1. Introduction
After an injury, many biological pathways become activated and respond to the harmful stimulus. Stem-cell-based therapy is an effective approach to facilitate wound healing and tissue regeneration, primarily via capitalizing on the paracrine activity of stem cells, such as the release of a variety of cytokines, chemokines, and growth factors that activate host cells, promote the homing of other cells to the site of injury and, thus, enhance the host’s intrinsic repair mechanisms [
1,
2,
3]. The beneficial effect of this cell therapy ultimately depends on the number of implanted cells at the target site, their viability, their engraftment into damaged tissue and their ability to promote tissue regeneration. Human Adipose-Derived Stem Cells (hADSC) represent an interesting stem cell candidate for tissue regeneration as they secrete bioactive molecules [
4], regulate the local immune response, and compared with stem cells harvested via bone marrow aspiration, hADSC from fat tissue are easily and abundantly available and carry a relatively lower donor site morbidity since they can be harvested using minimally invasive techniques (i.e., liposuction) [
5,
6]. The trophic and immunomodulatory activities exerted by hADSC would suggest that hADSC may serve as site-regulated ‘drugstores’ upon implantation [
7]. Nevertheless, the challenges associated with preserving stem cells in a culture and facilitating their engraftment in the damaged tissue remain [
8,
9]. Additionally, another challenge in clinical practice concerns the poor long-term maintenance of stem cell functions [
10,
11].
Recently, biomaterials have been proposed and developed to overcome these bottlenecks and support cell attachment and retention at the desired site [
3,
9]. Specifically, advances in biomaterial science combined with a more detailed knowledge about extracellular matrix (ECM) biology and the role of environmental factors in tissue formation and shaping have led to the development of biocompatible scaffolds tailored to provide appropriate structural support and, in some cases, biological and mechanical stimuli to promote tissue regeneration [
12,
13,
14]. Naturally derived polymeric materials, including polypeptides (e.g., collagen) and polysaccharides (e.g., hyaluronic acid), have been extensively explored as scaffolding materials [
15,
16]. Native ECM can also serve this function after decellularization [
8].
In addition to their modest mechanical strength, naturally derived polymers are typically digested over time by enzymes, following a complex and often not controllable degradation kinetics process [
8]. Differently, synthetic polymers can be manufactured with higher reproducibility, a wider range of mechanical properties and degradation kinetics [
17], and a surface amenable to chemical modification [
18,
19,
20]. Further, synthetic polymers are cheaper than natural polymers are and have longer storage time [
18]. The most widely used and clinically approved synthetic polymers are poly (ε-caprolactone) (PCL), poly (lactic acid) (PLA), poly (glycolic acid) (PGA), and its copolymers poly (lactic-co-glycolic acid) (PLGA) [
18]. Polyethylene Terephthalate (PET) is also a promising scaffolding biomaterial, which has been used, for example, to realize vascular grafts [
21,
22]. However, PET is a non-biodegradable linear polyester with notable mechanical properties and biological features, including biocompatibility, biostability, and the promotion of tissue growth [
22,
23]. Despite that, the high hydrophobicity of PET restricts its application in medicine [
24]. On the downside, synthetic polymers do not support cell attachment as natural polymers and may trigger a local immune response and toxicity [
25]. To overcome these limitations, hybrid scaffolds combining natural polymers (i.e., collagen, gelatin, or calcium phosphate) and synthetic polymers have been proposed to improve hydrophilicity, cell attachment, and biodegradability [
26]. The success of a scaffold largely depends on its proper integration at the implantation site and the realization of conditions supporting the implanted cells, while favoring the intimate interaction with resident and infiltrating cells [
8].
Moreover, based on the importance of stem cell transplantation in clinical practice, multiple laboratories have extensively tested different culturing conditions and microenvironments to favor cell nesting and replication, as well as guide cell differentiation. A spheroid-based culture system helps establish a physico-chemical microenvironment such as that in vivo by facilitating cell–cell and cell–matrix interactions, thus overcoming the limitations of conventional monolayer cell cultures [
27]. In addition, spheroids of mesenchymal stem cells (MSCs) tend to preserve their intrinsic phenotypic properties via cell–extracellular matrix interactions [
27]. However, spheroid cell culture systems are also characterized by having hypoxic and necrotic cores [
27]. Several strategies are available to support the arrangement of cells into spheroids, including the use of biocompatible hydrogel scaffolds made of alginate, fibrin, collagen, or hyaluronic acid [
28,
29,
30]; thin biocompatible films made of chitosan or graphene [
27,
31,
32]; micrometric particles whose incorporation into spheroids controls the cell culture condition [
33,
34,
35]. In particular, hydrogels have been extensively used to replicate the typical in vivo environment to improve cell viability, preserve stemness, and favor angiogenesis. On the other hand, biofilms have been shown to increase adhesion and proliferation, while preserving the differentiation potential. Finally, microparticles have been documented to control mechano-transduction mechanisms within the spheroid to eventually improve viability and proliferation [
27].
