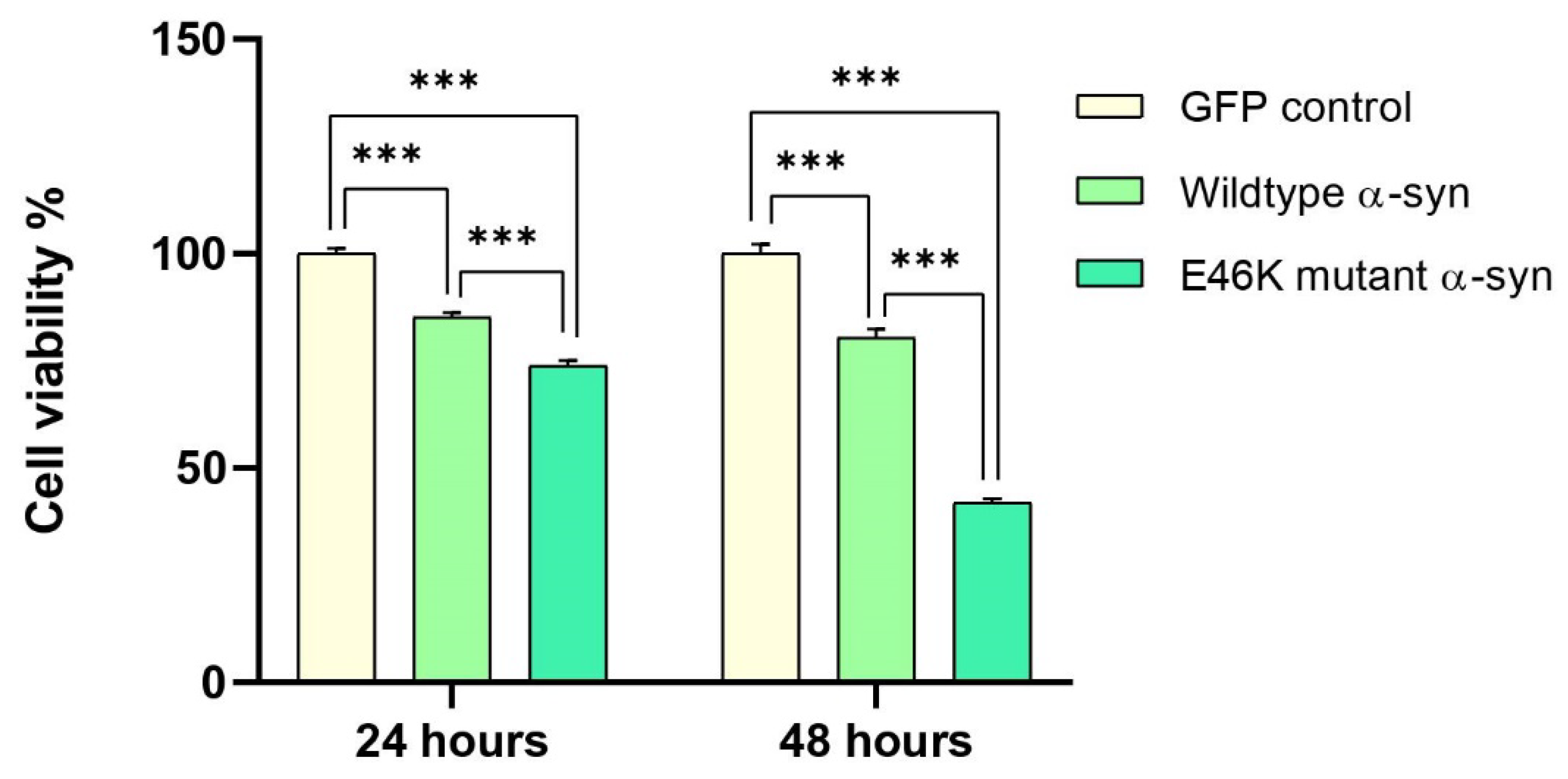
2.4. Elevated α-Syn mRNA Levels Exacerbate Cellular Toxicity in RA-Differentiated Cholinergic Cells
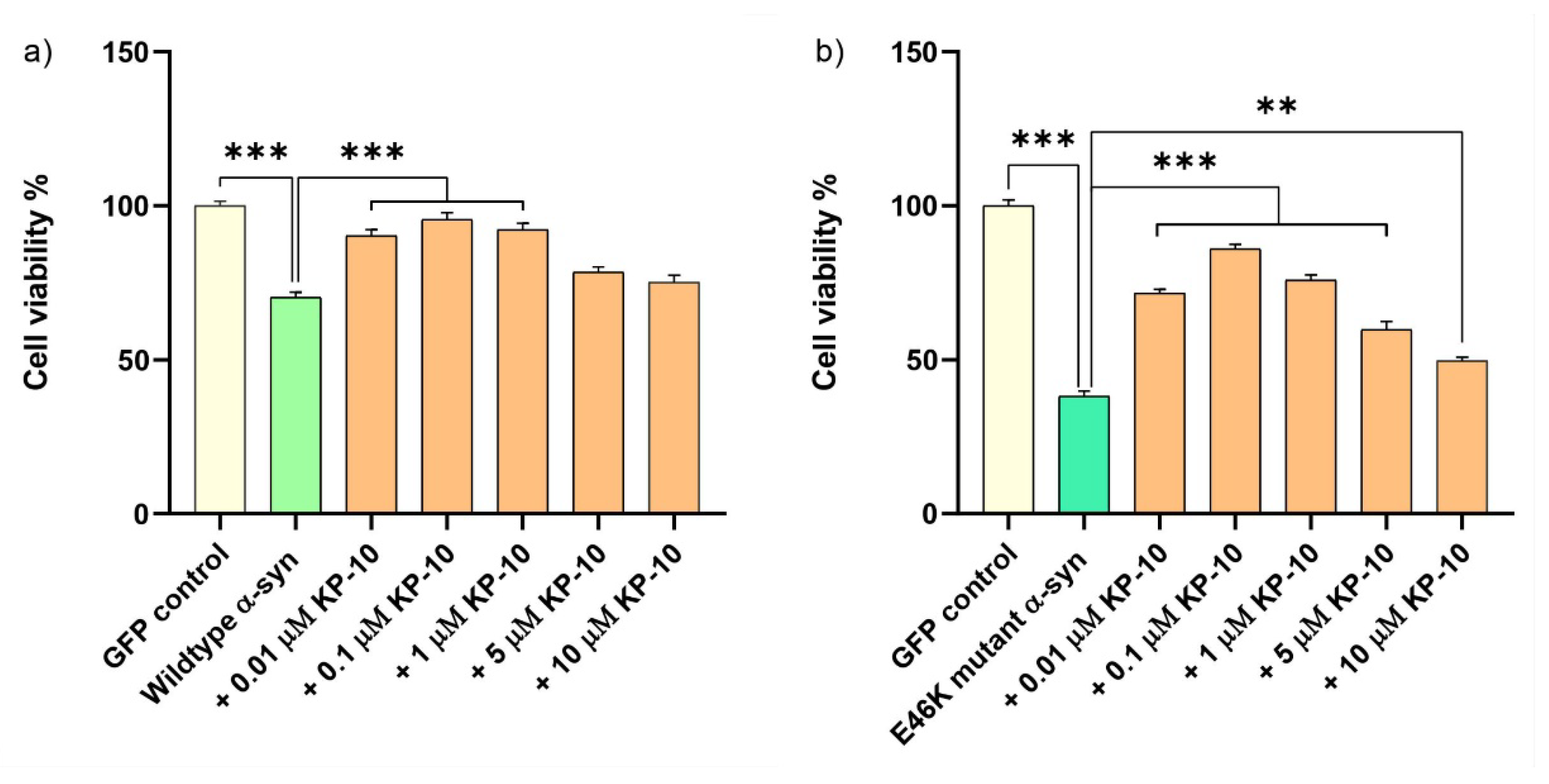
2.5. KP-10 Mitigates α-Syn-Induced Toxicity in RA-Differentiated Cholinergic Cells
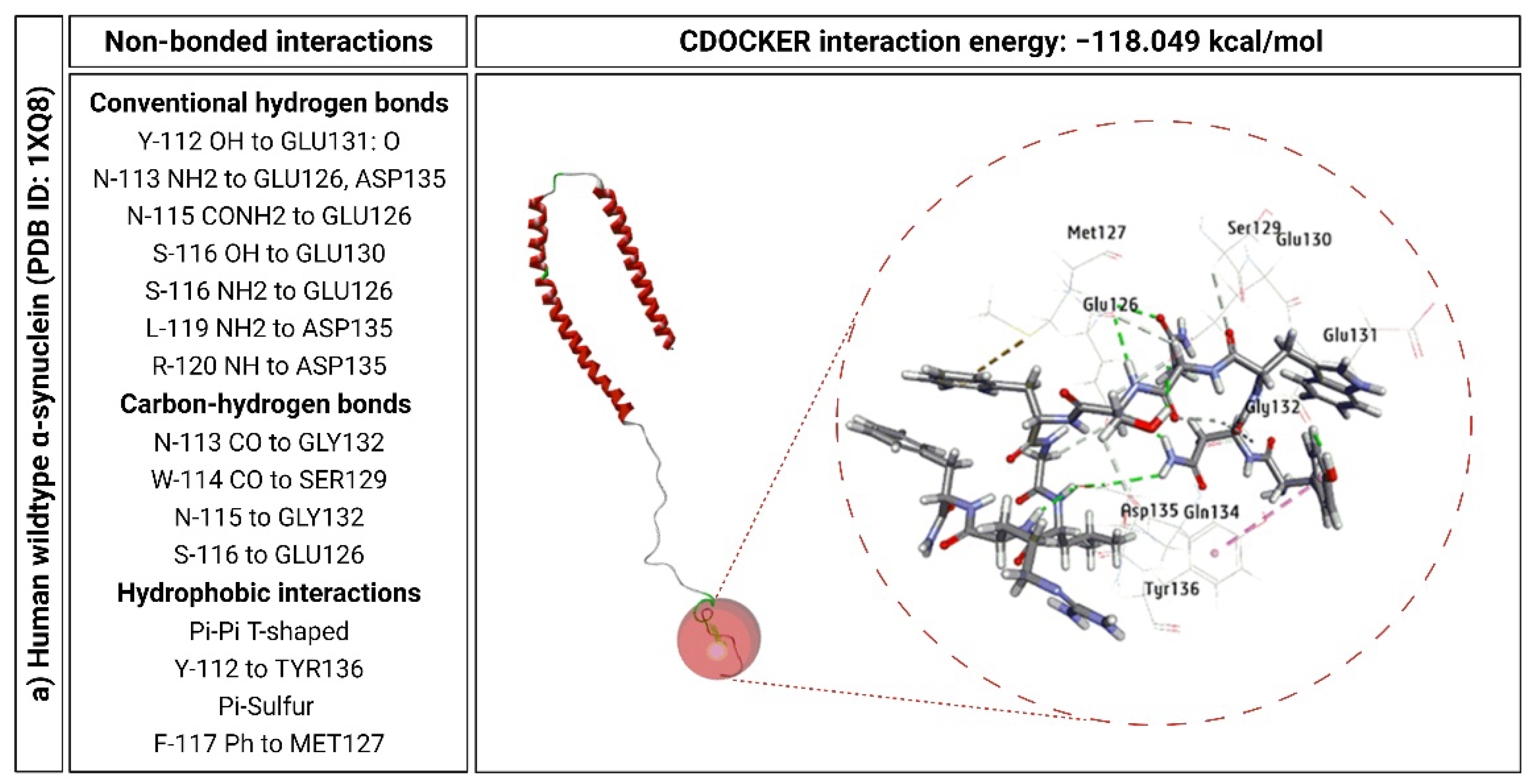
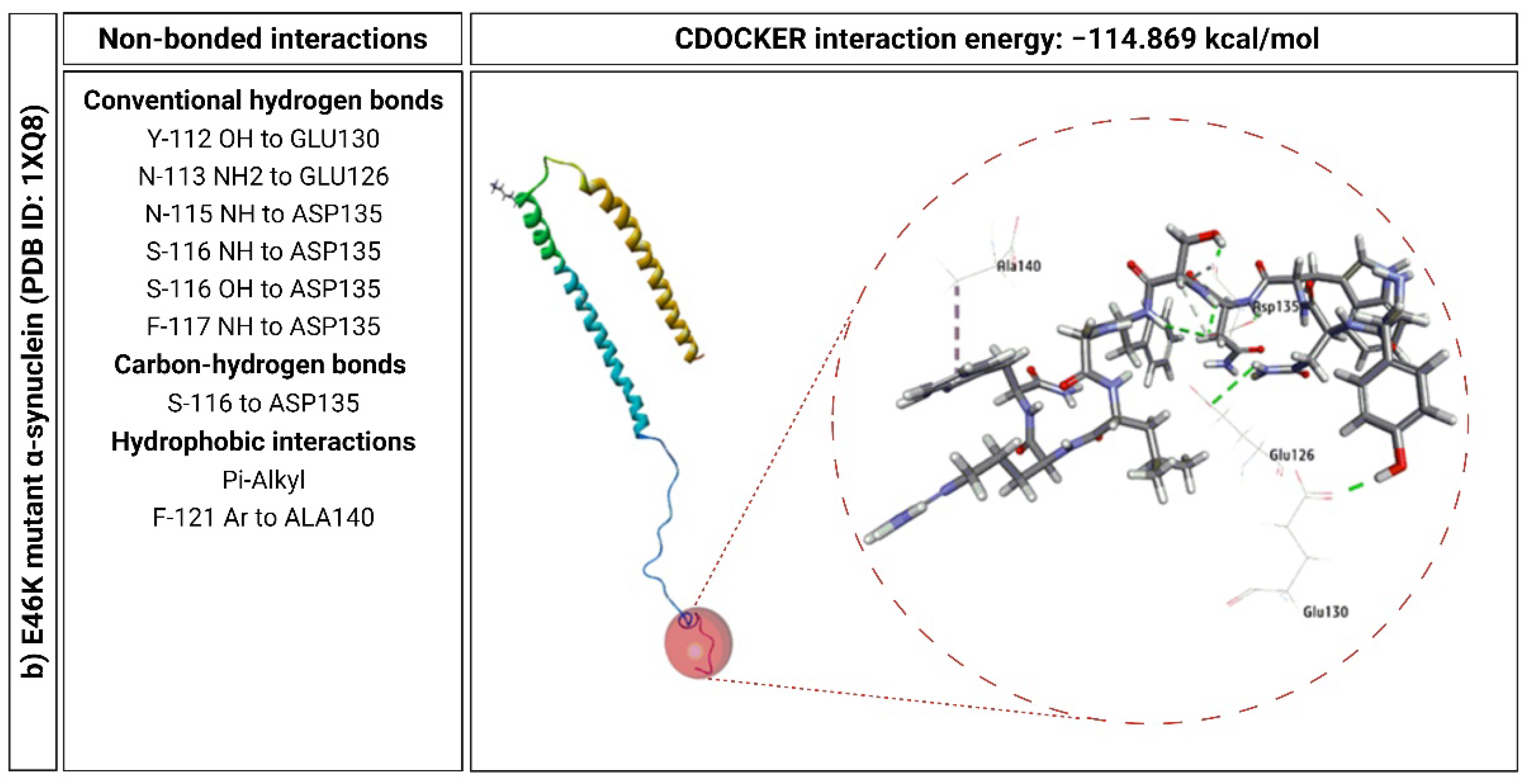
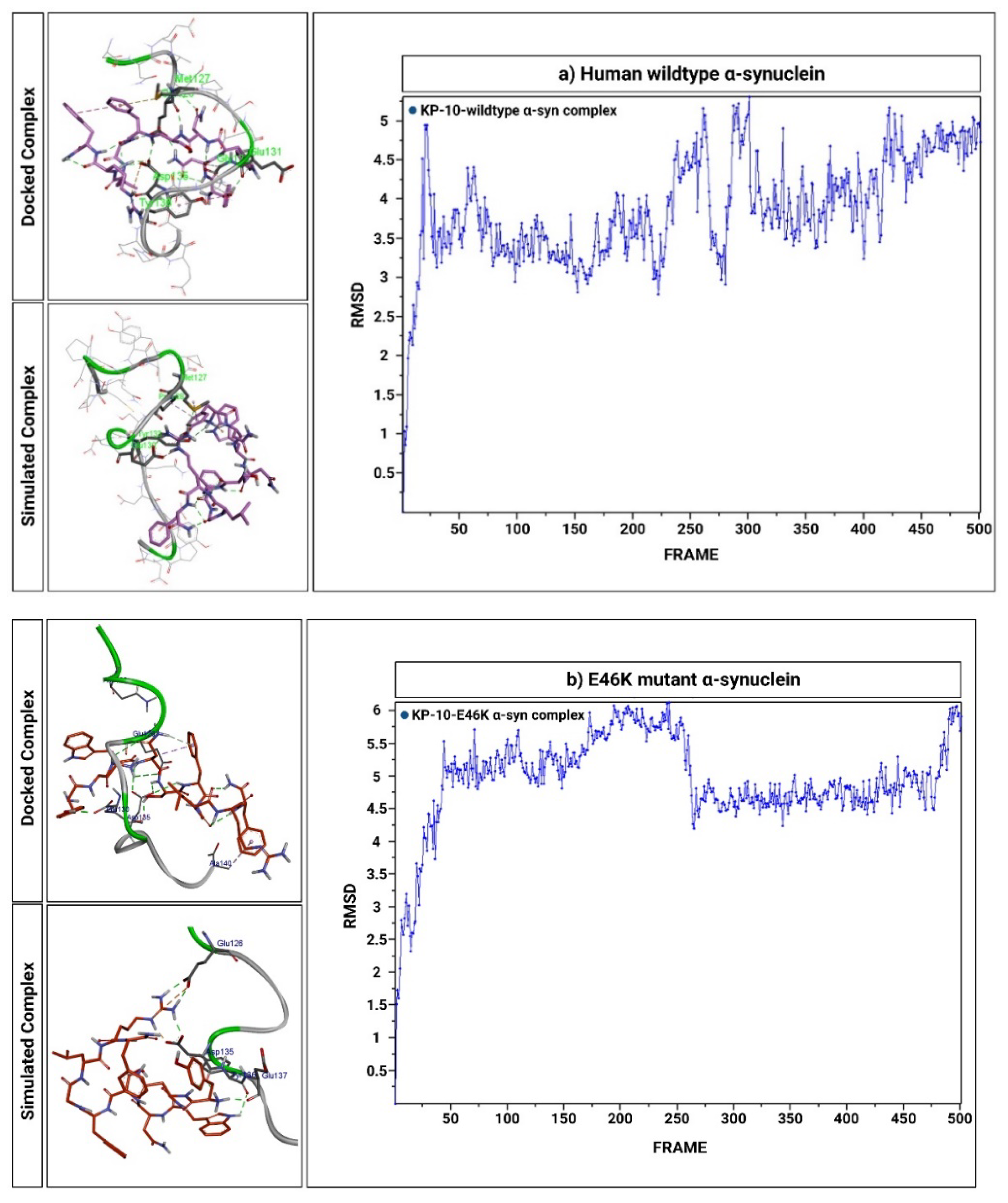
2.6. KP-10 Binds to the C-Terminal Residues of Wild-Type and E46K Mutant α-Syn In Silico
3. Discussion
4. Materials and Methods
4.1. KP-10
4.2. α-Syn Plasmids
4.3. Cell Culture
4.4. Differentiation of SHSY-5Y into Cholinergic Neurons
4.5. Plasmid Transfections
4.6. Immunofluorescent Staining
4.7. Cell Viability Assay
4.8. RNA Isolation and cDNA Synthesis
4.9. Quantification of α-Syn mRNA Using qPCR
4.10. In Silico Molecular Docking
4.11. Explicit-Solvent MD Simulations
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pepeu, G.; Giovannini, M.G. The fate of the brain cholinergic neurons in neurodegenerative diseases. Brain Res. 2017, 1670, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Tortosa, E.; Newell, K.; Irizarry, M.C.; Sanders, J.L.; Hyman, B.T. α-Synuclein immunoreactivity in dementia with Lewy bodies: Morphological staging and comparison with ubiquitin immunostaining. Acta Neuropathol. 2000, 99, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Soga, T.; Okano, H.J.; Parhar, I. α-Synuclein-mediated neurodegeneration in Dementia with Lewy bodies: The pathobiology of a paradox. Cell Biosci. 2021, 11, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of α-synuclein causes parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Colom-Cadena, M.; Gelpi, E.; Charif, S.; Belbin, O.; Blesa, R.; Martí, M.J.; Clarimón, J.; Lleó, A. Confluence of α-Synuclein, Tau, and β-Amyloid Pathologies in Dementia With Lewy Bodies. J. Neuropathol. Exp. Neurol. 2013, 72, 1203–1212. [Google Scholar] [CrossRef] [Green Version]
