1. Introduction
Corneal endothelial cells (CECs), embryologically originating from cranial neural crest cells (NCCs), form a single layer of hexagonal cells. CECs are responsible for barrier function between the corneal stroma and aqueous humor as they pump fluid out of the stroma to prevent edematous haze and actively transport protein and nutrients through the endothelial layer. CECs play a crucial role in controlling corneal transparency via tight-junction protein, zona occludens-1 (ZO-1), and sodium-potassium pump Na
+/K
+ ATPase (ATP1A1) [
1,
2,
3,
4].
CECs show no signs of regeneration in vivo because of contact inhibition at the G1 phase of the cell cycle [
5,
6]. Due to their lack of regeneration capability, pathological conditions such as disease and injury can cause cell loss in the corneal endothelium and consequent dysfunction of both pump and barrier. The CEC density of adult human cornea is approximately 2500 cells/mm
2, and the cell density decreases at a rate of 0.6% per year. [
5]. Corneal endothelial dystrophies and surgery-related trauma are the primary factors contributing to a decrease in endothelial cell density (ECD). If ECD decreases below 500 cells/mm
2, the cornea deteriorates physiologically, leading to bullous keratopathy and corneal endothelial dysfunction (CED). Transplantation of a donor cornea containing healthy CECs is the only option to reverse these conditions. However, corneal graft rejection and a lack of suitable donor tissue have been primary obstacles to preventing blindness in patients with CED. [
7,
8]
Previously, several attempts have been made to expand human CECs from human donor corneas in vitro [
9]; however, robust expansion of CECs remains a huge challenge due to the limited regenerative capacity of donor CECs [
9,
10]. In addition, cells from donors with older ages exhibit high heterogeneity, grow more slowly, and show senescence features compared with those from young donors [
11,
12]. Due to recent advances in induced pluripotent stem cell (iPSC) technology, however, it is now possible to generate an unlimited supply of CECs from iPSCs, and several groups have generated CECs from human iPSCs [
13,
14,
15,
16,
17,
18]. Zhao et al. generated CECs from human-fibroblast-derived iPSCs using small molecules associated with Wnt, TGF-β/BMP, and Rho-associated protein kinase (ROCK) signaling [
16]. Ali et al. generated CECs from peripheral blood mononuclear cell (PBMC)-originating iPSCs through NCCs by inhibiting Wnt and TGF-β/BMP signaling (dual SMAD and Wnt inhibition) [
13]. Cryopreserved and thawed corneal endothelial-like cells differentiated from iPSCs were transplanted into a monkey with corneal edema, showing improvement in the edema [
17].
We developed a small-molecule-based method for induction of CECs from fibroblast-originated iPSCs and further proposed a highly efficient method to induce CECs from NCCs by replacing the coating material and ROCK inhibitor. We also evaluated the efficacy and safety of iPSC-derived CEC transplantation in a CED rabbit model.
3. Discussion
Corneal transplantation using healthy CECs is the only option to treat CED. However, many issues remain to be resolved, such as a lack of donor tissue, allograft rejection, primary graft failure, and continual loss of ECD. Expanding the number of primary human CECs isolated from donor tissues also remains a challenge because of the limited regenerative capacity of CECs, due to their poor mitotic activity [
20]. Therefore, there has been considerable interest in the development of in vitro expandable cell sources for endothelial regeneration.
In the present study, we describe the derivation of CECs from fibroblast-originated iPSCs, using a stepwise differentiation protocol based on small-molecule-based guidance. We demonstrated that iPSCs differentiate into NCCs, and NCCs can be converted into CECs, with hexagonal, tightly packed morphologies, expressing established markers of mature CECs. Furthermore, using in vivo experiments, we demonstrated significant corneal clarity restoration after iPSC-derived CEC transplantation in a CED animal model.
