
The IL-1 Family and Its Role in Atherosclerosis

 , , and
, , and
Abstract
:1. Introduction
2. Systemic Inflammation and Cardiovascular Risk
3. Athero-Inflammation, Cholesterol, and the NLRP3 Inflammasome
4. The IL-1 Family
4.1. History
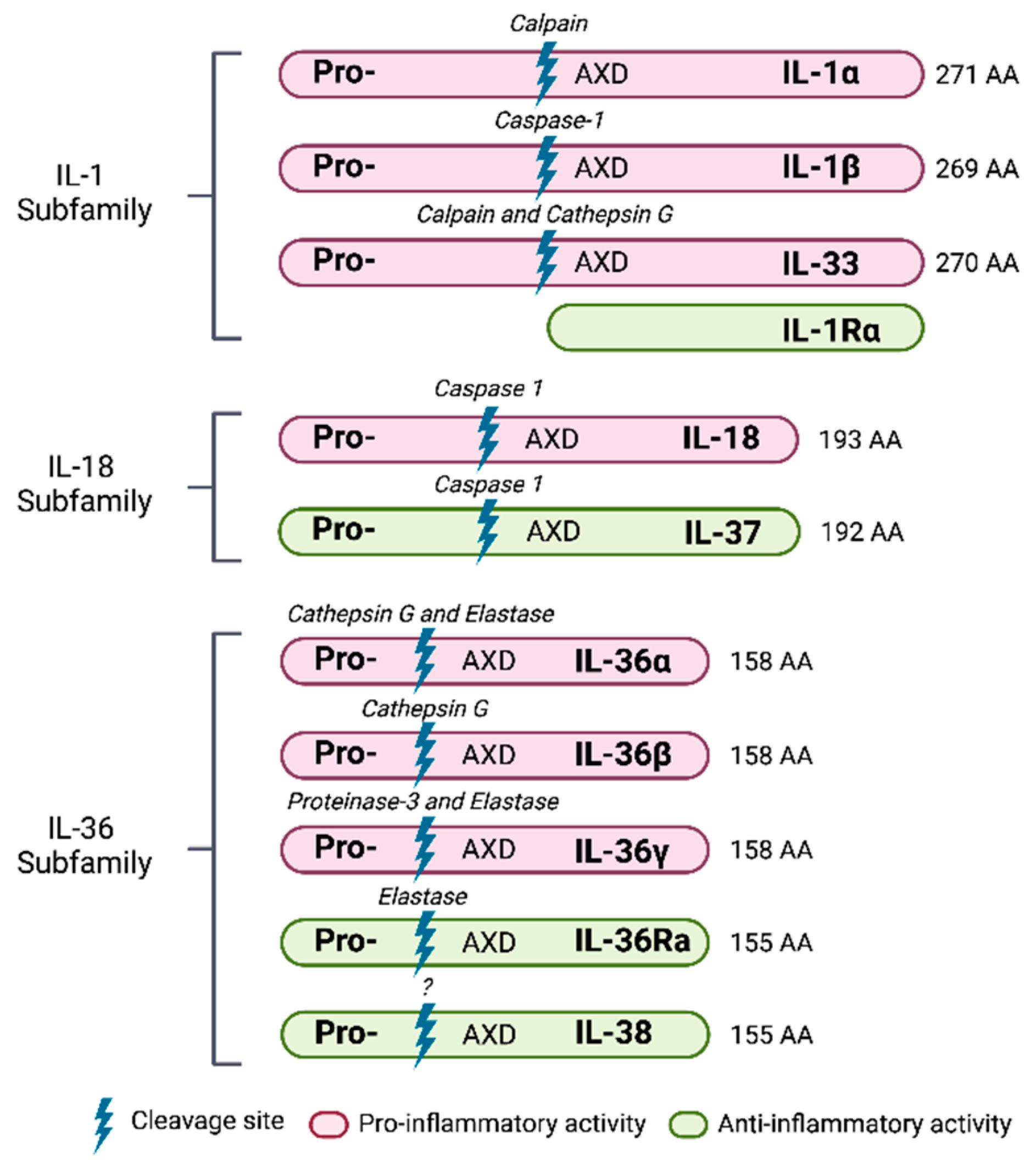
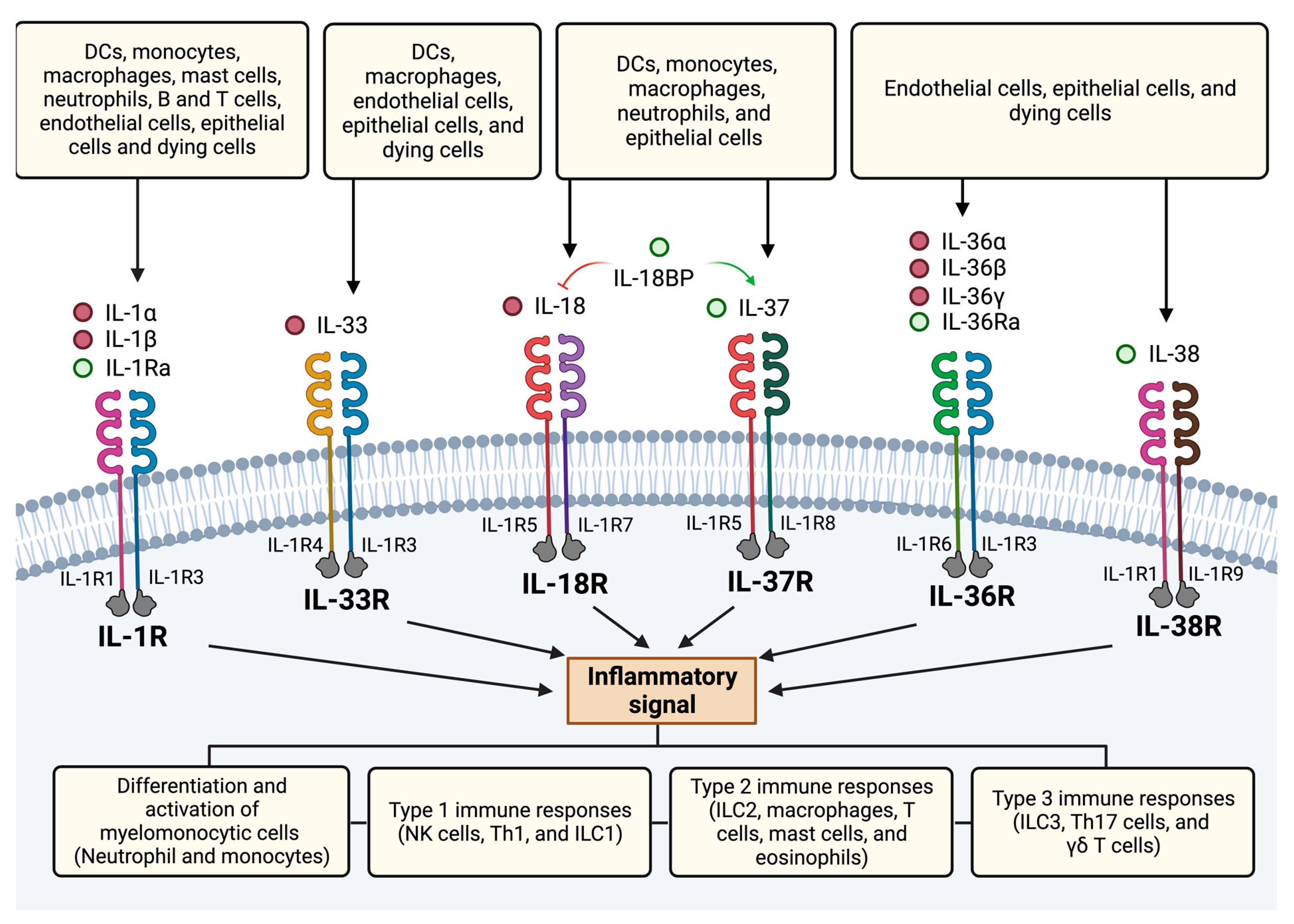
4.2. Members of the IL-1 Family
4.2.1. Cytokines
4.2.2. Receptors
4.3. Role in Disease
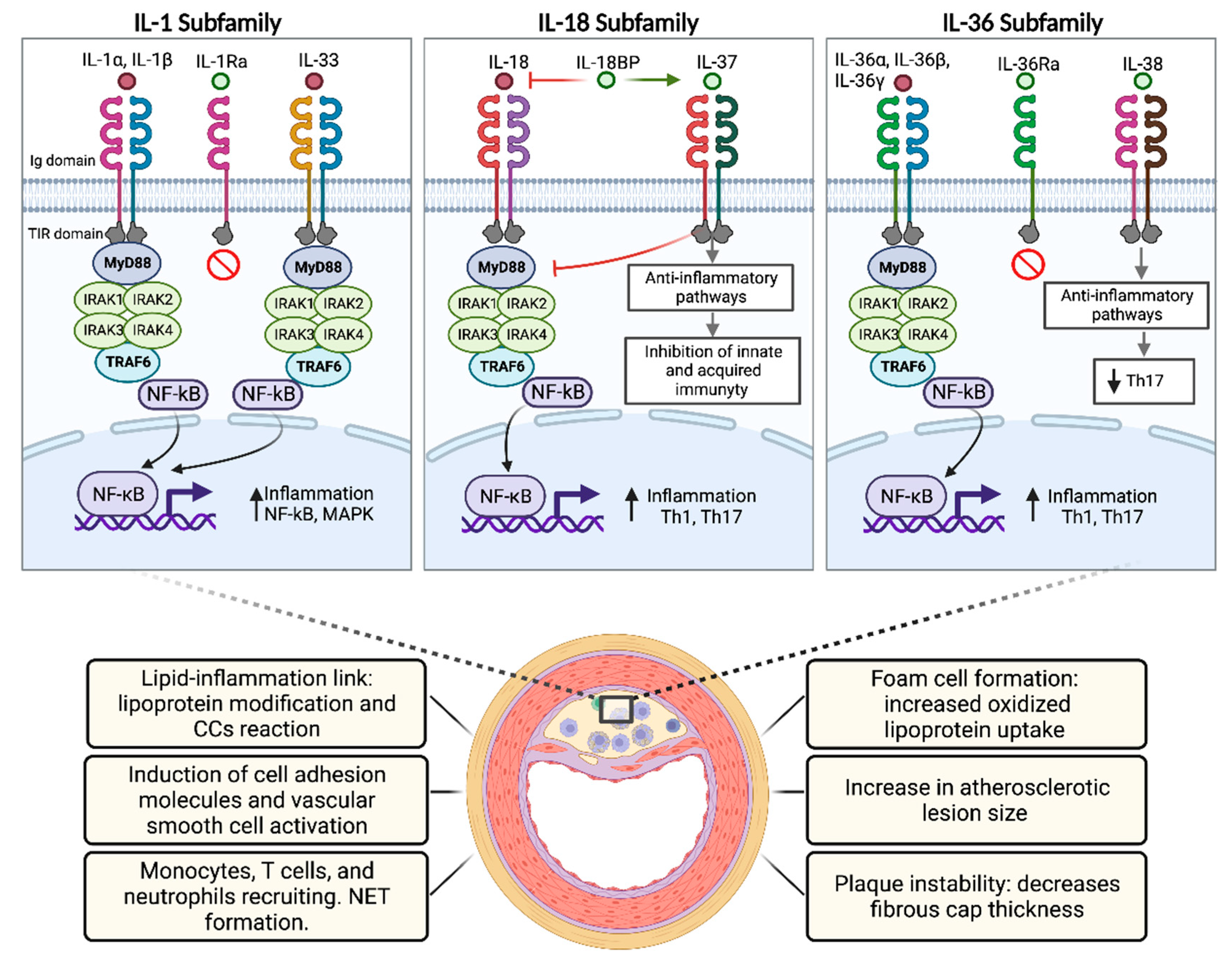
5. IL-1 Subfamily
5.1. IL-1 Alpha
5.2. IL-1 Beta
5.3. IL-33
5.4. IL-1 Receptor Antagonist
5.5. Role of the IL-1 Family in Atherosclerosis
6. IL-18 Subfamily
6.1. IL-18
6.2. IL-37
6.3. Role of the IL-18 Family in Atherosclerosis
7. IL-36 Subfamily
7.1. IL-36 α, β, γ
7.2. IL-36Ra
7.3. IL-38
7.4. Role of the IL-36 Subfamily in Atherosclerosis
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dai, H.; Much, A.A.; Maor, E.; Asher, E.; Younis, A.; Xu, Y.; Lu, Y.; Liu, X.; Shu, J.; Bragazzi, N.L. Global, regional, and national burden of ischaemic heart disease and its attributable risk factors, 1990–2017: Results from the Global Burden of Disease Study 2017. Eur. Heart J. Qual. Care Clin. Outcomes 2022, 8, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Rayner, K.J. Cell Death in the Vessel Wall: The Good, the Bad, the Ugly. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e75–e81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef]
- Doring, Y.; Drechsler, M.; Soehnlein, O.; Weber, C. Neutrophils in atherosclerosis: From mice to man. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 288–295. [Google Scholar] [CrossRef] [Green Version]
- Andersson, C.; Johnson, A.D.; Benjamin, E.J.; Levy, D.; Vasan, R.S. 70-year legacy of the Framingham Heart Study. Nat. Rev. Cardiol. 2019, 16, 687–698. [Google Scholar] [CrossRef]
- Hoogeveen, R.C.; Ballantyne, C.M. Residual Cardiovascular Risk at Low LDL: Remnants, Lipoprotein(a), and Inflammation. Clin. Chem. 2021, 67, 143–153. [Google Scholar] [CrossRef]
- Allard-Ratick, M.P.; Kindya, B.R.; Khambhati, J.; Engels, M.C.; Sandesara, P.B.; Rosenson, R.S.; Sperling, L.S. HDL: Fact, fiction, or function? HDL cholesterol and cardiovascular risk. Eur. J. Prev. Cardiol. 2021, 28, 166–173. [Google Scholar] [CrossRef]
- Farnier, M.; Zeller, M.; Masson, D.; Cottin, Y. Triglycerides and risk of atherosclerotic cardiovascular disease: An update. Arch. Cardiovasc. Dis. 2021, 114, 132–139. [Google Scholar] [CrossRef]
- Martinez, G.; Rigotti, A.; Acevedo, M.; Navarrete, C.; Rosales, J.; Giugliano, R.P.; Corbalan, R. Cholesterol levels and the association of statins with in-hospital mortality of myocardial infarction patients insights from a Chilean registry of myocardial infarction. Clin. Cardiol. 2013, 36, 305–311. [Google Scholar] [CrossRef]
- Huynh, T.; Montigny, M.; Iftikhar, U.; Gagnon, R.; Eisenberg, M.; Lauzon, C.; Mansour, S.; Rinfret, S.; Afilalo, M.; Nguyen, M.; et al. Recurrent Cardiovascular Events in Survivors of Myocardial Infarction With ST-Segment Elevation (from the AMI-QUEBEC Study). Am. J. Cardiol. 2018, 121, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.S.; Capers, Q.t.; Savage, M.; Gulati, M.; Potts, J.; Mohamed, M.O.; Nagaraja, V.; Patwala, A.; Heatlie, G.; Kontopantelis, E.; et al. Unplanned hospital readmissions after acute myocardial infarction: A nationwide analysis of rates, trends, predictors and causes in the United States between 2010 and 2014. Coron. Artery Dis. 2020, 31, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Blake, G.J.; Ridker, P.M. C-reactive protein and other inflammatory risk markers in acute coronary syndromes. J. Am. Coll. Cardiol. 2003, 41, 37S–42S. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Cushman, M.; Stampfer, M.J.; Tracy, R.P.; Hennekens, C.H. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N. Engl. J. Med. 1997, 336, 973–979. [Google Scholar] [CrossRef]
- Ridker, P.M.; Buring, J.E.; Shih, J.; Matias, M.; Hennekens, C.H. Prospective study of C-reactive protein and the risk of future cardiovascular events among apparently healthy women. Circulation 1998, 98, 731–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liuzzo, G.; Biasucci, L.M.; Gallimore, J.R.; Grillo, R.L.; Rebuzzi, A.G.; Pepys, M.B.; Maseri, A. The prognostic value of C-reactive protein and serum amyloid a protein in severe unstable angina. N. Engl. J. Med. 1994, 331, 417–424. [Google Scholar] [CrossRef]
- Morrow, D.A.; Rifai, N.; Antman, E.M.; Weiner, D.L.; McCabe, C.H.; Cannon, C.P.; Braunwald, E. C-reactive protein is a potent predictor of mortality independently of and in combination with troponin T in acute coronary syndromes: A TIMI 11A substudy. Thrombolysis in Myocardial Infarction. J. Am. Coll. Cardiol. 1998, 31, 1460–1465. [Google Scholar] [CrossRef] [Green Version]
- Heeschen, C.; Hamm, C.W.; Bruemmer, J.; Simoons, M.L. Predictive value of C-reactive protein and troponin T in patients with unstable angina: A comparative analysis. CAPTURE Investigators. Chimeric c7E3 AntiPlatelet Therapy in Unstable angina REfractory to standard treatment trial. J. Am. Coll. Cardiol. 2000, 35, 1535–1542. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, B.; Toss, H.; Siegbahn, A.; Venge, P.; Wallentin, L.; FRISC Study Group. Markers of myocardial damage and inflammation in relation to long-term mortality in unstable coronary artery disease. N. Engl. J. Med. 2000, 343, 1139–1147. [Google Scholar] [CrossRef]
- Mueller, C.; Buettner, H.J.; Hodgson, J.M.; Marsch, S.; Perruchoud, A.P.; Roskamm, H.; Neumann, F.J. Inflammation and long-term mortality after non-ST elevation acute coronary syndrome treated with a very early invasive strategy in 1042 consecutive patients. Circulation 2002, 105, 1412–1415. [Google Scholar] [CrossRef]
- Hwang, Y.C.; Morrow, D.A.; Cannon, C.P.; Liu, Y.; Bergenstal, R.; Heller, S.; Mehta, C.; Cushman, W.; Bakris, G.L.; Zannad, F.; et al. High-sensitivity C-reactive protein, low-density lipoprotein cholesterol and cardiovascular outcomes in patients with type 2 diabetes in the EXAMINE (Examination of Cardiovascular Outcomes with Alogliptin versus Standard of Care) trial. Diabetes Obes. Metab. 2018, 20, 654–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayerl, C.; Lukasser, M.; Sedivy, R.; Niederegger, H.; Seiler, R.; Wick, G. Atherosclerosis research from past to present—on the track of two pathologists with opposing views, Carl von Rokitansky and Rudolf Virchow. Virchows Arch. 2006, 449, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Libby, P.; Schonbeck, U.; Yan, Z.Q. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ. Res. 2002, 91, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Hansson, G.K. From Focal Lipid Storage to Systemic Inflammation: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 1594–1607. [Google Scholar] [CrossRef]
- Pothineni, N.V.K.; Subramany, S.; Kuriakose, K.; Shirazi, L.F.; Romeo, F.; Shah, P.K.; Mehta, J.L. Infections, atherosclerosis, and coronary heart disease. Eur. Heart J. 2017, 38, 3195–3201. [Google Scholar] [CrossRef]