- Swirski, M.; Miners, J.S.; De Silva, R.; Lashley, T.; Ling, H.; Holton, J.; Revesz, T.; Love, S. Evaluating the relationship between amyloid-β and α-synuclein phosphorylated at Ser129 in dementia with Lewy bodies and Parkinson’s disease. Alzheimer’s Res. Ther. 2014, 6, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Weinreb, P.H.; Lansbury Jr, P.T. The core Alzheimer’s peptide NAC forms amyloid fibrils which seed and are seeded by β-amyloid: Is NAC a common trigger or target in neurodegenerative disease? Chem. Biol. 1995, 2, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Jensen, P.H.; Sørensen, E.S.; Petersen, T.E.; Gliemann, J.; Rasmussen, L.K. Residues in the synuclein consensus motif of the α-synuclein fragment, NAC, participate in transglutaminase-catalysed cross-linking to Alzheimer-disease amyloid βA4 peptide. Biochem. J. 1995, 310, 91–94. [Google Scholar] [CrossRef]
- Jensen, P.H.; Højrup, P.; Hager, H.; Nielsen, M.S.; Jacobsen, L.; Olesen, O.F.; Gliemann, J.; Jakes, R. Binding of Aβ to α- and β-synucleins: Identification of segments in α-synuclein/NAC precursor that bind Aβ and NAC. Biochem. J. 1997, 323, 539–546. [Google Scholar] [CrossRef]
- Lloyd, G.M.; Dhillon, J.-K.S.; Gorion, K.-M.M.; Riffe, C.; Fromholt, S.E.; Xia, Y.; Giasson, B.I.; Borchelt, D.R. Collusion of α-synuclein and Aβ aggravating co-morbidities in a novel prion-type mouse model. Mol. Neurodegener. 2021, 16, 1–17. [Google Scholar] [CrossRef]
- Stuendl, A.; Kunadt, M.; Kruse, N.; Bartels, C.; Moebius, W.; Danzer, K.M.; Mollenhauer, B.; Schneider, A. Induction of α-synuclein aggregate formation by CSF exosomes from patients with Parkinson’s disease and dementia with Lewy bodies. Brain 2016, 139, 481–494. [Google Scholar] [CrossRef] [Green Version]
- Ngolab, J.; Trinh, I.; Rockenstein, E.; Mante, M.; Florio, J.; Trejo, M.; Masliah, D.; Adame, A.; Masliah, E.; Rissman, R.A. Brain-derived exosomes from dementia with Lewy bodies propagate α-synuclein pathology. Acta Neuropathol. Commun. 2017, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ottolini, D.; Calì, T.; Szabo, I.; Brini, M. Alpha-synuclein at the intracellular and the extracellular side: Functional and dysfunctional implications. Biol. Chem. 2017, 398, 77–100. [Google Scholar] [CrossRef]