Here, a microfabricated network of poly(lactic-co-glycolic acid) filaments, organized as orthogonal warps and wefts in a polymeric fabric, is demonstrated and tested for its biocompatibility with hADSC. Taking advantage of the accurate control in size, shape, and surface proprieties offered by top-down fabrication approaches such as soft lithography [
36], three different configurations of PLGA fabrics were realized, with warp (or weft) pitch distances of 5, 10, and 20 μm, respectively. The resulting PLGA microstructured fabrics are biodegradable and mechanically flexible to favor cell adhesion and tissue integration [
36,
37]. A critical comparison with conventional substrates for in vitro cell culturing (2D Petri dish) and in vivo cell transplantation (2D collagen Type I layer) is also presented.
3. Discussion
The flexible microfilament network was designed to realize a physical support for stem cell transplantation and stimulate tissue regeneration post-injury. The presented top-down fabrication strategy can precisely tailor the geometrical and mechanical features of microfilament networks, where the width and thickness of the PLGA filaments, as well as their separation distance and arrangement, can be independently tuned during the fabrication process by simply using a different silicon master template.
The micrometric PLGA fibers are very thin and flexible and are arranged to form wide regular openings of 5 × 5, 10 × 10, and 20 × 20 µm. This unique architecture allows the fabrics to be extremely flexible and establish a close interaction with the stem cells, such as, for instance, those documented in a previous manuscript by the authors [
36]. The flexibility per se without the openings (i.e., geometry) would not be enough to establish such an intimate interaction with the cultured cells.
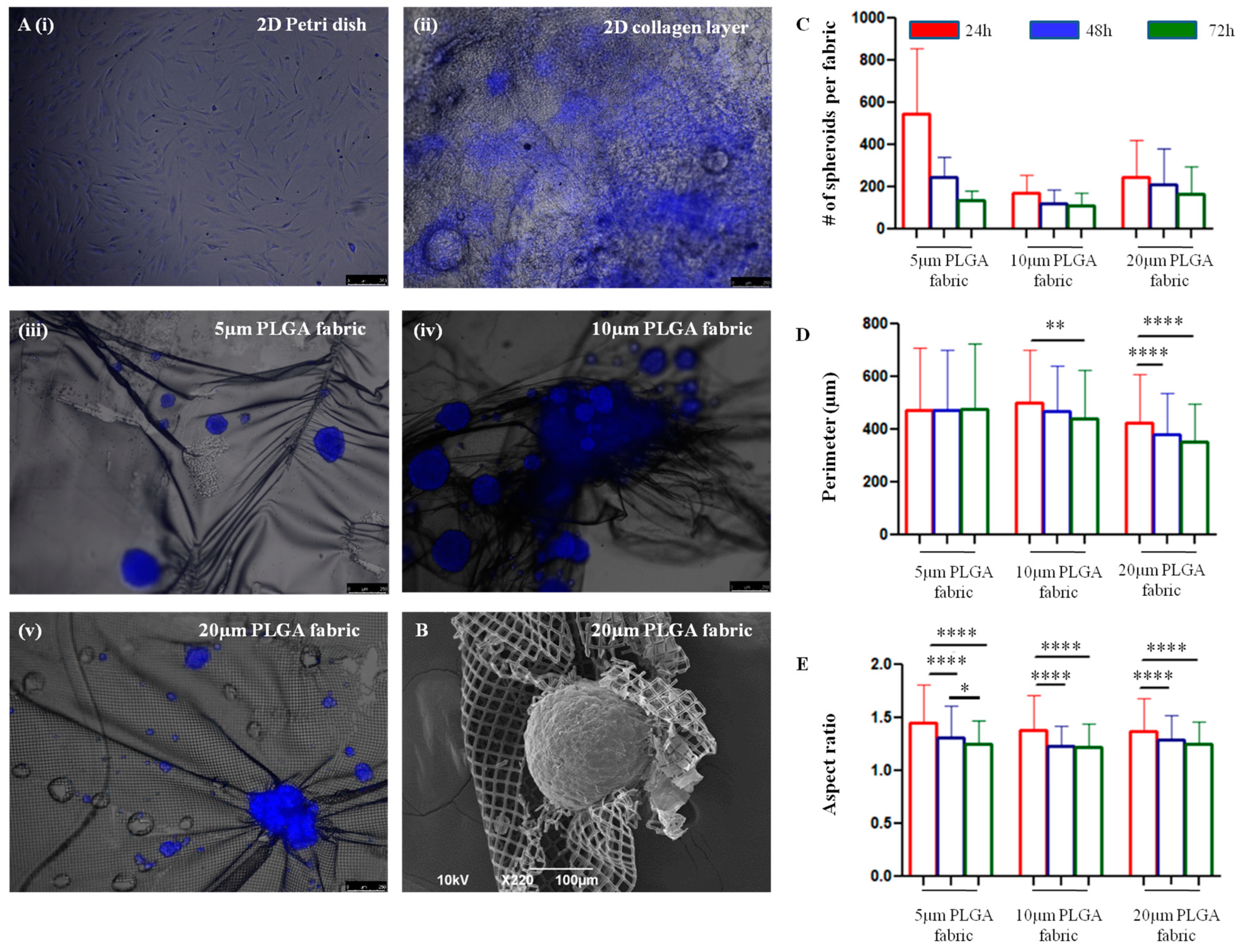
Interestingly, the microfilament networks allow hADSC to spontaneously form 3D spheroidal-like structures.