- Arida, A.; Protogerou, A.D.; Kitas, G.D.; Sfikakis, P.P. Systemic Inflammatory Response and Atherosclerosis: The Paradigm of Chronic Inflammatory Rheumatic Diseases. Int. J. Mol. Sci. 2018, 19, 1890. [Google Scholar] [CrossRef] [Green Version]
- Wright, S.D.; Burton, C.; Hernandez, M.; Hassing, H.; Montenegro, J.; Mundt, S.; Patel, S.; Card, D.J.; Hermanowski-Vosatka, A.; Bergstrom, J.D.; et al. Infectious agents are not necessary for murine atherogenesis. J. Exp. Med. 2000, 191, 1437–1442. [Google Scholar] [CrossRef] [Green Version]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [Green Version]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef] [Green Version]
- Grebe, A.; Latz, E. Cholesterol crystals and inflammation. Curr. Rheumatol. Rep. 2013, 15, 313. [Google Scholar] [CrossRef]
- Weber, K.; Schilling, J.D. Lysosomes integrate metabolic-inflammatory cross-talk in primary macrophage inflammasome activation. J. Biol. Chem. 2014, 289, 9158–9171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.Z.; Xu, Z.Q.; Han, B.Z.; Su, D.F.; Liu, C. NLRP3 inflammasome and its inhibitors: A review. Front. Pharmacol. 2015, 6, 262. [Google Scholar] [CrossRef] [Green Version]
- Sharma, B.R.; Kanneganti, T.D. NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Overview of the interleukin-1 family of ligands and receptors. Semin. Immunol. 2013, 25, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Goldin, N.P.; Wolff, S.M. Demonstration and characterization of two distinct human leukocytic pyrogens. J. Exp. Med. 1974, 139, 1369–1381. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Renfer, L.; Wolff, S.M. Human leukocytic pyrogen: Purification and development of a radioimmunoassay. Proc. Natl. Acad. Sci. USA 1977, 74, 4624–4627. [Google Scholar] [CrossRef] [Green Version]
- Auron, P.E.; Webb, A.C.; Rosenwasser, L.J.; Mucci, S.F.; Rich, A.; Wolff, S.M.; Dinarello, C.A. Nucleotide sequence of human monocyte interleukin 1 precursor cDNA. Proc. Natl. Acad. Sci. USA 1984, 81, 7907–7911. [Google Scholar] [CrossRef] [Green Version]
- Lomedico, P.T.; Gubler, U.; Hellmann, C.P.; Dukovich, M.; Giri, J.G.; Pan, Y.C.; Collier, K.; Semionow, R.; Chua, A.O.; Mizel, S.B. Cloning and expression of murine interleukin-1 cDNA in Escherichia coli. Nature 1984, 312, 458–462. [Google Scholar] [CrossRef]
- March, C.J.; Mosley, B.; Larsen, A.; Cerretti, D.P.; Braedt, G.; Price, V.; Gillis, S.; Henney, C.S.; Kronheim, S.R.; Grabstein, K.; et al. Cloning, sequence and expression of two distinct human interleukin-1 complementary DNAs. Nature 1985, 315, 641–647. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Rosenwasser, L.J.; Wolff, S.M. Demonstration of a circulating suppressor factor of thymocyte proliferation during endotoxin fever in humans. J. Immunol. 1981, 127, 2517–2519. [Google Scholar] [PubMed]
- Arend, W.P.; Joslin, F.G.; Massoni, R.J. Effects of immune complexes on production by human monocytes of interleukin 1 or an interleukin 1 inhibitor. J. Immunol. 1985, 134, 3868–3875. [Google Scholar] [PubMed]
- Prieur, A.M.; Kaufmann, M.T.; Griscelli, C.; Dayer, J.M. Specific interleukin-1 inhibitor in serum and urine of children with systemic juvenile chronic arthritis. Lancet 1987, 2, 1240–1242. [Google Scholar] [CrossRef]
- Seckinger, P.; Williamson, K.; Balavoine, J.F.; Mach, B.; Mazzei, G.; Shaw, A.; Dayer, J.M. A urine inhibitor of interleukin 1 activity affects both interleukin 1 alpha and 1 beta but not tumor necrosis factor alpha. J. Immunol. 1987, 139, 1541–1545. [Google Scholar] [PubMed]
- Eisenberg, S.P.; Evans, R.J.; Arend, W.P.; Verderber, E.; Brewer, M.T.; Hannum, C.H.; Thompson, R.C. Primary structure and functional expression from complementary DNA of a human interleukin-1 receptor antagonist. Nature 1990, 343, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Okamura, H.; Tsutsi, H.; Komatsu, T.; Yutsudo, M.; Hakura, A.; Tanimoto, T.; Torigoe, K.; Okura, T.; Nukada, Y.; Hattori, K.; et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature 1995, 378, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. The IL-1 family of cytokines and receptors in rheumatic diseases. Nat. Rev. Rheumatol. 2019, 15, 612–632. [Google Scholar] [CrossRef] [PubMed]
- Rivers-Auty, J.; Daniels, M.J.D.; Colliver, I.; Robertson, D.L.; Brough, D. Redefining the ancestral origins of the interleukin-1 superfamily. Nat. Commun. 2018, 9, 1156. [Google Scholar] [CrossRef] [Green Version]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The interleukin-1 family: Back to the future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Towne, J.E.; Renshaw, B.R.; Douangpanya, J.; Lipsky, B.P.; Shen, M.; Gabel, C.A.; Sims, J.E. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36alpha, IL-36beta, and IL-36gamma) or antagonist (IL-36Ra) activity. J. Biol. Chem. 2011, 286, 42594–42602. [Google Scholar] [CrossRef]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khazim, K.; Azulay, E.E.; Kristal, B.; Cohen, I. Interleukin 1 gene polymorphism and susceptibility to disease. Immunol. Rev. 2018, 281, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Carriere, V.; Roussel, L.; Ortega, N.; Lacorre, D.A.; Americh, L.; Aguilar, L.; Bouche, G.; Girard, J.P. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 282–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moussion, C.; Ortega, N.; Girard, J.P. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: A novel ‘alarmin’? PLoS ONE 2008, 3, e3331. [Google Scholar] [CrossRef] [Green Version]
- Gabay, C.; Lamacchia, C.; Palmer, G. IL-1 pathways in inflammation and human diseases. Nat. Rev. Rheumatol. 2010, 6, 232–241. [Google Scholar] [CrossRef]
- Yasuda, K.; Nakanishi, K.; Tsutsui, H. Interleukin-18 in Health and Disease. Int. J. Mol. Sci. 2019, 20, 649. [Google Scholar] [CrossRef] [Green Version]
- Rex, D.A.B.; Agarwal, N.; Prasad, T.S.K.; Kandasamy, R.K.; Subbannayya, Y.; Pinto, S.M. A comprehensive pathway map of IL-18-mediated signalling. J. Cell Commun. Signal. 2020, 14, 257–266. [Google Scholar] [CrossRef]
- Boraschi, D.; Lucchesi, D.; Hainzl, S.; Leitner, M.; Maier, E.; Mangelberger, D.; Oostingh, G.J.; Pfaller, T.; Pixner, C.; Posselt, G.; et al. IL-37: A new anti-inflammatory cytokine of the IL-1 family. Eur. Cytokine Netw. 2011, 22, 127–147. [Google Scholar] [CrossRef] [Green Version]
- Nold, M.F.; Nold-Petry, C.A.; Zepp, J.A.; Palmer, B.E.; Bufler, P.; Dinarello, C.A. IL-37 is a fundamental inhibitor of innate immunity. Nat. Immunol. 2010, 11, 1014–1022. [Google Scholar] [CrossRef]
- Nicklin, M.J.; Barton, J.L.; Nguyen, M.; FitzGerald, M.G.; Duff, G.W.; Kornman, K. A sequence-based map of the nine genes of the human interleukin-1 cluster. Genomics 2002, 79, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Todorovic, V. Interleukin-36: Structure, Signaling and Function. Adv. Exp. Med. Biol. 2021, 21, 191–210. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Peng, X.; Li, Y.; Li, M. Role of IL-38 and its related cytokines in inflammation. Mediat. Inflamm. 2015, 2015, 807976. [Google Scholar] [CrossRef] [Green Version]