- Clinton, L.K.; Blurton-Jones, M.; Myczek, K.; Trojanowski, J.Q.; LaFerla, F.M. Synergistic Interactions between Aβ, Tau, and -Synuclein: Acceleration of Neuropathology and Cognitive Decline. J. Neurosci. 2010, 30, 7281–7289. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-J.; Bae, E.-J.; Lee, S.-J. Extracellular α-synuclein—a novel and crucial factor in Lewy body diseases. Nat. Rev. Neurol. 2014, 10, 92–98. [Google Scholar] [CrossRef]
- Bassil, F.; Brown, H.J.; Pattabhiraman, S.; Iwasyk, J.E.; Maghames, C.M.; Meymand, E.S.; Cox, T.O.; Riddle, D.M.; Zhang, B.; Trojanowski, J.Q.; et al. Amyloid-Beta (Aβ) Plaques Promote Seeding and Spreading of Alpha-Synuclein and Tau in a Mouse Model of Lewy Body Disorders with Aβ Pathology. Neuron 2020, 105, 260–275. [Google Scholar] [CrossRef]
- Mills, E.G.; O’Byrne, K.T.; Comninos, A.N. Kisspeptin as a Behavioral Hormone. Semin. Reprod. Med. 2019, 37, 056–063. [Google Scholar] [CrossRef]
- Rumpler, E.; Skrapits, K.; Takács, S.; Göcz, B.; Trinh, S.H.; Rácz, G.; Matolcsy, A.; Kozma, Z.; Ciofi, P.; Dhillo, W.S.; et al. Characterization of kisspeptin neurons in the human rostral hypothalamus. Neuroendocrinology 2021, 111, 249–262. [Google Scholar] [CrossRef]
- Ibos, K.E.; Bodnár, E.; Bagosi, Z.; Bozsó, Z.; Tóth, G.; Szabó, G.; Csabafi, K. Kisspeptin-8 Induces Anxiety-Like Behavior and Hypolocomotion by Activating the HPA Axis and Increasing GABA Release in the Nucleus Accumbens in Rats. Biomedicines 2021, 9, 112. [Google Scholar] [CrossRef]
- Tanaka, M.; Csabafi, K.; Telegdy, G. Neurotransmissions of antidepressant-like effects of kisspeptin-13. Regul. Pept. 2013, 180, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Milton, N.G.N.; Chilumuri, A.; Rocha-Ferreira, E.; Nercessian, A.N.; Ashioti, M. Kisspeptin Prevention of Amyloid-β Peptide Neurotoxicity in Vitro. ACS Chem. Neurosci. 2012, 3, 706–719. [Google Scholar] [CrossRef] [Green Version]