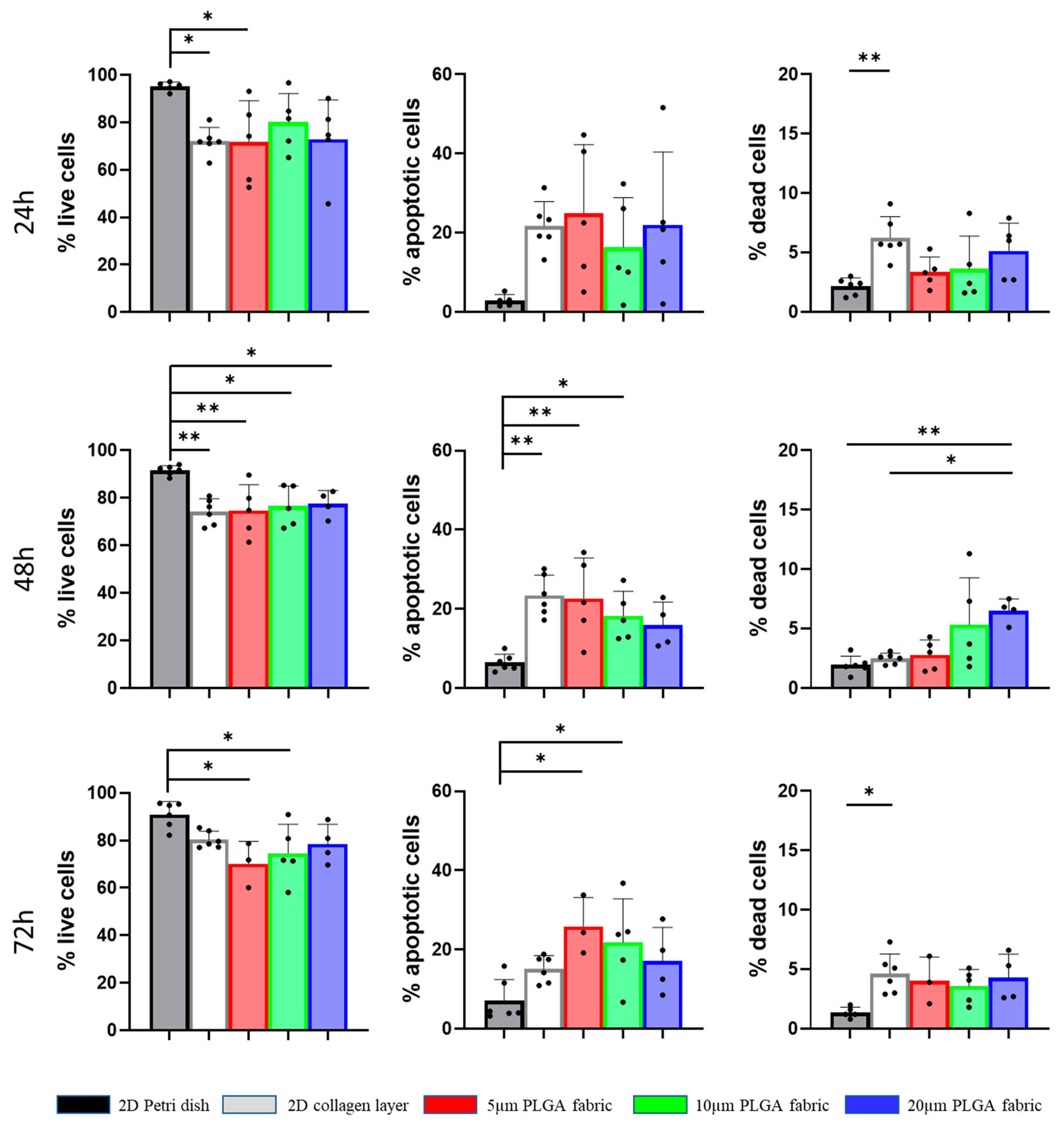
The relationship between the number of seeded cells and the number of spheroids formed over time on top of the three microfilament networks depends on different factors. The number of spheroids detected at 24 h was higher in the 5 μm PLGA fabric relative to those in the other configurations, probably because the smaller openings could more efficiently retain the stem cells and prevent them from dropping on the bottom of the dish. Anyway, at 72 h the number of spheroids anchored on top of 5 μm PLGA fabric was lower than the one observed on the 20 μm PLGA fabric, eventually suggesting a stronger attachment and interaction of the stem cell spheroids with the latter configuration. Hence, spheroids seemed to adhere less stably to the 5 μm PLGA fabric. Moreover, the perimeter did not change over time. Interestingly, spheroids tend to shrink their shape and become more spherical with an aspect ratio tending to one, as compared to the more elongated shape observed at 24 h. Over time, the survival of hADSC spheroids was not compromised, as compared to that of the 2D collagen type I layer, despite them being organized in a 3D assembly, which better represents natural tissue organization, but tend to decrease the supply of nutrients and the elimination of metabolic by-products [
27].