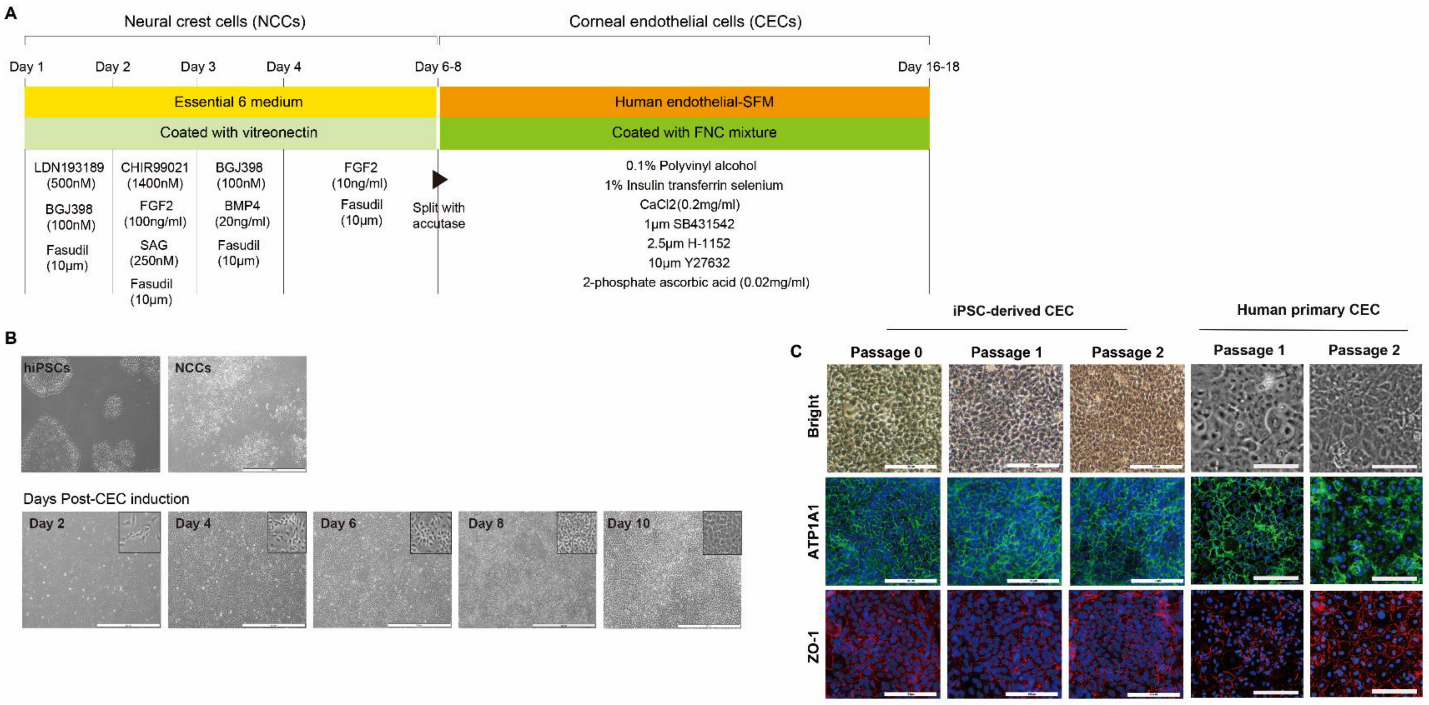
The corneal endothelium is derived from cranial NCCs in vivo [
21,
22]. The neural crest represents an embryonic migratory cell population that is able to distinguish itself into a variety of cell types. NCCs can differentiate into specific cell types, depending on their location, once they reach their target tissues by migrating throughout the body [
23]. First, we aimed to differentiate iPSCs into NCCs. To generate iPSC-derived NCCs, we took advantage of a recently published 6-day protocol that exploits lineage-specific differentiation of human PSCs to multipotent NCCs using small-molecule compounds: LDN193189 (selective inhibitor of the BMP type I receptors ALK-2 and ALK-3), BGJ398 (potent and selective FGFR inhibitor for FGFR1/2/3), fasudil (selective ROCK inhibitor), and CHIR99021 (a potent inhibitor of GSK3, inhibiting GSK3β, GSK3α, and a Wnt activator) [
19,
24].