- Chilumuri, A.; Ashioti, M.; Nercessian, A.N.; Milton, N.G.N. Immunolocalization of Kisspeptin Associated with Amyloid-β Deposits in the Pons of an Alzheimer’s Disease Patient. J. Neurodegener. Dis. 2013, 2013, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messager, S.; Chatzidaki, E.E.; Ma, D.; Hendrick, A.G.; Zahn, D.; Dixon, J.; Thresher, R.R.; Malinge, I.; Lomet, D.; Carlton, M.B.L.; et al. Kisspeptin directly stimulates gonadotropin-releasing hormone release via G protein-coupled receptor 54. Proc. Natl. Acad. Sci. USA 2005, 102, 1761–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chilumuri, A.; Milton, N.G.N. The Role of Neurotransmitters in Protection against Amyloid-β Toxicity by KiSS-1 Overexpression in SH-SY5Y Neurons. ISRN Neurosci. 2013, 2013, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagliafierro, L.; Chiba-Falek, O. Up-regulation of SNCA gene expression: Implications to synucleinopathies. Neurogenetics 2016, 17, 145–157. [Google Scholar] [CrossRef] [Green Version]
- Neystat, M.; Lynch, T.; Przedborski, S.; Kholodilov, N.; Rzhetskaya, M.; Burke, R.E. α-Synuclein expression in substantia nigra and cortex in Parkinson’s disease. Mov. Disord. 1999, 14, 417–422. [Google Scholar] [CrossRef]
- Taylor, J.-P.; Collerton, D.; LeBeau, F.; Perry, E. Cholinergic Pathology in Dementia with Lewy Bodies. In Dementia with Lewy Bodies; Springer: Berlin/Heidelberg, Germany, 2017; pp. 23–39. [Google Scholar] [CrossRef]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The SH-SY5Y cell line in Parkinson’s disease research: A systematic review. Mol. Neurodegener. 2017, 12, 1–11. [Google Scholar] [CrossRef] [Green Version]
- de Medeiros, L.M.; De Bastiani, M.A.; Rico, E.P.; Schonhofen, P.; Pfaffenseller, B.; Wollenhaupt-Aguiar, B.; Grun, L.; Barbé-Tuana, F.; Zimmer, E.R.; Castro, M.A.A.; et al. Cholinergic Differentiation of Human Neuroblastoma SH-SY5Y Cell Line and Its Potential Use as an In vitro Model for Alzheimer’s Disease Studies. Mol. Neurobiol. 2019, 56, 7355–7367. [Google Scholar] [CrossRef]