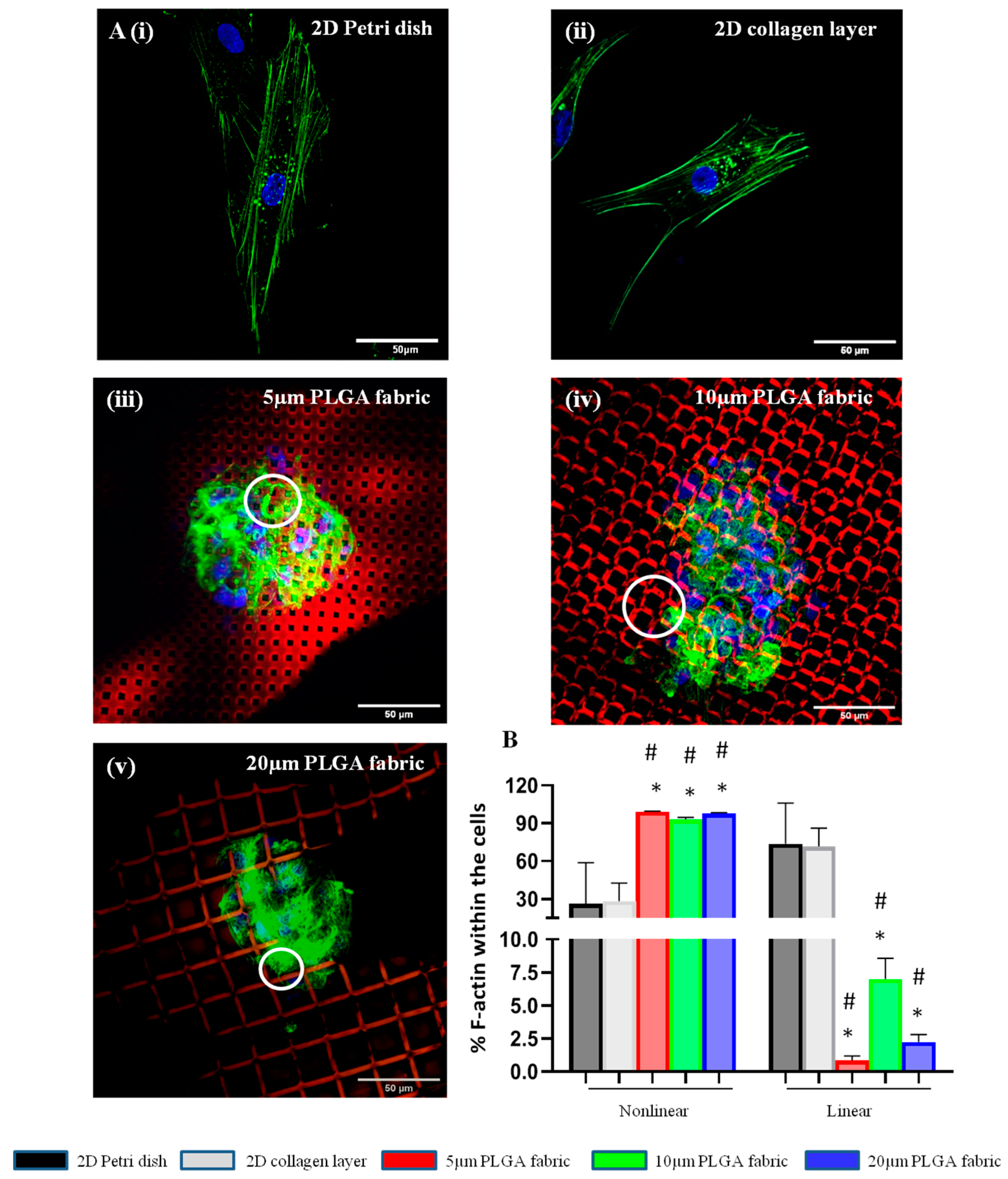
Spheroid formation is induced via cytoskeleton rearrangement. Indeed, actin filaments forming the cytoskeleton are responsible for mechanical support and for dictating the cells’ shape, which are often critical to their functions [
38]. In 2D culture systems, either a 2D Petri dish or a flat collagen layer, the actin cytoskeleton formed linear filaments within the body of the cells. Conversely, the peculiar surface geometry of the microfilament network imposes structural constraints on the cell, whereby the cytoskeleton forms and reorganizes to confer a 3D cell shape with multidirectional actin arrangements in nonlinear structures. Importantly, morphological variations in stem cells acquired through cell–substrate and cell–cell interactions could influence their paracrine activity [
39]. However, the influence of materials on cells’ behavior is intricate, and less is known on how cells should be handled to achieve the optimal cell function, and thereby, a therapeutic outcome in vivo [
40]. Anyway, based on our preliminary study investigating the secretome of hADSC cultured on different substrates, we found that the microfilament network geometry provides a unique microenvironment that is able to enhance hADSC paracrine secretion.