To subsequently produce CECs from iPSC-derived NCCs, we took advantage of an additional 10-day cornea endothelial induction protocol using three small-molecule compounds: SB431542, H-1152, and Y27632. During eye development, separation of the lens from the surface ectoderm is essential [
25]. This separation leads to reduced signaling activity of growth factors, such as FGF2 and TGF-β, thus making it necessary to suppress the TGF-β signaling pathway with SB431542 and to remove FGF2 from the culture medium during CEC induction from NCCs. Okumura et al. demonstrated that inhibition of ROCK signaling with the small molecule Y27632 inhibited CEC apoptosis, increased CEC proliferation, and enhanced endothelial wound healing in vitro and in vivo [
25,
26]. H-1152, a more potent ROCK inhibitor, exhibited a larger stimulatory effect on CEC migration, proliferation, and wound healing, compared with Y27632. Thus, we used a combination of H-1152 and Y27632 to block ROCK activity more efficiently during CEC induction.
As shown, we used iPSCs to generate CECs by modifying a previously published procedure. Several groups have generated CECs from iPSCs through NCC [
13,
14,
15,
16]. Fukuta et al. established a 19-day protocol to generate CECs from human primary-fibroblast-derived iPSCs via NCC under CEC-conditioned, chemically defined media, with addition of Y27632 on the first day of CEC induction [
14]. The combination of CHIR99021 and SB431542 with a minimum growth factor (insulin) efficiently induced NCCs from PSCs at a rate of 70–80%. The iPSC-derived CECs exhibited polygonal morphology, expressed the endothelial marker ZO-1, and showed increased expression of COL4A1 and COL8A1 [
14]. Zhao et al. generated CECs from human foreskin-fibroblast-derived iPSCs using a small-molecule-based protocol, in which the simultaneous inhibition of TGF-β, BMP, and Wnt signaling resulted in elevated expression of early eye field transcription factors (PAX6, LHX2); an increase in Wnt signaling in the ocular niche environment is required for the formation of ocular NCCs [
16]. First, they derived eye field stem cells (EFSCs) from PSCs by the addition of SB431542, LDN193189, and IWP2 (for 7 days); they then directed EFSCs toward ocular NCCs with the addition of CHIR99021 (7 days) and subsequently differentiated ocular NCCs to CECs via the addition of SB431542 and H-1152 (7 days; 21 days in total) [
16]. Ali et al. generated CECs from PBMC-originated PSCs through NCCs using dual SMAD inhibitor medium containing Noggin and SB431542 for 2 days, followed by corneal medium containing Dickkopf related protein-2 (DKK-2, a potent Wnt inhibitor) for 13 days (totally 20 days) [
13]. Consistent with the aforementioned studies, to generate CECs from iPSCs through NCCs, NCCs were treated with SB431542, H-1152, and Y27632 for 10 days in our study. Wagoner et al. generated CECs from adult dermal-fibroblast-derived iPSCs through NCCs. NCCs were differentiated from iPSCs by adding CHIR99021 and SB431542 for 5 to 16 days, and CECs were differentiated from NCCs by adding B27, PDGF-BB, and DKK-2 to endothelial medium for 25 to 96 days [
15]. Wagoner et al. also found that the iPSC–CEC differentiation time may be reduced by restricting NCC induction to 3–8 days, demonstrating that use of early-stage NCCs can generate CECs in only 25 days [
15]. Similarly, a recently published protocol was used to produce CECs directly from iPSCs by omitting NCC differentiation. These cells expressed characteristic molecules found in CECs, such as N-cadherin and PITX2 [
17].