- Korecka, J.A.; van Kesteren, R.; Blaas, E.; Spitzer, S.O.; Kamstra, J.; Smit, A.B.; Swaab, D.; Verhaagen, J.; Bossers, K. Phenotypic Characterization of Retinoic Acid Differentiated SH-SY5Y Cells by Transcriptional Profiling. PLoS ONE 2013, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Datki, Z.; Juhász, A.; Gálfi, M.; Soós, K.; Papp, R.; Zádori, D.; Penke, B. Method for measuring neurotoxicity of aggregating polypeptides with the MTT assay on differentiated neuroblastoma cells. Brain Res. Bull. 2003, 62, 223–229. [Google Scholar] [CrossRef]
- Delenclos, M.; Burgess, J.D.; Lamprokostopoulou, A.; Outeiro, T.F.; Vekrellis, K.; McLean, P.J. Cellular models of alpha-synuclein toxicity and aggregation. J. Neurochem. 2019, 150, 566–576. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Ulusoy, A.; Innamorato, N.G.; Sahin, G.; Rábano, A.; Kirik, D.; Cuadrado, A.I. α-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet. 2012, 21, 3173–3192. [Google Scholar] [CrossRef] [Green Version]
- Pandey, N.; Schmidt, R.E.; Galvin, J.E. The alpha-synuclein mutation E46K promotes aggregation in cultured cells. Exp. Neurol. 2006, 197, 515–520. [Google Scholar] [CrossRef]
- Tiraboschi, P.; Hansen, L.A.; Alford, M.; Sabbagh, M.N.; Schoos, B.; Masliah, E.; Thal, L.J.; Corey-Bloom, J. Cholinergic dysfunction in diseases with Lewy bodies. Neurology 2000, 54, 407–411. [Google Scholar] [CrossRef]
- Fujishiro, H.; Umegaki, H.; Isojima, D.; Akatsu, H.; Iguchi, A.; Kosaka, K. Depletion of cholinergic neurons in the nucleus of the medial septum and the vertical limb of the diagonal band in dementia with Lewy bodies. Acta Neuropathol. 2006, 111, 109–114. [Google Scholar] [CrossRef]
- Tozzi, A.; de Iure, A.; Bagetta, V.; Tantucci, M.; Durante, V.; Quiroga-Varela, A.; Costa, C.; Di Filippo, M.; Ghiglieri, V.; Latagliata, E.C.; et al. Alpha-Synuclein Produces Early Behavioral Alterations via Striatal Cholinergic Synaptic Dysfunction by Interacting With GluN2D N -Methyl-D-Aspartate Receptor Subunit. Biol. Psychiatry 2016, 79, 402–414. [Google Scholar] [CrossRef]
- Vekrellis, K.; Stefanis, L. Targeting intracellular and extracellular alpha-synuclein as a therapeutic strategy in Parkinson’s disease and other synucleinopathies. Expert Opin. Ther. Targets 2012, 16, 421–432. [Google Scholar] [CrossRef]