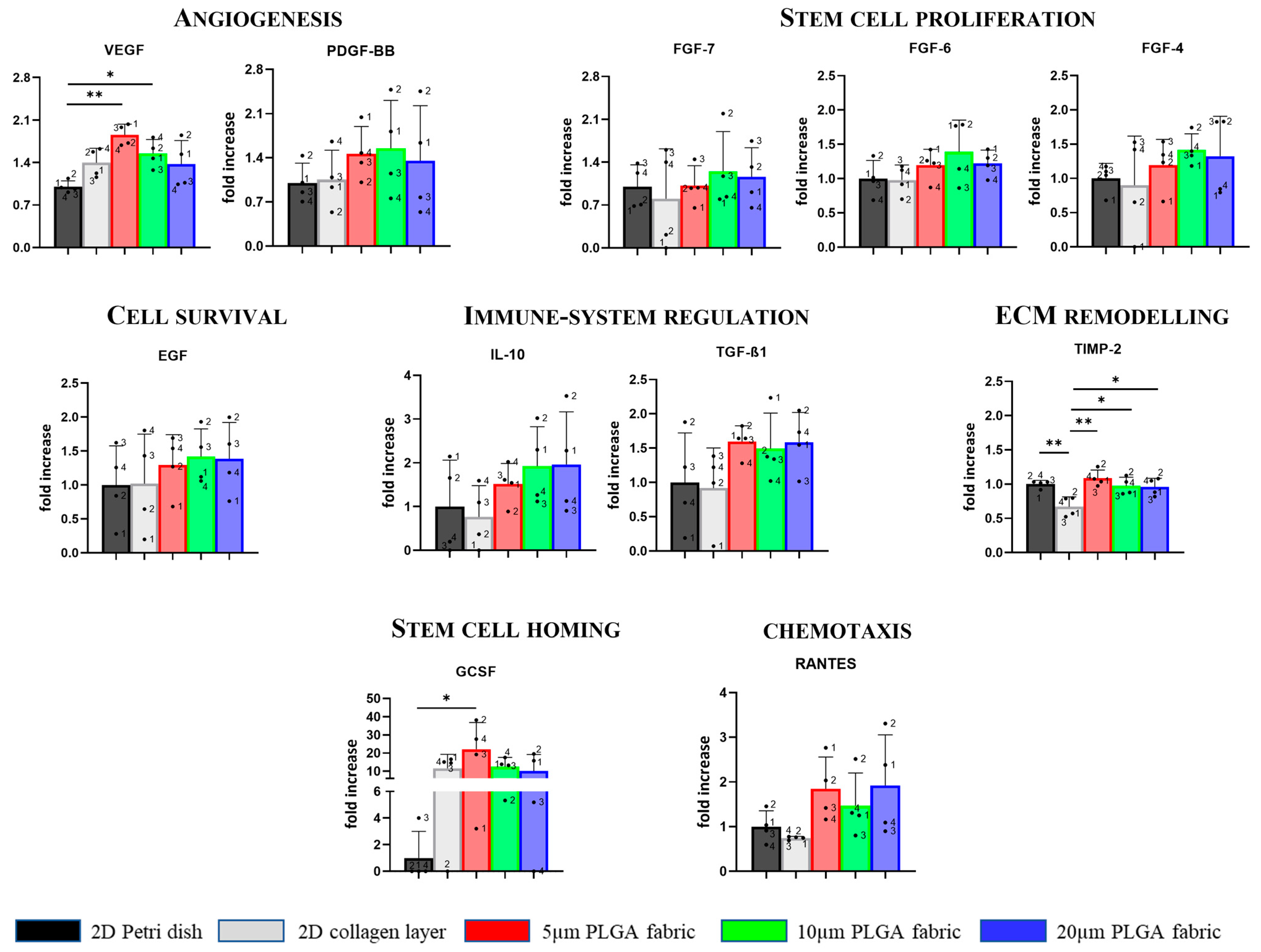
Notably, the highest rate of VEGF secretion was found for the 5 μm PLGA fabric, demonstrating its potential to promote angiogenesis [
41,
42]. Moreover, it has emerged that highest amount of GCSF was found for the 5 μm PLGA fabric involved in stem cell mobilization. Thus, all three microstructure networks facilitated, with statistically significant differences, the secretion level of TIMP-2 relative to that in the 2D collagen layer condition. Conversely, TIMP-2 secretion was not upregulated in the collagen scaffold, making collagen a less useful substrate in these application since TIMPs—as endogenous regulators of the metalloproteinase (MMPs), which play a pivotal role in all stages of wound healing via modifying the wound matrix, thereby allowing for cell migration and tissue remodeling—may be fundamental to wound resolution [
43,
44,
45]. The result that was just mentioned could be an another advantage of the proposed PLGA scaffolds.