For induction of CECs from NCCs, we adopted a 10-day protocol with modifications [
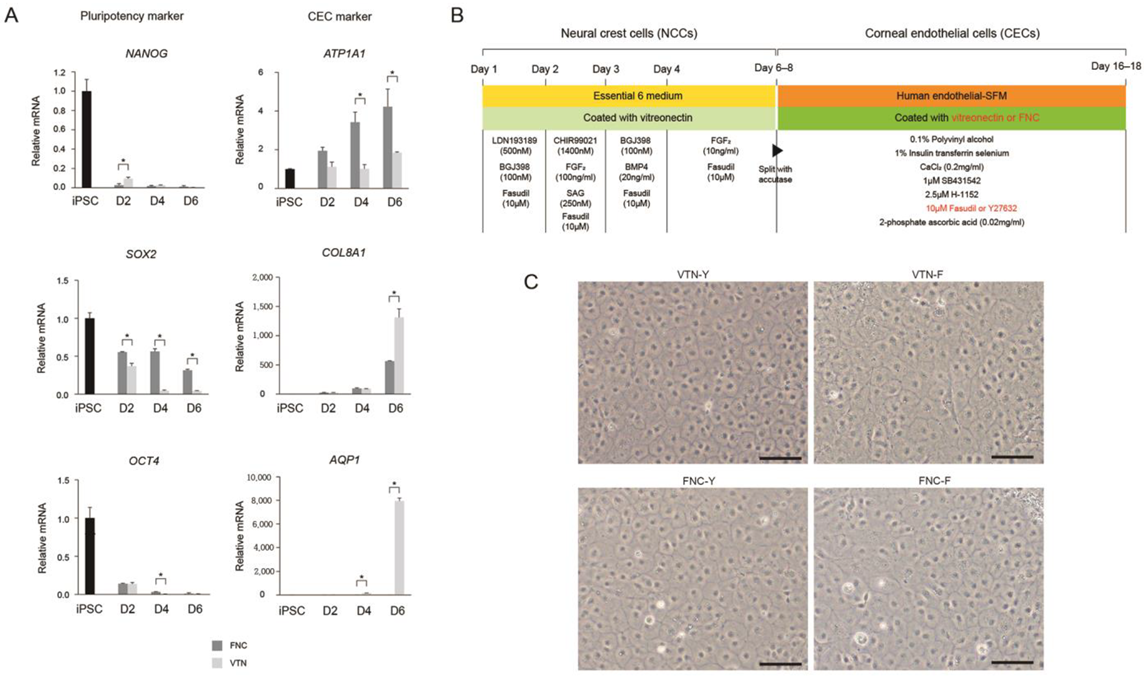
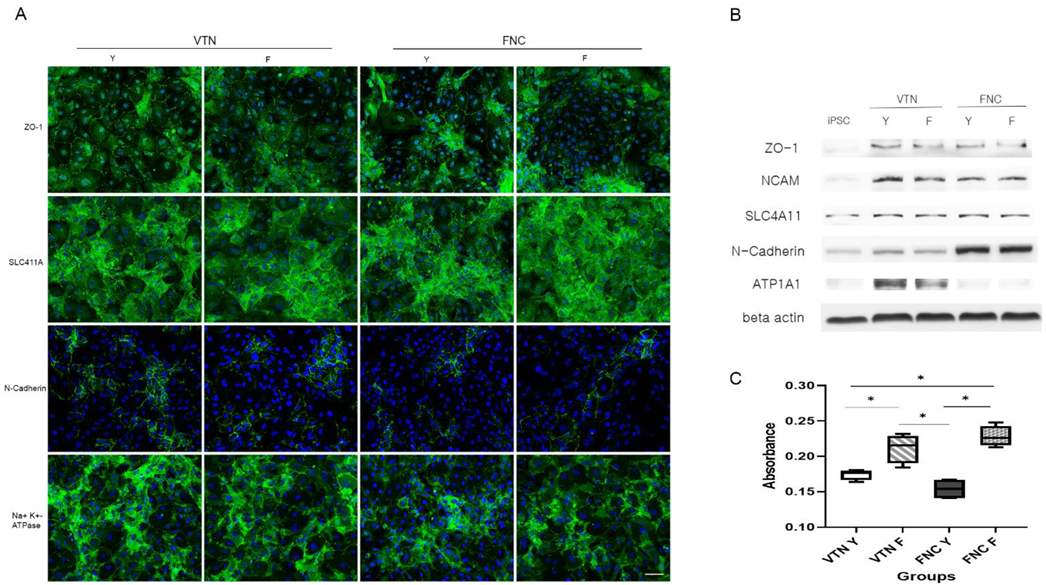
13]; there was no difference between expression levels of CE-associated markers at days 10 and 20 (data not shown). High expression of CE markers, and more importantly, lack of NCC marker expression, strongly encourages differentiation of NCCs into a CEC lineage. It is worth noting that we developed the 17-day protocol for the generation of CECs from iPSCs using a small-molecule-based protocol. Moreover, the combination of the VTN coating material and fasudil, instead of FNC mixture and Y27632 afforded the best results in terms of CEC differentiation’s in vitro and in vivo efficacy, showing improvements in corneal clarity.