- Paleologou, K.; Irvine, G.; El-Agnaf, O. α-Synuclein aggregation in neurodegenerative diseases and its inhibition as a potential therapeutic strategy. Biochem. Soc. Trans. 2005, 33, 1106–1110. [Google Scholar] [CrossRef]
- Brundin, P.; Dave, K.D.; Kordower, J.H. Therapeutic approaches to target alpha-synuclein pathology. Exp. Neurol. 2017, 298, 225–235. [Google Scholar] [CrossRef]
- Stefanis, L.; Emmanouilidou, E.; Pantazopoulou, M.; Kirik, D.; Vekrellis, K.; Tofaris, G.K. How is alpha-synuclein cleared from the cell? J. Neurochem. 2019, 150, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Kakish, J.; Lee, D.; Lee, J.S. Drugs That Bind to α-Synuclein: Neuroprotective or Neurotoxic? ACS Chem. Neurosci. 2015, 6, 1930–1940. [Google Scholar] [CrossRef] [PubMed]
- Allison, J.R.; Rivers, R.C.; Christodoulou, J.C.; Vendruscolo, M.; Dobson, C.M. A Relationship between the Transient Structure in the Monomeric State and the Aggregation Propensities of α-Synuclein and β-Synuclein. Biochemistry 2014, 53, 7170–7183. [Google Scholar] [CrossRef] [PubMed]
- Periquet, M.; Fulga, T.; Myllykangas, L.; Schlossmacher, M.; Feany, M.B. Aggregated -Synuclein Mediates Dopaminergic Neurotoxicity In Vivo. J. Neurosci. 2007, 27, 3338–3346. [Google Scholar] [CrossRef]
- Wu, K.-P.; Baum, J. Detection of Transient Interchain Interactions in the Intrinsically Disordered Protein α-Synuclein by NMR Paramagnetic Relaxation Enhancement. J. Am. Chem. Soc. 2010, 132, 5546–5547. [Google Scholar] [CrossRef] [Green Version]
- El-Agnaf, O.M.; Jakes, R.; Curran, M.D.; Middleton, D.; Ingenito, R.; Bianchi, E.; Pessi, A.; Neill, D.; Wallace, A. Aggregates from mutant and wild-type α-synuclein proteins and NAC peptide induce apoptotic cell death in human neuroblastoma cells by formation of β-sheet and amyloid-like filaments. FEBS Lett. 1998, 440, 71–75. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, T.; Raj, R.; Dubey, A.; Kumar, D.; Chaturvedi, R.K.; Sharma, S.K.; Priya, S. Fast kinetics of environmentally induced α-synuclein aggregation mediated by structural alteration in NAC region and result in structure dependent cytotoxicity. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566. [Google Scholar] [CrossRef]