- Mills, K.H.; Dungan, L.S.; Jones, S.A.; Harris, J. The role of inflammasome-derived IL-1 in driving IL-17 responses. J. Leukoc. Biol. 2013, 93, 489–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffatt, M.F.; Gut, I.G.; Demenais, F.; Strachan, D.P.; Bouzigon, E.; Heath, S.; von Mutius, E.; Farrall, M.; Lathrop, M.; Cookson, W.; et al. A large-scale, consortium-based genomewide association study of asthma. N. Engl. J. Med. 2010, 363, 1211–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef] [PubMed]
- Boraschi, D.; Tagliabue, A. The interleukin-1 receptor family. Semin. Immunol. 2013, 25, 394–407. [Google Scholar] [CrossRef]
- Li, X.; Qin, J. Modulation of Toll-interleukin 1 receptor mediated signaling. J. Mol. Med. 2005, 83, 258–266. [Google Scholar] [CrossRef]
- Boraschi, D.; Italiani, P.; Weil, S.; Martin, M.U. The family of the interleukin-1 receptors. Immunol. Rev. 2018, 281, 197–232. [Google Scholar] [CrossRef]
- Garlanda, C.; Anders, H.J.; Mantovani, A. TIR8/SIGIRR: An IL-1R/TLR family member with regulatory functions in inflammation and T cell polarization. Trends Immunol. 2009, 30, 439–446. [Google Scholar] [CrossRef]
- Mora, J.; Schlemmer, A.; Wittig, I.; Richter, F.; Putyrski, M.; Frank, A.C.; Han, Y.; Jung, M.; Ernst, A.; Weigert, A.; et al. Interleukin-38 is released from apoptotic cells to limit inflammatory macrophage responses. J. Mol. Cell. Biol. 2016, 8, 426–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef] [PubMed]
- Eastgate, J.A.; Symons, J.A.; Wood, N.C.; Grinlinton, F.M.; di Giovine, F.S.; Duff, G.W. Correlation of plasma interleukin 1 levels with disease activity in rheumatoid arthritis. Lancet 1988, 2, 706–709. [Google Scholar] [CrossRef]
- Dalbeth, N.; Gosling, A.L.; Gaffo, A.; Abhishek, A. Gout. Lancet 2021, 397, 1843–1855. [Google Scholar] [CrossRef]
- Vazquez-Del Mercado, M.; Garcia-Gonzalez, A.; Munoz-Valle, J.F.; Garcia-Iglesias, T.; Martinez-Bonilla, G.; Bernard-Medina, G.; Sanchez-Ortiz, A.; Ornelas-Aguirre, J.M.; Salazar-Paramo, M.; Gamez-Nava, J.I.; et al. Interleukin 1beta (IL-1beta), IL-10, tumor necrosis factor-alpha, and cellular proliferation index in peripheral blood mononuclear cells in patients with ankylosing spondylitis. J. Rheumatol. 2002, 29, 522–526. [Google Scholar] [PubMed]
- Pascual, V.; Allantaz, F.; Arce, E.; Punaro, M.; Banchereau, J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J. Exp. Med. 2005, 201, 1479–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horneff, G.; Peitz, J.; Kekow, J.; Foell, D. Canakinumab for first line steroid-free treatment in a child with systemic-onset juvenile idiopathic arthritis. Scand. J. Rheumatol. 2017, 46, 500–501. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Dinarello, C.A. Treating rheumatological diseases and co-morbidities with interleukin-1 blocking therapies. Rheumatology 2015, 54, 2134–2144. [Google Scholar] [CrossRef] [Green Version]
- So, A.K.; Martinon, F. Inflammation in gout: Mechanisms and therapeutic targets. Nat. Rev. Rheumatol. 2017, 13, 639–647. [Google Scholar] [CrossRef]
- Roldan, R.; Ruiz, A.M.; Miranda, M.D.; Collantes, E. Anakinra: New therapeutic approach in children with Familial Mediterranean Fever resistant to colchicine. Jt. Bone Spine 2008, 75, 504–505. [Google Scholar] [CrossRef]
- Gonzalez, L.; Bulnes, J.F.; Orellana, M.P.; Munoz Venturelli, P.; Martinez Rodriguez, G. The Role of Colchicine in Atherosclerosis: From Bench to Bedside. Pharmaceutics 2022, 14, 1395. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, H.M.; Throne, M.L.; Amar, N.J.; Sebai, M.; Kivitz, A.J.; Kavanaugh, A.; Weinstein, S.P.; Belomestnov, P.; Yancopoulos, G.D.; Stahl, N.; et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: Results from two sequential placebo-controlled studies. Arthritis Rheum. 2008, 58, 2443–2452. [Google Scholar] [CrossRef]
- Reddy, S.; Jia, S.; Geoffrey, R.; Lorier, R.; Suchi, M.; Broeckel, U.; Hessner, M.J.; Verbsky, J. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N. Engl. J. Med. 2009, 360, 2438–2444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksentijevich, I.; Masters, S.L.; Ferguson, P.J.; Dancey, P.; Frenkel, J.; van Royen-Kerkhoff, A.; Laxer, R.; Tedgard, U.; Cowen, E.W.; Pham, T.H.; et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N. Engl. J. Med. 2009, 360, 2426–2437. [Google Scholar] [CrossRef] [Green Version]
- Ishikura, T.; Kanai, T.; Uraushihara, K.; Iiyama, R.; Makita, S.; Totsuka, T.; Yamazaki, M.; Sawada, T.; Nakamura, T.; Miyata, T.; et al. Interleukin-18 overproduction exacerbates the development of colitis with markedly infiltrated macrophages in interleukin-18 transgenic mice. J. Gastroenterol. Hepatol. 2003, 18, 960–969. [Google Scholar] [CrossRef]
- Sivakumar, P.V.; Westrich, G.M.; Kanaly, S.; Garka, K.; Born, T.L.; Derry, J.M.; Viney, J.L. Interleukin 18 is a primary mediator of the inflammation associated with dextran sulphate sodium induced colitis: Blocking interleukin 18 attenuates intestinal damage. Gut 2002, 50, 812–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteleone, G.; Trapasso, F.; Parrello, T.; Biancone, L.; Stella, A.; Iuliano, R.; Luzza, F.; Fusco, A.; Pallone, F. Bioactive IL-18 expression is up-regulated in Crohn’s disease. J. Immunol. 1999, 163, 143–147. [Google Scholar] [PubMed]
- Furuya, D.; Yagihashi, A.; Komatsu, M.; Masashi, N.; Tsuji, N.; Kobayashi, D.; Watanabe, N. Serum interleukin-18 concentrations in patients with inflammatory bowel disease. J. Immunother. 2002, 25 (Suppl. S1), S65–S67. [Google Scholar] [CrossRef]
- Williams, M.A.; O’Callaghan, A.; Corr, S.C. IL-33 and IL-18 in Inflammatory Bowel Disease Etiology and Microbial Interactions. Front. Immunol. 2019, 10, 1091. [Google Scholar] [CrossRef] [Green Version]
- Sedhom, M.A.; Pichery, M.; Murdoch, J.R.; Foligne, B.; Ortega, N.; Normand, S.; Mertz, K.; Sanmugalingam, D.; Brault, L.; Grandjean, T.; et al. Neutralisation of the interleukin-33/ST2 pathway ameliorates experimental colitis through enhancement of mucosal healing in mice. Gut 2013, 62, 1714–1723. [Google Scholar] [CrossRef]
- Malik, A.; Sharma, D.; Zhu, Q.; Karki, R.; Guy, C.S.; Vogel, P.; Kanneganti, T.D. IL-33 regulates the IgA-microbiota axis to restrain IL-1alpha-dependent colitis and tumorigenesis. J. Clin. Investig. 2016, 126, 4469–4481. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.; Dodel, R.C.; Eastwood, B.J.; Bales, K.R.; Gao, F.; Lohmuller, F.; Muller, U.; Kurz, A.; Zimmer, R.; Evans, R.M.; et al. Association of an interleukin 1 alpha polymorphism with Alzheimer’s disease. Neurology 2000, 55, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Mrak, R.E.; Griffinbc, W.S. The role of activated astrocytes and of the neurotrophic cytokine S100B in the pathogenesis of Alzheimer’s disease. Neurobiol. Aging 2001, 22, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Piancone, F.; La Rosa, F.; Marventano, I.; Saresella, M.; Clerici, M. The Role of the Inflammasome in Neurodegenerative Diseases. Molecules 2021, 26, 953. [Google Scholar] [CrossRef]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 2019, 15, 459–472. [Google Scholar] [CrossRef]