Since fetal bovine serum (FBS) can cause an acute immune response due to any xeno-contamination after transplantation, we replaced FBS with polyvinyl alcohol (PVA). FBS used in cell culture may include contaminants which result in the presence of xeno antigens that cause graft versus host response [
27]. Contamination may also result in variations between batches [
28]. Based upon our results, we suggest that PVA can be a replacement for FBS.
Further studies are necessary to elucidate the functional characteristics and therapeutic potential of iPSC-derived CECs in clinical trials. Moreover, precise measurement of the population purity of a sub-lineage cell type must be determined with cell sorting based on the expression of stable and cell-type-specific markers, although an absolute marker for CECs is yet to be discovered. Functional studies should be directed to testing iPSC-derived CECs for their ability to pump water and metabolites and to survive the energy demands of ATP-dependent pump activities [
29,
30].
4. Materials and Methods
4.1. Ethics
This study was approved by the Institutional Review Board (IRB: 2020-1373) at Asan Medical Center. Experiments on live vertebrates were conducted in strict accordance with the relevant national and international guidelines regarding animal handling as mandated by the Institutional Animal Care and Use Committee (IACUC) of the University of Ulsan College of Medicine (Seoul, Republic of Korea). The committee reviewed and approved our animal study protocol (2019-12-184).
4.2. Generation of Induced Pluripotent Stem Cells
Human fibroblasts were obtained from a 7-year-old male patient and were transduced with the CytoTune-iPS Reprogramming Kit (Life Technologies, Grand Island, NY, USA) according to the manufacturer’s instructions. Fibroblasts (2 × 10
4 cells/cm2) were plated into 24-well culture plates, coated with gelatin containing Stempro 34 medium (Life Technologies), and supplemented with L-ascorbic acid (S4641, Sigma-Aldrich, St. Louis, MO, USA), stem cell factor (Peprotech, Rocky Hill, NJ, USA), and bFGF (Peprotech) 7 days before transduction. Transduced cells were seeded onto mouse embryonic fibroblast (MEF)-coated plates 1 week after transduction. After 3 weeks, colonies were selected and expanded in stem MACS medium (Miltenyi Biotec, Bergisch Gladbach, Germany) on plates coated with vitronectin (VTN; Life Technologies) [
19].
4.3. Teratoma Formation
Experiments for teratoma formation were performed by injection of human iPSC or iPSC-derived CECs [1 × 10
6 iPSC or iPSC-derived CECs in 150 μL of culture medium (StemMACS or CEC differentiation medium)] into the bilateral femoral region of 7-week-old NOD-SCID Gamma mice (n = 2, NSG, The Jackson Laboratory, Bar Harbor, ME, USA) using a 1 mL syringe (Korea Vaccine Co., Seoul, Republic of Korea) [
19]. After 2 months, the mice were euthanized and teratomas isolated, sectioned, and histologically characterized using H & E staining.
4.4. Neural Crest Cell Differentiation
For NCC differentiation, iPSCs were plated on a VTN-coated surface. After iPSCs reached approximately 30–40% confluence, the medium was switched to a neural crest induction E6 medium (Life Technologies) supplemented with 500 nM LDN19318 (Selleckchem, Houston, TX, USA), 100 nM BGJ398 (Selleckchem), and 10 μM fasudil (Adooq, Irvine, CA, USA). One day after differentiation, the medium was switched to E6 medium, supplemented with 1400 nM CHIR99021 (Peprotech), 100 ng/mL FGF2 (Peprotech), 250 nM SAG (Selleckchem), and 10 μM fasudil. The following day (day two), the medium was switched again to E6 medium supplemented with 100 nM BGJ398, 20 ng/mL BMP4 (Peprotech), and 10 μM fasudil. On day three, the medium was switched to E6 medium, supplemented with 10 ng/mL FGF2 and 10 μM fasudil. This medium was replaced each day for four days.