Furthermore, the fabrics seemed to ameliorate—although in a not statistically significant fashion—the secretion of TFG-β1 (5 μm fabric), IL-10, RANTES (5 and 20 μm fabrics), participating in immune modulation and chemotaxis processes. A similar effect was observed for both FGF-family and EGF (10 μm fabric) that are implicated in stem cell proliferation and cell survival, respectively. Indeed there is a trend for the increased production of these factors on PLGA fabrics as compared to those of the collagen scaffold. Finally, despite the lack of other statistically significant differences with the collagen layer, the induction of proper spheroid formation using the PLGA microstructured fabrics presented—without any type of induction (i.e., active molecules)—is crucial since it has been reported that the 3D microenvironment (i.e., spheroids) offers a cellular niche that reproduces the native tissue, in which the cells maintain their natural stemness, as compared to those of the traditional two-dimensional (2D) in vitro models [
46]. The side-by-side comparison with conventional substrates for cell culturing (Petri dish) and collagen layers (which can be considered a sort of gold standard for cell implantation [
4]) showed that PLGA fabrics (5 and 10 µm) can significantly favor the secretion of the VEGF factor: the master regulator of vascular growth. Additionally, only a few other minor changes were observed between the PLGA fabrics and the Petri dish and collagen scaffold. Therefore, considering that plastic dish cannot be implanted and that PLGA fabrics can be further optimized and derivatized to expose specific cell adhesion molecules, these preliminary results are encouraging since all these processes are of particular interest in tissue regeneration and repair.
4. Materials and Methods
4.1. Materials
Polydimethylsiloxane (PDMS) (Sylgard 184) was purchased from Dow Corning (Midland, MI, USA). Poly-(vinyl alcohol) (PVA), poly-(lactic-co-glycolic) acid (PLGA) (50:50), Rhodamine B (RhB), and Acetonitrile (ACN) were obtained from Sigma Aldrich (St. Louis, MO, USA). Curcumin (CURC) was acquired from Alfa Aesar (Haverhill, MA, USA). Collagen Cell Carrier (CCC 10 × 10, geklebter Rand) was obtained from Viscofan Bioengineering (Weinheim, Germany). High-glucose Dulbecco’s modified Eagle’s Minimal Essential Medium (DMEM), heat-inactivated fetal bovine serum (FBS), Penicillin, Streptomycin, L-Glutamine solution, Dulbecco’s Phosphate-Buffered Saline (PBS), Trypsin-EDTA solution, Paraformaldehyde (PFA), and Tumor Necrosis Factor-α (TNF-α) were acquired from Sigma Aldrich (St. Louis, MO, USA). Human Recombinant Basic Fibroblast Growth Factor (bFGF) was obtained from Merck (Darmstadt, Germany), whereas Trypan Blue Stain (0,4%) was purchased from GIBCO (Invitrogen Corporation, Giuliano Milanese, Milan, Italy). 2′-[4-ethoxyphenyl]-5-[4-methyl-1-piperazinyl]-2,5′-bi-1H-benzimidazole trihydrochloridetrihydrate (Hoechst 33342) and 2 mg/mL Bovine Serum Albumin Standard Ampules were derived from ThermoFischer (Waltham, MA, USA). The Bicinchoninic Protein Assay kit (BCA) was obtained from Euroclone (Pero, Milan, Italy). Cytokine Array-Human Cytokine Antibody Array (Membrane, 80 targets), PE anti-CD44 antibody, FITC anti-90/Thyl antibody, and APC Anti-CD105 antibody were acquired from Abcam (Cambridge, UK). FITC Annexin V/Dead Cell Apoptosis kit, Alexa Fluor 488 Phalloidinwere sourced from Invitrogen (Waltham, MA, USA).