- Colotta, F.; Re, F.; Muzio, M.; Bertini, R.; Polentarutti, N.; Sironi, M.; Giri, J.G.; Dower, S.K.; Sims, J.E.; Mantovani, A. Interleukin-1 type II receptor: A decoy target for IL-1 that is regulated by IL-4. Science 1993, 261, 472–475. [Google Scholar] [CrossRef]
- Smith, D.E.; Hanna, R.; Della, F.; Moore, H.; Chen, H.; Farese, A.M.; MacVittie, T.J.; Virca, G.D.; Sims, J.E. The soluble form of IL-1 receptor accessory protein enhances the ability of soluble type II IL-1 receptor to inhibit IL-1 action. Immunity 2003, 18, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Kurt-Jones, E.A.; Beller, D.I.; Mizel, S.B.; Unanue, E.R. Identification of a membrane-associated interleukin 1 in macrophages. Proc. Natl. Acad. Sci. USA 1985, 82, 1204–1208. [Google Scholar] [CrossRef] [Green Version]
- Werman, A.; Werman-Venkert, R.; White, R.; Lee, J.K.; Werman, B.; Krelin, Y.; Voronov, E.; Dinarello, C.A.; Apte, R.N. The precursor form of IL-1alpha is an intracrine proinflammatory activator of transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 2434–2439. [Google Scholar] [CrossRef]
- Cohen, I.; Rider, P.; Carmi, Y.; Braiman, A.; Dotan, S.; White, M.R.; Voronov, E.; Martin, M.U.; Dinarello, C.A.; Apte, R.N. Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 2574–2579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Paolo, N.C.; Shayakhmetov, D.M. Interleukin 1alpha and the inflammatory process. Nat. Immunol. 2016, 17, 906–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.; Lee, Y.; Kim, E.; Kwak, A.; Ryoo, S.; Bae, S.H.; Azam, T.; Kim, S.; Dinarello, C.A. The Interleukin-1alpha Precursor is Biologically Active and is Likely a Key Alarmin in the IL-1 Family of Cytokines. Front. Immunol. 2013, 4, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stutz, A.; Golenbock, D.T.; Latz, E. Inflammasomes: Too big to miss. J. Clin. Investig. 2009, 119, 3502–3511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef]
- Wesche, H.; Resch, K.; Martin, M.U. Effects of IL-1 receptor accessory protein on IL-1 binding. FEBS Lett. 1998, 429, 303–306. [Google Scholar] [CrossRef] [Green Version]
- Brough, D.; Rothwell, N.J. Caspase-1-dependent processing of pro-interleukin-1beta is cytosolic and precedes cell death. J. Cell Sci. 2007, 120, 772–781. [Google Scholar] [CrossRef] [Green Version]
- Fantuzzi, G.; Ku, G.; Harding, M.W.; Livingston, D.J.; Sipe, J.D.; Kuida, K.; Flavell, R.A.; Dinarello, C.A. Response to local inflammation of IL-1 beta-converting enzyme- deficient mice. J. Immunol. 1997, 158, 1818–1824. [Google Scholar]
- Joosten, L.A.; Netea, M.G.; Fantuzzi, G.; Koenders, M.I.; Helsen, M.M.; Sparrer, H.; Pham, C.T.; van der Meer, J.W.; Dinarello, C.A.; van den Berg, W.B. Inflammatory arthritis in caspase 1 gene-deficient mice: Contribution of proteinase 3 to caspase 1-independent production of bioactive interleukin-1beta. Arthritis Rheum. 2009, 60, 3651–3662. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Gautier, V.; Cayrol, C.; Farache, D.; Roga, S.; Monsarrat, B.; Burlet-Schiltz, O.; Gonzalez de Peredo, A.; Girard, J.P. Extracellular IL-33 cytokine, but not endogenous nuclear IL-33, regulates protein expression in endothelial cells. Sci. Rep. 2016, 6, 34255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demyanets, S.; Stojkovic, S.; Huber, K.; Wojta, J. The Paradigm Change of IL-33 in Vascular Biology. Int. J. Mol. Sci. 2021, 22, 13288. [Google Scholar] [CrossRef] [PubMed]
- Bouffi, C.; Rochman, M.; Zust, C.B.; Stucke, E.M.; Kartashov, A.; Fulkerson, P.C.; Barski, A.; Rothenberg, M.E. IL-33 markedly activates murine eosinophils by an NF-kappaB-dependent mechanism differentially dependent upon an IL-4-driven autoinflammatory loop. J. Immunol. 2013, 191, 4317–4325. [Google Scholar] [CrossRef] [Green Version]
- Grotenboer, N.S.; Ketelaar, M.E.; Koppelman, G.H.; Nawijn, M.C. Decoding asthma: Translating genetic variation in IL33 and IL1RL1 into disease pathophysiology. J. Allergy Clin. Immunol. 2013, 131, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Matsuda, A.; Yanagisawa, K.; Hirota, T.; Akahoshi, M.; Inomata, N.; Ebe, K.; Tanaka, K.; Sugiura, H.; Nakashima, K.; et al. Functional SNPs in the distal promoter of the ST2 gene are associated with atopic dermatitis. Hum. Mol. Genet. 2005, 14, 2919–2927. [Google Scholar] [CrossRef] [Green Version]
- Chackerian, A.A.; Oldham, E.R.; Murphy, E.E.; Schmitz, J.; Pflanz, S.; Kastelein, R.A. IL-1 receptor accessory protein and ST2 comprise the IL-33 receptor complex. J. Immunol. 2007, 179, 2551–2555. [Google Scholar] [CrossRef] [Green Version]
- Demyanets, S.; Kaun, C.; Pentz, R.; Krychtiuk, K.A.; Rauscher, S.; Pfaffenberger, S.; Zuckermann, A.; Aliabadi, A.; Groger, M.; Maurer, G.; et al. Components of the interleukin-33/ST2 system are differentially expressed and regulated in human cardiac cells and in cells of the cardiac vasculature. J. Mol. Cell Cardiol. 2013, 60, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Kang, J.; Zhang, C.; Zhang, X. The role of IL-33/ST2L signals in the immune cells. Immunol. Lett. 2015, 164, 11–17. [Google Scholar] [CrossRef]
- Pinto, S.M.; Subbannayya, Y.; Rex, D.A.B.; Raju, R.; Chatterjee, O.; Advani, J.; Radhakrishnan, A.; Keshava Prasad, T.S.; Wani, M.R.; Pandey, A. A network map of IL-33 signaling pathway. J. Cell Commun. Signal. 2018, 12, 615–624. [Google Scholar] [CrossRef]
- Zhao, W.; Hu, Z. The enigmatic processing and secretion of interleukin-33. Cell Mol. Immunol. 2010, 7, 260–262. [Google Scholar] [CrossRef] [Green Version]
- Arend, W.P.; Welgus, H.G.; Thompson, R.C.; Eisenberg, S.P. Biological properties of recombinant human monocyte-derived interleukin 1 receptor antagonist. J. Clin. Investig. 1990, 85, 1694–1697. [Google Scholar] [CrossRef] [PubMed]
- Moyer, C.F.; Sajuthi, D.; Tulli, H.; Williams, J.K. Synthesis of IL-1 alpha and IL-1 beta by arterial cells in atherosclerosis. Am. J. Pathol. 1991, 138, 951–960. [Google Scholar] [PubMed]
- Abbate, A.; Toldo, S.; Marchetti, C.; Kron, J.; Van Tassell, B.W.; Dinarello, C.A. Interleukin-1 and the Inflammasome as Therapeutic Targets in Cardiovascular Disease. Circ. Res. 2020, 126, 1260–1280. [Google Scholar] [CrossRef] [PubMed]