4.5. Corneal Endothelial Cell Differentiation
For CEC differentiation on day seven, iPSC-derived NCCs were detached using accutase (Life Technologies) and seeded on 35-mm plates coated with FNC mixture (United States Biological, Salem, MA, USA). The medium was switched to human CEC induction medium consisting of human endothelial-SFM supplemented with 0.1% polyvinyl alcohol (PVA; Sigma-Aldrich), 1% insulin-transferrin-selenium (Life Technologies), 0.2 mg/mL CaCl2 (Sigma-Aldrich), 0.02 mg/mL 2-phosphate ascorbic acid, 1 μM SB431542 (Selleckchem), 2.5 μM ROCK inhibitor H-1152 (Tocris, Abington, UK), and 10 μM Y27632 (Sigma-Aldrich) for an additional 10 days. The medium was changed every other day for those 10 days. Differentiated cells were passaged when they reached approximately 80% confluency.
To compare the effects of coating materials between the FNC mixture and VTN on CEC differentiation, iPSC-derived NCCs were split with accutase and seeded on a 35-mm plate coated with VTN instead of the FNC mixture. Quantitative real-time RT-PCR (qRT-PCR) measured concentrations of pluripotent markers (NANOG, OCT4, and SOX2), and CEC-associated markers (ATP1A1, COL8A1, and AQP1) at different time points during differentiation of iPSCs into CECs (iPSC and at 2-, 4-, and 6-days post-CEC induction). Furthermore, to compare the effects of two ROCK inhibitors (Y27632 versus fasudil) on CEC differentiation, iPSC-derived NCCs were split and seeded on 35-mm plates coated with the FNC mixture or VTN. They were differentiated with human endothelial-SFM, supplemented with 0.1% PVA, 1% insulin-transferrin-selenium, 0.2 mg/mL CaCl2, 0.02 mg/mL 2-phosphate ascorbic acid, 1 μM SB431542, 2.5 μM ROCK inhibitor H-1152, and fasudil instead of Y27632. CEC markers at 10 days post-CEC induction were measured via immunocytochemistry for ZO-1, SLC4A11, N-Cadherin, and ATP1A1, and via Western blot analysis for ZO-1, NCAM, SLC4A11, N-Cadherin, and ATP1A1.
4.6. Production of iPSC (ZsGreen)-Derived Corneal Endothelial Cells
iPSCs were transduced with Lentivirus (Vector pHIV-ZsGreen carrying the ZsGreen gene). The lentiviral particles were produced by co-transfecting the lentiviral vector and the packaging plasmids (psPAX2 and VSV-G envelope pMD2.G, addgene) into HEK293FT cells. HEK293FT cells were seeded at 90% confluency in P60 culture dish one day before transfection. On the day of transfection, pHIV-ZsGreen, psPAX2, and VSV-G envelope pMD2.G plasmids were mixed in a medium at a ratio of 2:2:1 and treated with Lipofectamine TM Transfection Reagent (Invitrogen, Carlsbad, CA, USA). The lentiviral-particle-containing supernatant was collected 2 days after transfection and concentrated using Lenti-X™ Concentrator (Takara, Kyoto, Japan) according to the manufacturer’s protocol. iPSCs were seeded in VTN-coated 12-well plates at 2 × 104 cells. The next day, the lentiviral concentrate was diluted to 0.1% in culture medium and treated to iPSCs twice at one-day intervals. Expression of ZsGreen in iPSCs was confirmed by fluorescence microscopy 5 days after transduction.