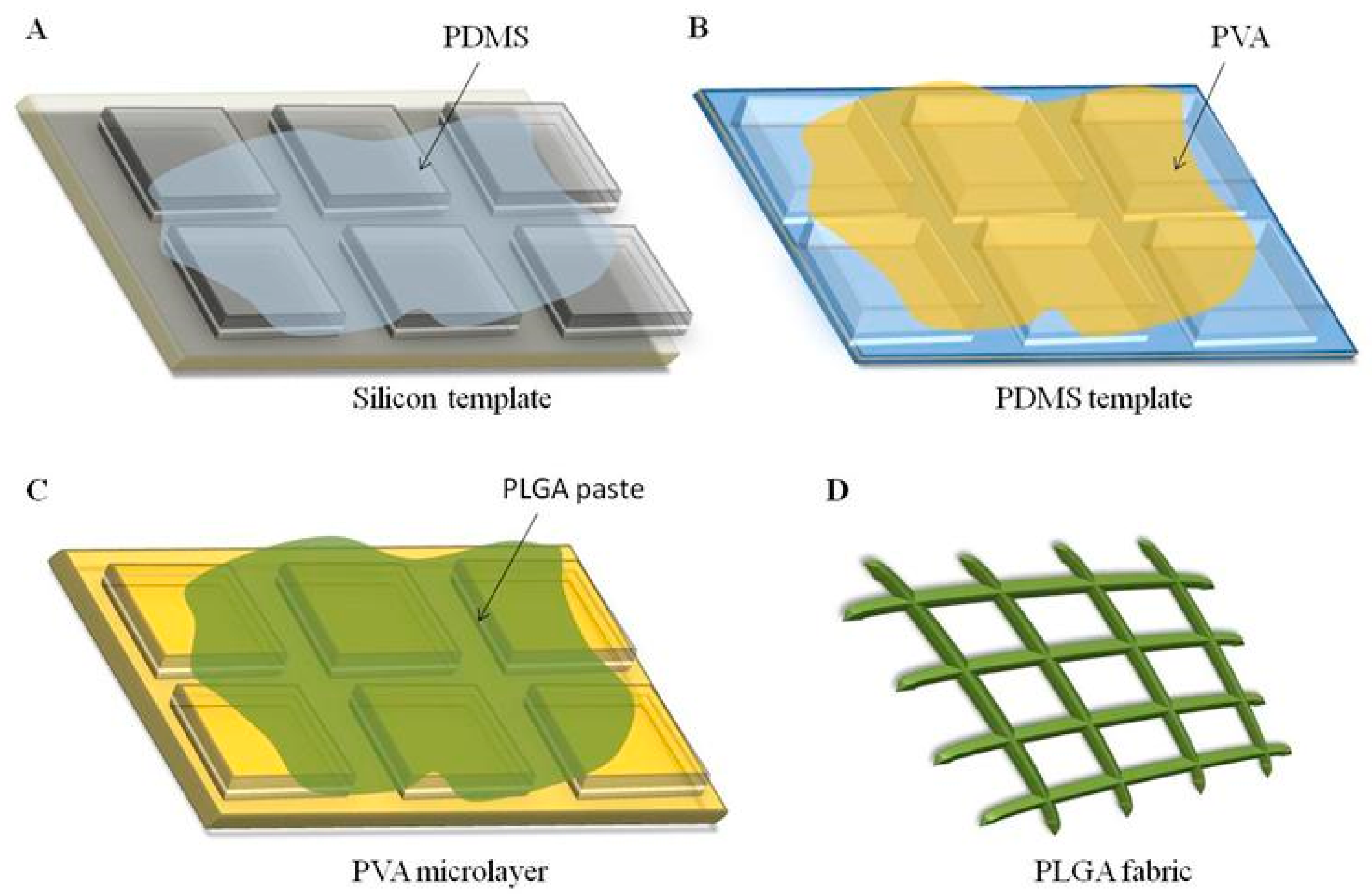
4.2. Fabrication of the PLGA Microstructured Fabrics
A soft lithography fabrication approach was employed to realize a regular network of PLGA microscopic filaments arranged as warps and wefts, presenting a rectangular cross section and perpendicularly intersecting with pitch distances of 5, 10, and 20 μm. The fabrication technique involved multiple sequential steps. First, a silicon master template was generated via direct laser writing (DLW), which engraves a matrix of square pillars on a 5-inch silicon wafer, whose edge length uniquely identifies the microfilament arrangement, pitch distance (5, 10, or 20 μm), and cross section. In the 5 and 10 μm network configurations, the width of the microscopic filaments (i.e., the distance between adjacent square pillars) is 5 μm, while for the 20 μm network configuration, this distance reduces to 3 μm. For all the configurations, the thickness of the microscopic filaments (i.e., the height of the pillars) is 5 μm. Second, a polydimethylsiloxane (PDMS) solution was deposited over the silicon template. Third, after 4 h of polymerization at 60 °C, the PDMS template, which reproduces the negative geometry of the original silicon template, was replicated into a sacrificial template via a PVA solution (3.5% w/w in water) being poured on top of it. After 1.5 h at 60 °C, the polymerized PVA template displayed the same matrix of pillars as the original silicon master template did. Fourth, a polymeric paste of PLGA was uniformly dispersed to accurately fill up the ridges between the matrix of pillars over the PVA template. Fifth, following solvent evaporation, the PLGA microfilament network was released via dissolving the PVA microlayer in water. Note that the three microfilament network configurations present different pillar and ridge sizes, and thus, require different amounts of PLGA for the same fabricated area. Specifically, the 5, 10, and 20 μm networks were obtained by using 24.5, 18.2, and 5.0 mg of PLGA, respectively. For all the configurations, PLGA was first dissolved in acetonitrile (ACN), and then spread over the whole PVA template.
To prepare RhB-loaded microscopic filaments, 2 μL of an RhB solution (2 mg/mL in ACN) was added to the PLGA paste before spreading it on top of the PVA template. To prepare curcumin-loaded microscopic filaments, 25 μL and 15 μL of a curcumin solution (10 mg/mL in ACN) were added to the PLGA paste before spreading it on the PVA template for the 5 and 10 μm configurations and for the 20 μm configuration, respectively.