- Lipton, B.A.; Parthasarathy, S.; Ord, V.A.; Clinton, S.K.; Libby, P.; Rosenfeld, M.E. Components of the protein fraction of oxidized low density lipoprotein stimulate interleukin-1 alpha production by rabbit arterial macrophage-derived foam cells. J. Lipid Res. 1995, 36, 2232–2242. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.C.; Talib, S.; Figg, N.L.; Bennett, M.R. Vascular smooth muscle cell apoptosis induces interleukin-1-directed inflammation: Effects of hyperlipidemia-mediated inhibition of phagocytosis. Circ. Res. 2010, 106, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Kirii, H.; Niwa, T.; Yamada, Y.; Wada, H.; Saito, K.; Iwakura, Y.; Asano, M.; Moriwaki, H.; Seishima, M. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Bhaskar, V.; Yin, J.; Mirza, A.M.; Phan, D.; Vanegas, S.; Issafras, H.; Michelson, K.; Hunter, J.J.; Kantak, S.S. Monoclonal antibodies targeting IL-1 beta reduce biomarkers of atherosclerosis in vitro and inhibit atherosclerotic plaque formation in Apolipoprotein E-deficient mice. Atherosclerosis 2011, 216, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Kamari, Y.; Shaish, A.; Shemesh, S.; Vax, E.; Grosskopf, I.; Dotan, S.; White, M.; Voronov, E.; Dinarello, C.A.; Apte, R.N.; et al. Reduced atherosclerosis and inflammatory cytokines in apolipoprotein-E-deficient mice lacking bone marrow-derived interleukin-1alpha. Biochem. Biophys. Res. Commun. 2011, 405, 197–203. [Google Scholar] [CrossRef]
- Kamari, Y.; Werman-Venkert, R.; Shaish, A.; Werman, A.; Harari, A.; Gonen, A.; Voronov, E.; Grosskopf, I.; Sharabi, Y.; Grossman, E.; et al. Differential role and tissue specificity of interleukin-1alpha gene expression in atherogenesis and lipid metabolism. Atherosclerosis 2007, 195, 31–38. [Google Scholar] [CrossRef]
- Isoda, K.; Sawada, S.; Ishigami, N.; Matsuki, T.; Miyazaki, K.; Kusuhara, M.; Iwakura, Y.; Ohsuzu, F. Lack of interleukin-1 receptor antagonist modulates plaque composition in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1068–1073. [Google Scholar] [CrossRef] [Green Version]
- Elhage, R.; Maret, A.; Pieraggi, M.T.; Thiers, J.C.; Arnal, J.F.; Bayard, F. Differential effects of interleukin-1 receptor antagonist and tumor necrosis factor binding protein on fatty-streak formation in apolipoprotein E-deficient mice. Circulation 1998, 97, 242–244. [Google Scholar] [CrossRef] [PubMed]
- Devlin, C.M.; Kuriakose, G.; Hirsch, E.; Tabas, I. Genetic alterations of IL-1 receptor antagonist in mice affect plasma cholesterol level and foam cell lesion size. Proc. Natl. Acad. Sci. USA 2002, 99, 6280–6285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, M.R.; Moehle, C.W.; Johnson, J.L.; Yang, Z.; Lee, J.K.; Jackson, C.L.; Owens, G.K. Genetic inactivation of IL-1 signaling enhances atherosclerotic plaque instability and reduces outward vessel remodeling in advanced atherosclerosis in mice. J. Clin. Investig. 2012, 122, 70–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, D.; Baylis, R.A.; Durgin, B.G.; Newman, A.A.C.; Alencar, G.F.; Mahan, S.; St Hilaire, C.; Muller, W.; Waisman, A.; Francis, S.E.; et al. Interleukin-1beta has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat. Med. 2018, 24, 1418–1429. [Google Scholar] [CrossRef] [PubMed]
- Burger, F.; Baptista, D.; Roth, A.; da Silva, R.F.; Montecucco, F.; Mach, F.; Brandt, K.J.; Miteva, K. NLRP3 Inflammasome Activation Controls Vascular Smooth Muscle Cells Phenotypic Switch in Atherosclerosis. Int. J. Mol. Sci. 2021, 23, 340. [Google Scholar] [CrossRef]
- Gage, J.; Hasu, M.; Thabet, M.; Whitman, S.C. Caspase-1 deficiency decreases atherosclerosis in apolipoprotein E-null mice. Can. J. Cardiol. 2012, 28, 222–229. [Google Scholar] [CrossRef]
- Usui, F.; Shirasuna, K.; Kimura, H.; Tatsumi, K.; Kawashima, A.; Karasawa, T.; Hida, S.; Sagara, J.; Taniguchi, S.; Takahashi, M. Critical role of caspase-1 in vascular inflammation and development of atherosclerosis in Western diet-fed apolipoprotein E-deficient mice. Biochem. Biophys. Res. Commun. 2012, 425, 162–168. [Google Scholar] [CrossRef]
- Hendrikx, T.; Jeurissen, M.L.; van Gorp, P.J.; Gijbels, M.J.; Walenbergh, S.M.; Houben, T.; van Gorp, R.; Pottgens, C.C.; Stienstra, R.; Netea, M.G.; et al. Bone marrow-specific caspase-1/11 deficiency inhibits atherosclerosis development in Ldlr(-/-) mice. FEBS J. 2015, 282, 2327–2338. [Google Scholar] [CrossRef]
- Menu, P.; Pellegrin, M.; Aubert, J.F.; Bouzourene, K.; Tardivel, A.; Mazzolai, L.; Tschopp, J. Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. 2011, 2, e137. [Google Scholar] [CrossRef] [Green Version]
- van der Heijden, T.; Kritikou, E.; Venema, W.; van Duijn, J.; van Santbrink, P.J.; Slutter, B.; Foks, A.C.; Bot, I.; Kuiper, J. NLRP3 Inflammasome Inhibition by MCC950 Reduces Atherosclerotic Lesion Development in Apolipoprotein E-Deficient Mice-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1457–1461. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Xie, W.L.; Kong, W.W.; Chen, D.; Qu, P. Expression of the NLRP3 Inflammasome in Carotid Atherosclerosis. J. Stroke Cerebrovasc. Dis. 2015, 24, 2455–2466. [Google Scholar] [CrossRef] [PubMed]
- Paramel Varghese, G.; Folkersen, L.; Strawbridge, R.J.; Halvorsen, B.; Yndestad, A.; Ranheim, T.; Krohg-Sorensen, K.; Skjelland, M.; Espevik, T.; Aukrust, P.; et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J. Am. Heart Assoc. 2016, 5, e003031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altaf, A.; Qu, P.; Zhao, Y.; Wang, H.; Lou, D.; Niu, N. NLRP3 inflammasome in peripheral blood monocytes of acute coronary syndrome patients and its relationship with statins. Coron. Artery Dis. 2015, 26, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef] [Green Version]
- Tsimikas, S.; Duff, G.W.; Berger, P.B.; Rogus, J.; Huttner, K.; Clopton, P.; Brilakis, E.; Kornman, K.S.; Witztum, J.L. Pro-inflammatory interleukin-1 genotypes potentiate the risk of coronary artery disease and cardiovascular events mediated by oxidized phospholipids and lipoprotein(a). J. Am. Coll. Cardiol. 2014, 63, 1724–1734. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Pascual-Figal, D.A.; Bayes-Genis, A.; Asensio-Lopez, M.C.; Hernandez-Vicente, A.; Garrido-Bravo, I.; Pastor-Perez, F.; Diez, J.; Ibanez, B.; Lax, A. The Interleukin-1 Axis and Risk of Death in Patients with Acutely Decompensated Heart Failure. J. Am. Coll. Cardiol. 2019, 73, 1016–1025. [Google Scholar] [CrossRef]
- Silvain, J.; Kerneis, M.; Zeitouni, M.; Lattuca, B.; Galier, S.; Brugier, D.; Mertens, E.; Procopi, N.; Suc, G.; Salloum, T.; et al. Interleukin-1beta and Risk of Premature Death in Patients with Myocardial Infarction. J. Am. Coll. Cardiol. 2020, 76, 1763–1773. [Google Scholar] [CrossRef]
- Herder, C.; de Las Heras Gala, T.; Carstensen-Kirberg, M.; Huth, C.; Zierer, A.; Wahl, S.; Sudduth-Klinger, J.; Kuulasmaa, K.; Peretz, D.; Ligthart, S.; et al. Circulating Levels of Interleukin 1-Receptor Antagonist and Risk of Cardiovascular Disease: Meta-Analysis of Six Population-Based Cohorts. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1222–1227. [Google Scholar] [CrossRef] [Green Version]
- Abbate, A.; Kontos, M.C.; Abouzaki, N.A.; Melchior, R.D.; Thomas, C.; Van Tassell, B.W.; Oddi, C.; Carbone, S.; Trankle, C.R.; Roberts, C.S.; et al. Comparative safety of interleukin-1 blockade with anakinra in patients with ST-segment elevation acute myocardial infarction (from the VCU-ART and VCU-ART2 pilot studies). Am. J. Cardiol. 2015, 115, 288–292. [Google Scholar] [CrossRef]