4.7. Quantitative Real-Time RT-PCR
Total RNA from iPSCs, iPSC-derived NCCs, and iPSC-derived CECs was extracted using TRIzol reagent (Invitrogen). Complementary DNA was synthesized using a kit (Superscript III; Invitrogen) and qRT-PCR performed using the Power SYBR Green PCR Master Mix (Applied Biosystems, CA, USA) with a Step One ABI Real-Time PCR System (Applied Biosystems). Pluripotent markers (
NANOG,
OCT4, and
SOX2), neural crest markers (
p75NTR and
PAX3), and CEC markers (
ATP1A1,
COL8A1, and
AQP1) across multiple time points during differentiation of iPSCs into CECs (iPSC, NCC, and at 2, 4, 6, 8, and 10 days post-CEC induction) were measured. mRNA levels were normalized to GAPDH; primer sequences are listed in
Table S1.
4.8. CCK-8 Assay
Cells from each group were seeded in a 96-well culture plate at a density of 104 cells/well in 100 μL of culture medium and then incubated at 37 °C for 24 h. Then, we added 10 μL of CCK-8 solution (Dojindo, Munich, Germany) to each well of the plate and incubated for 2 h in the incubator. The absorbance was measured at 450 nm using a microplate reader (Spectra Max 190, Molecular Devices, San Jose, CA, USA).
4.9. Immunocytochemical Staining
Human primary CECs or iPSC-derived CECs were fixed with 4% paraformaldehyde overnight at 4 °C, washed with PBS/0.05% Tween 20 (PBST) buffer, permeabilized in 0.5% Triton X-100, and blocked in PBST containing 1% bovine serum albumin (Sigma-Aldrich). Samples were incubated with primary antibodies against human anti-ZO-1 (1:250; Life Technologies), Na+/K+ ATPase α1 (1:200, Santa Cruz Biotechnology, Dallas, TX, USA), SLC4A11 (1:250, Novus, Centennial, CO, USA), and N-cadherin (1:200, Cell signaling technology, Danvers, MA, USA) overnight at 4 °C, followed by goat anti-mouse IgG Alexa Fluor 488-conjugated and donkey anti-rabbit IgG Alexa Fluor 647-conjugated secondary antibodies (1:500; Abcam, Cambridge, UK). Nuclei were counterstained with 4’, 6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich) for 10 min and fluorescence signals detected with either a fluorescence microscope (EVOS, Life Technologies) or a laser-scanning confocal microscope (Olympus, Tokyo, Japan).
4.10. Stability of iPSC-Derived Corneal Endothelial-like Cells
To determine whether iPSC-CECs maintained the function of CECs after freeze and thaw cycles, ATP1A1, COL8A1, and AQP1 expression in iPSC-derived CECs was measured before and after the cycles using qRT-PCR. Additionally, to determine whether iPSC-derived CECs returned to iPSCs, iPSC-derived CECs were cultured on human endothelial-SFM for 2 days and the medium switched to Stem MACS iPS-Brew XF medium (Milteny Biotec) for 6 days. Pluripotent marker (NANOG, OCT4, and SOX2) expression levels were quantified using qRT-PCR 6 days after the change to iPS medium and compared with the levels in iPSCs and iPSC-derived CECs. mRNA levels were normalized to GAPDH.
4.11. Cell Transplantation Experiments
New Zealand white rabbits (n = 20, 1.8–2.2 kg) were placed in standard rabbit cages and housed under good environmental conditions. Room temperature was maintained at 24 °C with a 12 h light/dark cycle. After 7 days of acclimation, animals underwent the Descemet’s membrane stripping and cell injection procedures, performed by an experienced physician (HL). After intramuscular injections of 5 mg/kg tiletamine/zolazepam (Zoletil 50, Virbac Korea, Seoul, Republic of Korea) and 2 mg/kg xylazine hydrochloride (Rompun, Bayer Korea, Seoul, Republic of Korea), the anterior chamber was filled with a viscoelastic through a paracentesis incision, and the Descemet’s membrane was scored and stripped off from the posterior stroma in a circular pattern under the area of the epithelial marking using a reverse Sinskey hook. The diameter of the stripped membrane was measured for all the corneas of the experimental group and the value was 8.94 ± 0.18 mm. The viscoelastic was extensively removed by irrigating the anterior chamber using balanced salted solution (BSS). Then, a direct injection of cells was performed into the anterior chamber.