- Ruzza, P.; Siligardi, G.; Hussain, R.; Marchiani, A.; Islami, M.; Bubacco, L.; Delogu, G.; Fabbri, D.; Dettori, M.A.; Sechi, M.; et al. Ceftriaxone Blocks the Polymerization of α-Synuclein and Exerts Neuroprotective Effects in Vitro. ACS Chem. Neurosci. 2014, 5, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Jia, C.; Ma, X.; Liu, Z.; Gu, J.; Zhang, X.; Li, D.; Zhang, S. Different Heat Shock Proteins Bind α-Synuclein With Distinct Mechanisms and Synergistically Prevent Its Amyloid Aggregation. Front. Neurosci. 2019, 13, 1124. [Google Scholar] [CrossRef] [Green Version]
- Cox, D.; Whiten, D.R.; Brown, J.W.P.; Horrocks, M.H.; Gil, R.S.; Dobson, C.M.; Klenerman, D.; van Oijen, A.M.; Ecroyd, H. The small heat shock protein Hsp27 binds α-synuclein fibrils, preventing elongation and cytotoxicity. J. Biol. Chem. 2018, 293, 4486–4497. [Google Scholar] [CrossRef] [Green Version]
- Tavassoly, O.; Kakish, J.; Nokhrin, S.; Dmitriev, O.; Lee, J.S. The use of nanopore analysis for discovering drugs which bind to α-synuclein for treatment of Parkinson’s disease. Eur. J. Med. Chem. 2014, 88, 42–54. [Google Scholar] [CrossRef]
- Ahmad, B.; Lapidus, L.J. Curcumin Prevents Aggregation in α-Synuclein by Increasing Reconfiguration Rate. J. Biol. Chem. 2012, 287, 9193–9199. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Rajamani, S.; Kaylor, J.; Han, S.; Zhou, F.; Fink, A.L. The Flavonoid Baicalein Inhibits Fibrillation of α-Synuclein and Disaggregates Existing Fibrils. J. Biol. Chem. 2004, 279, 26846–26857. [Google Scholar] [CrossRef] [Green Version]
- Lorenzen, N.; Nielsen, S.B.; Yoshimura, Y.; Vad, B.S.; Andersen, C.B.; Betzer, C.; Kaspersen, J.D.; Christiansen, G.; Pedersen, J.S.; Jensen, P.H.; et al. How Epigallocatechin Gallate Can Inhibit α-Synuclein Oligomer Toxicity in Vitro. J. Biol. Chem. 2014, 289, 21299–21310. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Porat-Shliom, Y.; Pei, Z.; Cheng, Y.; Xiang, L.; Sommers, K.; Li, Q.; Gillardon, F.; Hengerer, B.; Berlinicke, C.; et al. Baicalein reduces E46K α-synuclein aggregation in vitro and protects cells against E46K α-synuclein toxicity in cell models of familiar Parkinsonism. J. Neurochem. 