- El Sayed, H.; Kerensky, R.; Stecher, M.; Mohanty, P.; Davies, M. A randomized phase II study of Xilonix, a targeted therapy against interleukin 1alpha, for the prevention of superficial femoral artery restenosis after percutaneous revascularization. J. Vasc. Surg. 2016, 63, 133–141.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micallef, M.J.; Ohtsuki, T.; Kohno, K.; Tanabe, F.; Ushio, S.; Namba, M.; Tanimoto, T.; Torigoe, K.; Fujii, M.; Ikeda, M.; et al. Interferon-gamma-inducing factor enhances T helper 1 cytokine production by stimulated human T cells: Synergism with interleukin-12 for interferon-gamma production. Eur. J. Immunol. 1996, 26, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Tao, X. Current Understanding of IL-37 in Human Health and Disease. Front. Immunol. 2021, 12, 696605. [Google Scholar] [CrossRef] [PubMed]
- McCurdy, S.; Liu, C.A.; Yap, J.; Boisvert, W.A. Potential role of IL-37 in atherosclerosis. Cytokine 2019, 122, 154169. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, N.; Yokota, A.; Kimura, T.; Kato, Z.; Fukao, T.; Shirakawa, M.; Ohnishi, H.; Tochio, H. An innate interaction between IL-18 and the propeptide that inactivates its precursor form. Sci. Rep. 2019, 9, 6160. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, S.; Otani, T.; Okura, R.; Ijiri, Y.; Motoda, R.; Kurimoto, M.; Orita, K. Expression and responsiveness of human interleukin-18 receptor (IL-18R) on hematopoietic cell lines. Leukemia 2000, 14, 1052–1059. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, T.; Takeda, K.; Tanaka, T.; Ohkusu, K.; Kashiwamura, S.; Okamura, H.; Akira, S.; Nakanishi, K. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: Synergism with IL-18 for IFN-gamma production. J. Immunol. 1998, 161, 3400–3407. [Google Scholar]
- Cimato, T.R.; Palka, B.A. Effects of statins on TH1 modulating cytokines in human subjects. PeerJ 2015, 3, e764. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, K. Unique Action of Interleukin-18 on T Cells and Other Immune Cells. Front. Immunol. 2018, 9, 763. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-18 is a unique cytokine that stimulates both Th1 and Th2 responses depending on its cytokine milieu. Cytokine Growth Factor Rev. 2001, 12, 53–72. [Google Scholar] [CrossRef]
- Puren, A.J.; Fantuzzi, G.; Dinarello, C.A. Gene expression, synthesis, and secretion of interleukin 18 and interleukin 1beta are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc. Natl. Acad. Sci. USA 1999, 96, 2256–2261. [Google Scholar] [CrossRef]
- Akita, K.; Ohtsuki, T.; Nukada, Y.; Tanimoto, T.; Namba, M.; Okura, T.; Takakura-Yamamoto, R.; Torigoe, K.; Gu, Y.; Su, M.S.; et al. Involvement of caspase-1 and caspase-3 in the production and processing of mature human interleukin 18 in monocytic THP.1 cells. J. Biol. Chem. 1997, 272, 26595–26603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardella, S.; Andrei, C.; Costigliolo, S.; Poggi, A.; Zocchi, M.R.; Rubartelli, A. Interleukin-18 synthesis and secretion by dendritic cells are modulated by interaction with antigen-specific T cells. J. Leukoc. Biol. 1999, 66, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, N.; Sukhova, G.K.; Libby, P.; Reynolds, R.S.; Young, J.L.; Schönbeck, U. Expression of interleukin (IL)-18 and functional IL-18 receptor on human vascular endothelial cells, smooth muscle cells, and macrophages: Implications for atherogenesis. J. Exp. Med. 2002, 195, 245–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novick, D.; Kim, S.H.; Fantuzzi, G.; Reznikov, L.L.; Dinarello, C.A.; Rubinstein, M. Interleukin-18 binding protein: A novel modulator of the Th1 cytokine response. Immunity 1999, 10, 127–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.Y.; Hisham, Y.; Shin, H.M.; Yeom, S.C.; Kim, S. Interleukin-18 Binding Protein in Immune Regulation and Autoimmune Diseases. Biomedicines 2022, 10, 1750. [Google Scholar] [CrossRef] [PubMed]
- Yellayi, S.; Braun, S.; Domingues, H.G.; Kityatana, N.; Murali, R.; Johnson, R.P.; Mansfield, K.; Westmoreland, S.V.; O’Neil, S.P. Cloning and characterization of rhesus IL-18 binding protein, a natural antagonist to IL-18. Cytokine 2010, 51, 232–239. [Google Scholar] [CrossRef]
- Nold-Petry, C.A.; Lo, C.Y.; Rudloff, I.; Elgass, K.D.; Li, S.; Gantier, M.P.; Lotz-Havla, A.S.; Gersting, S.W.; Cho, S.X.; Lao, J.C.; et al. IL-37 requires the receptors IL-18Rα and IL-1R8 (SIGIRR) to carry out its multifaceted anti-inflammatory program upon innate signal transduction. Nat. Immunol. 2015, 16, 354–365. [Google Scholar] [CrossRef]
- Cavalli, G.; Dinarello, C.A. Suppression of inflammation and acquired immunity by IL-37. Immunol. Rev. 2018, 281, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Fields, J.K.; Günther, S.; Sundberg, E.J. Structural Basis of IL-1 Family Cytokine Signaling. Front. Immunol. 2019, 10, 1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulau, A.M.; Nold, M.F.; Li, S.; Nold-Petry, C.A.; Fink, M.; Mansell, A.; Schwerd, T.; Hong, J.; Rubartelli, A.; Dinarello, C.A.; et al. Role of caspase-1 in nuclear translocation of IL-37, release of the cytokine, and IL-37 inhibition of innate immune responses. Proc. Natl. Acad. Sci. USA 2014, 111, 2650–2655. [Google Scholar] [CrossRef]
- Wald, D.; Qin, J.; Zhao, Z.; Qian, Y.; Naramura, M.; Tian, L.; Towne, J.; Sims, J.E.; Stark, G.R.; Li, X. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat. Immunol. 2003, 4, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Amo-Aparicio, J.; Neff, C.P.; Tengesdal, I.W.; Azam, T.; Palmer, B.E.; López-Vales, R.; Bufler, P.; Dinarello, C.A. Role for nuclear interleukin-37 in the suppression of innate immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 4456–4461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudloff, I.; Ung, H.K.; Dowling, J.K.; Mansell, A.; D’Andrea, L.; Ellisdon, A.M.; Whisstock, J.C.; Berger, P.J.; Nold-Petry, C.A.; Nold, M.F. Parsing the IL-37-Mediated Suppression of Inflammasome Function. Cells 2020, 9, 178. [Google Scholar] [CrossRef] [Green Version]
- Rudloff, I.; Cho, S.X.; Lao, J.C.; Ngo, D.; McKenzie, M.; Nold-Petry, C.A.; Nold, M.F. Monocytes and dendritic cells are the primary sources of interleukin 37 in human immune cells. J. Leukoc. Biol. 2017, 101, 901–911. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Nold-Petry, C.; Nold, M.; Fujita, M.; Li, S.; Kim, S.; Bufler, P. Suppression of innate inflammation and immunity by interleukin-37. Eur. J. Immunol. 2016, 46, 1067–1081. [Google Scholar] [CrossRef] [Green Version]
- McNamee, E.N.; Masterson, J.C.; Jedlicka, P.; McManus, M.; Grenz, A.; Collins, C.B.; Nold, M.F.; Nold-Petry, C.; Bufler, P.; Dinarello, C.A.; et al. Interleukin 37 expression protects mice from colitis. Proc. Natl. Acad. Sci. USA 2011, 108, 16711–16716. [Google Scholar] [CrossRef] [Green Version]
- Whitman, S.C.; Ravisankar, P.; Daugherty, A. Interleukin-18 enhances atherosclerosis in apolipoprotein E(-/-) mice through release of interferon-gamma. Circ. Res. 2002, 90, E34–E38. [Google Scholar] [CrossRef] [Green Version]