For the cell transplantation group, we injected iPSC-derived CECs (1 × 106 iPSC-CECs in 150 μL of PBS supplemented with 100 μM of Y27632) into the anterior chamber through a paracentesis incision and followed for up to 3 weeks (n = 6). We also injected iPSC-CECs or iPSC (ZsGreen)-CECs, produced via a modified protocol (VTN with fasudil) (1 × 106 iPSC-CECs in 150 μL of PBS supplemented with 100 μM of fasudil) into the anterior chamber and followed for up to 3 weeks (total n = 8; 4 for each iPSC-CECs). Two rabbits injected with iPSC-CECs were followed for up to 20 weeks. For the non-transplantation control group (control; n = 6), rabbits were evaluated for up to 3 weeks after Descemet’s membrane stripping. We assessed corneal opacity on a scale of 0–4 (0 = none; 1 = mild turbidity, iris texture evident; 2 = moderate turbidity, iris texture unclear; 3 = severe turbidity, pupil faint; and 4 = severe turbidity, pupil not visible) using a slit-lamp microscope, before and 3 weeks after transplantation. Whole rabbit corneas were harvested 3 weeks after cell transplantation. Transplanted iPSC (ZsGreen)-derived CECs expressing green fluorescent protein were observed using a fluorescence microscope (EVOS) with fixed excitation laser intensity (488 nm) and identical exposure times. All procedures conformed to the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research (ARVO Animal Policy).
4.12. Histological Staining and Analysis
All retrieved corneal tissues were fixed in 3.7% formaldehyde and embedded in paraffin. The paraffin blocks were sectioned into 4 μm thick slices. Paraffin sections were stained with H & E to assess corneal endothelial cell failure in Descemet’s-membrane-stripped rabbits. Three weeks following cell transplantation, H & E staining and immunohistochemistry were performed. Rabbits were sacrificed, and paraffin-embedded whole cornea tissues were incubated for 16 h at 37 °C with primary antibodies against ZO-1, Na+/K+ ATPase, CD11b (1:100, Abcam), CD4 (1:1000, Novus), and CD8 (1:1000, Abcam). The secondary antibodies included the corresponding Alexa-488, -555, or -647 fluorescent-labeled antibodies (1:1000, Abcam). Cell nuclei were counterstained with DAPI. The sections were viewed using an LSM780 confocal microscope (Carl Zeiss Meditec, Jena, Germany).
4.13. Western Blot Analysis
iPSC-derived CECs were harvested and lysed in RIPA lysis buffer (EPIS BIO, Sejong-si, Republic of Korea) containing phosphatase and protease inhibitors (Sigma-Aldrich) according to the manufacturer’s protocol. Cell lysates were separated on 8% SDS–polyacrylamide gels (SDS–PAGE) and subsequently transferred to PVDF membranes (Sigma-Aldrich) using a Trans-Blot-Cell (Bio-Rad Laboratories Inc., Richmond, CA, USA) as previously described. Non-specific protein binding was blocked with 3% BSA (Sigma-Aldrich) in PBST. Membranes were incubated with antibodies against ZO-1 antibodies (1:1000), NCAM (1:200, R & D systems, Minneapolis, MN, USA), SLC4A11 (1:250, Novus), N-cadherin (1:200, Cell signaling technology), and β-actin (1:1000; Cell signaling technology) overnight. Membranes were washed and incubated with an anti-mouse secondary antibody conjugated to horseradish peroxidase (GeneTex, Hsinchu City, Taiwan) at a 1:10,000 dilution. Detection was performed with the Gel DocTM XR+ (Bio-Rad Laboratories Inc.) according to the manufacturer’s instructions.
4.14. Statistical Analysis
All experiments were performed on at least three independent biological samples, and data are presented as means ± standard deviation (SD). Statistical analysis was performed with GraphPad Prism 6.0 software (GraphPad Software, San Diego, CA, USA). Comparisons of three or more data sets were performed using a one-way or two-way ANOVA followed by Bonferroni’s multiple comparison tests. Two-group comparisons were made using two-tailed Student’s t-tests. A value of p < 0.05 was considered statistically significant.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