2010, 114, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Mbefo, M.K.; Fares, M.-B.; Paleologou, K.; Oueslati, A.; Yin, G.; Tenreiro, S.; Pinto, M.; Outeiro, T.; Zweckstetter, M.; Masliah, E.; et al. Parkinson Disease Mutant E46K Enhances α-Synuclein Phosphorylation in Mammalian Cell Lines, in Yeast, and in Vivo. J. Biol. Chem. 2015, 290, 9412–9427. [Google Scholar] [CrossRef] [Green Version]
- Tiong, K.H.; Yunus, N.A.M.; Yiap, B.C.; Tan, E.L.; Ismail, R.; Ong, C.E. Inhibitory Potency of 8-Methoxypsoralen on Cytochrome P450 2A6 (CYP2A6) Allelic Variants CYP2A6*15, CYP2A6*16, CYP2A6*21 and CYP2A6*22: Differential Susceptibility Due to Different Sequence Locations of the Mutations. PLoS ONE 2014, 9, 1–8. [Google Scholar] [CrossRef]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA-a self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simon, C.; Soga, T.; Ahemad, N.; Bhuvanendran, S.; Parhar, I. Kisspeptin-10 Rescues Cholinergic Differentiated SHSY-5Y Cells from α-Synuclein-Induced Toxicity In Vitro. Int. J. Mol. Sci. 2022, 23, 5193. https://doi.org/10.3390/ijms23095193
Simon C, Soga T, Ahemad N, Bhuvanendran S, Parhar I. Kisspeptin-10 Rescues Cholinergic Differentiated SHSY-5Y Cells from α-Synuclein-Induced Toxicity In Vitro. International Journal of Molecular Sciences. 2022; 23(9):5193. https://doi.org/10.3390/ijms23095193
Chicago/Turabian StyleSimon, Christopher, Tomoko Soga, Nafees Ahemad, Saatheeyavaane Bhuvanendran, and Ishwar Parhar. 2022. "Kisspeptin-10 Rescues Cholinergic Differentiated SHSY-5Y Cells from α-Synuclein-Induced Toxicity In Vitro" International Journal of Molecular Sciences 23, no. 9: 5193. https://doi.org/10.3390/ijms23095193
APA StyleSimon, C., Soga, T., Ahemad, N., Bhuvanendran, S., & Parhar, I. (2022). Kisspeptin-10 Rescues Cholinergic Differentiated SHSY-5Y Cells from α-Synuclein-Induced Toxicity In Vitro. International Journal of Molecular Sciences, 23(9), 5193. https://doi.org/10.3390/ijms23095193