- Elyasi, A.; Voloshyna, I.; Ahmed, S.; Kasselman, L.J.; Behbodikhah, J.; De Leon, J.; Reiss, A.B. The role of interferon-γ in cardiovascular disease: An update. Inflamm. Res. 2020, 69, 975–988. [Google Scholar] [CrossRef]
- Moss, J.W.; Ramji, D.P. Interferon-γ: Promising therapeutic target in atherosclerosis. World J. Exp. Med. 2015, 5, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Schindler, H.; Lutz, M.B.; Röllinghoff, M.; Bogdan, C. The production of IFN-gamma by IL-12/IL-18-activated macrophages requires STAT4 signaling and is inhibited by IL-4. J. Immunol. 2001, 166, 3075–3082. [Google Scholar] [CrossRef] [PubMed]
- Tenger, C.; Sundborger, A.; Jawien, J.; Zhou, X. IL-18 accelerates atherosclerosis accompanied by elevation of IFN-gamma and CXCL16 expression independently of T cells. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 791–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, O.M.; Uday Kumar, P.; Harishankar, N.; Ravichandaran, L.; Bhatia, A.; Dhawan, V. Interleukin-18-induced cell adhesion molecule expression is associated with feedback regulation by PPAR-γ and NF-κB in Apo E-/- mice. Mol. Cell Biochem. 2017, 428, 119–128. [Google Scholar] [CrossRef]
- Bhat, O.M.; Kumar, P.U.; Giridharan, N.V.; Kaul, D.; Kumar, M.J.; Dhawan, V. Interleukin-18-induced atherosclerosis involves CD36 and NF-κB crosstalk in Apo E-/- mice. J. Cardiol. 2015, 66, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Bhat, O.M.; Kumar, P.U.; Rao, K.R.; Ahmad, A.; Dhawan, V. Terminalia arjuna prevents Interleukin-18-induced atherosclerosis via modulation of NF-κB/PPAR-γ-mediated pathway in Apo E-/- mice. Inflammopharmacology 2018, 26, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Joosten, L.A.; Lewis, E.; Jensen, D.R.; Voshol, P.J.; Kullberg, B.J.; Tack, C.J.; van Krieken, H.; Kim, S.H.; Stalenhoef, A.F.; et al. Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat. Med. 2006, 12, 650–656. [Google Scholar] [CrossRef]
- Pejnovic, N.; Vratimos, A.; Lee, S.H.; Popadic, D.; Takeda, K.; Akira, S.; Chan, W.L. Increased atherosclerotic lesions and Th17 in interleukin-18 deficient apolipoprotein E-knockout mice fed high-fat diet. Mol. Immunol. 2009, 47, 37–45. [Google Scholar] [CrossRef]
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Besnard, S.; Lesèche, G.; Chvatchko, Y.; Tedgui, A. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation 2001, 104, 1598–1603. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Sun, C.; Gerdes, N.; Liu, C.; Liao, M.; Liu, J.; Shi, M.A.; He, A.; Zhou, Y.; Sukhova, G.K.; et al. Interleukin 18 function in atherosclerosis is mediated by the interleukin 18 receptor and the Na-Cl co-transporter. Nat. Med. 2015, 21, 820–826. [Google Scholar] [CrossRef] [Green Version]
- Ji, Q.; Zeng, Q.; Huang, Y.; Shi, Y.; Lin, Y.; Lu, Z.; Meng, K.; Wu, B.; Yu, K.; Chai, M.; et al. Elevated plasma IL-37, IL-18, and IL-18BP concentrations in patients with acute coronary syndrome. Mediat. Inflamm. 2014, 2014, 165742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Li, Q.; Su, S.; Dong, W.; Zong, S.; Ma, Q.; Yang, X.; Zuo, D.; Zheng, S.; Meng, X.; et al. Interleukin 37 Suppresses M1 Macrophage Polarization Through Inhibition of the Notch1 and Nuclear Factor Kappa B Pathways. Front. Cell Dev. Biol. 2020, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- McCurdy, S.; Baumer, Y.; Toulmin, E.; Lee, B.H.; Boisvert, W.A. Macrophage-Specific Expression of IL-37 in Hyperlipidemic Mice Attenuates Atherosclerosis. J. Immunol. 2017, 199, 3604–3613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinetti-Gbaguidi, G.; Colin, S.; Staels, B. Macrophage subsets in atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 10–17. [Google Scholar] [CrossRef]
- Ballak, D.B.; van Diepen, J.A.; Moschen, A.R.; Jansen, H.J.; Hijmans, A.; Groenhof, G.J.; Leenders, F.; Bufler, P.; Boekschoten, M.V.; Müller, M.; et al. IL-37 protects against obesity-induced inflammation and insulin resistance. Nat. Commun. 2014, 5, 4711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhai, Y.; Ao, L.; Hui, H.; Fullerton, D.A.; Dinarello, C.A.; Meng, X. Interleukin-37 suppresses the inflammatory response to protect cardiac function in old endotoxemic mice. Cytokine 2017, 95, 55–63. [Google Scholar] [CrossRef]
- Liu, J.; Lin, J.; He, S.; Wu, C.; Wang, B.; Liu, J.; Duan, Y.; Liu, T.; Shan, S.; Yang, K.; et al. Transgenic Overexpression of IL-37 Protects Against Atherosclerosis and Strengthens Plaque Stability. Cell Physiol. Biochem. 2018, 45, 1034–1050. [Google Scholar] [CrossRef] [Green Version]
- Ji, Q.; Meng, K.; Yu, K.; Huang, S.; Huang, Y.; Min, X.; Zhong, Y.; Wu, B.; Liu, Y.; Nie, S.; et al. Exogenous interleukin 37 ameliorates atherosclerosis via inducing the Treg response in ApoE-deficient mice. Sci. Rep. 2017, 7, 3310. [Google Scholar] [CrossRef] [Green Version]
- Hermansson, A.; Johansson, D.K.; Ketelhuth, D.F.; Andersson, J.; Zhou, X.; Hansson, G.K. Immunotherapy with tolerogenic apolipoprotein B-100-loaded dendritic cells attenuates atherosclerosis in hypercholesterolemic mice. Circulation 2011, 123, 1083–1091. [Google Scholar] [CrossRef] [Green Version]
- Zhu, R.; Zhang, F.; Pan, C.; Yu, K.; Zhong, Y.; Zeng, Q. Role of IL-37- and IL-37-Treated Dendritic Cells in Acute Coronary Syndrome. Oxid. Med. Cell. Longev. 2021, 2021, 6454177. [Google Scholar] [CrossRef]
- Ballak, D.B.; Brunt, V.E.; Sapinsley, Z.J.; Ziemba, B.P.; Richey, J.J.; Zigler, M.C.; Johnson, L.C.; Gioscia-Ryan, R.A.; Culp-Hill, R.; Eisenmesser, E.Z.; et al. Short-term interleukin-37 treatment improves vascular endothelial function, endurance exercise capacity, and whole-body glucose metabolism in old mice. Aging Cell 2020, 19, e13074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blankenberg, S.; Tiret, L.; Bickel, C.; Peetz, D.; Cambien, F.; Meyer, J.; Rupprecht, H.J.; AtheroGene, I. Interleukin-18 is a strong predictor of cardiovascular death in stable and unstable angina. Circulation 2002, 106, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Hulthe, J.; McPheat, W.; Samnegard, A.; Tornvall, P.; Hamsten, A.; Eriksson, P. Plasma interleukin (IL)-18 concentrations is elevated in patients with previous myocardial infarction and related to severity of coronary atherosclerosis independently of C-reactive protein and IL-6. Atherosclerosis 2006, 188, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Jefferis, B.J.; Papacosta, O.; Owen, C.G.; Wannamethee, S.G.; Humphries, S.E.; Woodward, M.; Lennon, L.T.; Thomson, A.; Welsh, P.; Rumley, A.; et al. Interleukin 18 and coronary heart disease: Prospective study and systematic review. Atherosclerosis 2011, 217, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Everett, B.M.; Bansal, S.; Rifai, N.; Buring, J.E.; Ridker, P.M. Interleukin-18 and the risk of future cardiovascular disease among initially healthy women. Atherosclerosis 2009, 202, 282–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, S.R.; Novick, D.; Stock, C.J.; Sanders, J.; Brull, D.; Cooper, J.; Woo, P.; Miller, G.; Rubinstein, M.; Humphries, S.E. Free Interleukin (IL)-18 levels, and the impact of IL18 and IL18BP genetic variation, in CHD patients and healthy men. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2743–2749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, A.M.; Baliwag, J.; Chen, C.S.; Guzman, A.M.; Stoll, S.W.; Gudjonsson, J.E.; Ward, N.L.; Johnston, A. IL-36 promotes myeloid cell infiltration, activation, and inflammatory activity in skin. J. Immunol. 2014, 192, 6053–6061. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.C.; Xu, W.D.; Liu, X.Y.; Liu, X.Y.; Huang, A.F.; Su, L.C. Biology of IL-36 Signaling and Its Role in Systemic Inflammatory Diseases. Front. Immunol. 2019, 10, 2532. [Google Scholar] [CrossRef] [Green Version]
- Clancy, D.M.; Henry, C.M.; Sullivan, G.P.; Martin, S.J. Neutrophil extracellular traps can serve as platforms for processing and activation of IL-1 family cytokines. FEBS J. 2017, 284, 1712–1725. [Google Scholar] [CrossRef] [Green Version]
- Henry, C.M.; Sullivan, G.P.; Clancy, D.M.; Afonina, I.S.; Kulms, D.; Martin, S.J. Neutrophil-Derived Proteases Escalate Inflammation through Activation of IL-36 Family Cytokines. Cell Rep. 2016, 14, 708–722. [Google Scholar] [CrossRef] [Green Version]
- Byrne, J.; Baker, K.; Houston, A.; Brint, E. IL-36 cytokines in inflammatory and malignant diseases: Not the new kid on the block anymore. Cell Mol. Life Sci. 2021, 78, 6215–6227. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Huard, A.; Mora, J.; da Silva, P.; Brüne, B.; Weigert, A. IL-36 family cytokines in protective versus destructive inflammation. Cell Signal. 2020, 75, 109773. [Google Scholar] [CrossRef] [PubMed]
- Bridgewood, C.; Stacey, M.; Alase, A.; Lagos, D.; Graham, A.; Wittmann, M. IL-36γ has proinflammatory effects on human endothelial cells. Exp. Dermatol. 2017, 26, 402–408. [Google Scholar] [CrossRef]
- Vigne, S.; Palmer, G.; Lamacchia, C.; Martin, P.; Talabot-Ayer, D.; Rodriguez, E.; Ronchi, F.; Sallusto, F.; Dinh, H.; Sims, J.E.; et al. IL-36R ligands are potent regulators of dendritic and T cells. Blood 2011, 118, 5813–5823. [Google Scholar] [CrossRef]
- Macleod, T.; Doble, R.; McGonagle, D.; Wasson, C.W.; Alase, A.; Stacey, M.; Wittmann, M. Neutrophil Elastase-mediated proteolysis activates the anti-inflammatory cytokine IL-36 Receptor antagonist. Sci. Rep. 2016, 6, 24880. [Google Scholar] [CrossRef] [Green Version]
- van de Veerdonk, F.L.; Stoeckman, A.K.; Wu, G.; Boeckermann, A.N.; Azam, T.; Netea, M.G.; Joosten, L.A.; van der Meer, J.W.; Hao, R.; Kalabokis, V.; et al. IL-38 binds to the IL-36 receptor and has biological effects on immune cells similar to IL-36 receptor antagonist. Proc. Natl. Acad. Sci. USA 2012, 109, 3001–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blumberg, H.; Dinh, H.; Trueblood, E.S.; Pretorius, J.; Kugler, D.; Weng, N.; Kanaly, S.T.; Towne, J.E.; Willis, C.R.; Kuechle, M.K.; et al. Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J. Exp. Med. 2007, 204, 2603–2614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; McDonnell, P.C.; Lehr, R.; Tierney, L.; Tzimas, M.N.; Griswold, D.E.; Capper, E.A.; Tal-Singer, R.; Wells, G.I.; Doyle, M.L.; et al. Identification and initial characterization of four novel members of the interleukin-1 family. J. Biol. Chem. 2000, 275, 10308–10314. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Liu, J.; Gao, R.; Hu, Y.; Lu, L.; Liu, C.; Ai, L.; Pan, J.; Tian, L.; Fan, J. Interleukin-36γ aggravates macrophage foam cell formation and atherosclerosis progression in ApoE knockout mice. Cytokine 2021, 146, 155630. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Ling, X.Y.; Chen, D.L.; Zhang, X.Q.; Qiu, C.M. Interleukin-36 receptor antagonist attenuates atherosclerosis development by inhibiting NLRP3 inflammasome. J. Cell Physiol. 2020, 235, 9992–9996. [Google Scholar] [CrossRef]
- Kazemian, S.; Ahmadi, R.; Rafiei, A.; Azadegan-Dehkordi, F.; Khaledifar, A.; Abdollahpour-Alitappeh, M.; Bagheri, N. The Serum Levels of IL-36 in Patients with Coronary Artery Disease and Their Correlation with the Serum Levels of IL-32, IL-6, TNF-α, and Oxidative Stress. Int. Arch. Allergy Immunol. 2022, 183, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cytokine Name | Alternative Name | Subfamily | Main Role | References |
|---|---|---|---|---|
| IL-1α | IL-1F1 | IL-1 | Pro-inflammatory | [52,53] |
| IL-1β | IL-1F2 | IL-1 | Pro-inflammatory | [52,53] |
| IL-33 | IL-1F11 | IL-1 | Pro-inflammatory, type 2 immunity | [52,54,55] |
| IL-Ra | IL-1F3 | IL-1 | Antagonist of IL-1α and IL-1β | [53,56] |
| IL-18 | IL-1F4 | IL-18 | Pro-inflammatory, Type 1 immunity | [57,58] |
| IL-37 | IL-1F7 | IL-18 | Anti-inflammatory | [59,60] |
| IL-36Rα | IL-1F5 | IL-36 | Antagonist of IL-36α, β, γ | [61,62] |
| IL-36 α, β, γ | IL-1F6, 8, 9 | IL-36 | Pro-inflammatory | [61,62] |
| IL-38 | IL-F10 | IL-36 | Anti-inflammatory | [63] |
| Receptor | Alternative Name | Role | Ligands | References |
|---|---|---|---|---|
| IL-1R1 | IL-1RI | IL-1 binding receptor | IL-1 α, IL-1β, and IL-1Ra | [49,67,69] |
| IL-1R2 | IL-1RII | IL-1 decoy receptor | IL-1β | [49,67,69] |
| IL-1R3 | IL-1RAcP | IL-1 co-receptor | IL-1 α, IL-1β, IL-33, IL-36 | [49,67,69] |
| IL-1R4 | ST2/T1 | IL-33 binding receptor | IL-33 | [49,67,69] |
| IL-1R5 | IL-18Rα/IL-1Rrp1 | IL-18 binding receptor | IL-18 | [49,67,69] |
| IL-1R6 | IL-36R/IL-1Rrp2 | IL-36 binding receptor | IL-36 | [49,67,69] |
| IL-1R7 | IL-18Rβ | IL-18 co-receptor | IL-18 | [49,67,69] |
| IL-1R8 | TIR8/SIGIRR | IL-37 co-receptor/decoy receptor | IL-37 | [69,70] |
| IL-1R9 | APL/TIGIRR-2/IL-1RAPL | IL-38 co-receptor | IL-38 | [69,71] |
| IL-1R10 | TIGIRR, TIGIRR-1/IL-1RAPL2 | Orphan receptor | - | [69] |
| Cytokine | Animal Studies | Human Studies | References |
|---|---|---|---|
| IL-1α |
|
| [128,129,152] |
| IL-1β |
|
| [126,127,128,134,135,145,147,148,149] |
| IL-1Ra |
|
| [130,131,132,150,151] |
| IL-18 |
|
| [179,183,184,185,189,202,203,204,206] |
| IL-37 |
|
| [191,193,195,196,197,198,199,200] |
| IL-36 subfamily |
|
| [219,220,221] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González, L.; Rivera, K.; Andia, M.E.; Martínez Rodriguez, G. The IL-1 Family and Its Role in Atherosclerosis. Int. J. Mol. Sci. 2023, 24, 17. https://doi.org/10.3390/ijms24010017
González L, Rivera K, Andia ME, Martínez Rodriguez G. The IL-1 Family and Its Role in Atherosclerosis. International Journal of Molecular Sciences. 2023; 24(1):17. https://doi.org/10.3390/ijms24010017
Chicago/Turabian StyleGonzález, Leticia, Katherine Rivera, Marcelo E. Andia, and Gonzalo Martínez Rodriguez. 2023. "The IL-1 Family and Its Role in Atherosclerosis" International Journal of Molecular Sciences 24, no. 1: 17. https://doi.org/10.3390/ijms24010017
APA StyleGonzález, L., Rivera, K., Andia, M. E., & Martínez Rodriguez, G. (2023). The IL-1 Family and Its Role in Atherosclerosis. International Journal of Molecular Sciences, 24(1), 17. https://doi.org/10.3390/ijms24010017