4.3. Morphological Characterization of the Microfilament Networks
Scanning electron microscopy (SEM, JSM 6490, JEOL, Milan, Italy) was used to image all the microfilament network configurations. A portion of the PLGA network was placed on a silicon template and uniformly sputter coated with 10 nm of gold to increase the contrast and avoid damaging the sample. An acceleration voltage of 10 kV was employed for SEM imaging. Additionally, confocal microscopy (Nikon A1, Dexter, MI) was used to study the structure of all the network configurations, as well as the uniform distribution of the loaded molecules. To this end, CURC was considered for its natural green fluorescence, which under a fluorescent microscope was easily observed and quantified.
4.4. Biophysical Characterization of the hADSC Aggregates
Human Adipose-Derived Stem Cells (hADSC) were used to perform all the in vitro experiments. Cells were provided by the University of Navarra, Spain (Laboratory of Cell Therapy, Foundation for Applied Medical Research), and cultured at 37 °C in 5% CO2 in high-glucose DMEM supplemented with 10% FBS, 1% Penicillin–Streptavicin, 1% L-Glutamine, and bFGF (1 ng/mL), which from now on, is named the culture medium. For all the microfilament network configurations, 5 × 105 cells were seeded on top of two polymeric networks, which were located sufficiently apart one from the other and placed on the bottom of a Petri dish. Each microfilament network had a size of 2.5 × 2.5 cm2, which was derived via precisely cutting the original 4 × 4 cm2 large network released from the PVA template with a surgical scalpel. Cells were carefully positioned in separate drops and homogeneously distributed all over the surface of the microfilament network. A few minutes after seeding, the culture medium wetted the entire surface of the substrates, and the PVA substrate started to dissolve, releasing the final PLGA microfilament network. Approximately 8 h later, the microfilament network was covered with the culture medium to prevent the dehydration of cells throughout the analysis and relocated into another Petri dish, containing a fresh culture medium, to remove the unbound cells or the ones that adhered at the bottom of the initial Petri dish.
The interaction between hADSC and the PLGA network was examined. Cell nuclei were stained with Hoechst 33342 to accurately identify the cells distributed in 3D spheroidal-like structures. The physical features of these cell aggregates were investigated using a fluorescent microscopy (Leica 6000, Wetzlar, Germany) at different time points, namely 24, 48 and 72 h, and acquiring images over the entire network. The analysis resulted in a collection of multiple fluorescent images, which were first binarized, and then thresholded via the ‘Default’ threshold method using ImageJ. These steps converted the image into binary image masks that defined groups of pixels as objects. Based on these masks, the perimeter and aspect ratio of all the objects of interest (corresponding to the cell aggregates) were quantified. The perimeter was measured as the length of the contour of each object detected in the images, whereas the aspect ratio as the ratio between the major and minor axes of the same contour.
4.5. Isolation of Single hADSC from Aggregates
At predetermined time points, after gently washing them with PBS, hADSC were incubated with a Trypsin-EDTA solution for 10 min. The spheroids detached from the microfilament network were further dissociated into single cell suspensions via gently pipetting the solution to avoid cell damage for 5 min. The cell suspension was collected in PBS, filtered using 40 µm cell strainers, centrifuged for 6 min at 200× g, re-suspended in culture medium, and counted using an automated cell counter (ChemiDoc MP, BIORAD). As a comparison, a similar protocol was applied on hADSC monolayers formed on a regular plastic dish (2D Petri dish) and an ultra-thin collagen scaffold (2D collagen layer) made of pure collagen type I fibers, which was provided by Naturin-Viscofan (Germany). The resulting suspensions were then processed for cell viability study.
4.6. Cell Viability of hADSC
The viability of hADSC was determined using an Annexin V-FITC Apoptosis Detection Kit and flow cytometry. After washing with PBS, individual cell suspensions (106 cells/mL) were centrifuged and re-suspended in 1× Annexin Binding Buffer, which was prepared by adding 1 mL of 5× Annexin Binding Buffer to 4 mL deionized water. The single cell suspensions were incubated for 15 min at room temperature (RT) with 1.25 μL of Annexin V-FITC and Propidium Iodide (PI, at 100μg/mL). The working solution was prepared via diluting 1 mg/mL PI stock solution into 45 μL of 1× Annexin Binding Buffer. Specifically, 1 μL of 100μg/mL PI working solution was added to each 100 μL of cell suspension (1 × 105 cells). After the incubation period, 400 μL of 1× Annexin Binding Buffer was added to each sample, which were immediately stored on ice and vortexed right before analysis. Flow cytometry was performed using an FACS ARIA (Becton Dickinson, Franklin Lakes, NJ, USA), and data were analyzed using FACSDiva 9.0.1 software. The experiments were performed independently at least four times.
4.7. Cytoskeleton Organization of hADSC
Cells were stained with Hoechst 33342 before being seeding on top of the microfilament networks to avoid any dye percolation into the PLGA structure. Additionally, to increase the imaging contrast, the red fluorescent molecule, Rhodamine B (RhB), was distributed within the PLGA network. The resulting microfilament network, which was still supported by the PVA microlayer, was cut in 1 cm2 pieces and placed at the bottom of the wells in an 8-well plate. Similarly, 1 cm2 of flat collagen scaffold was positioned in each well. Cell suspension (104 cells/200µL) was seeded on top of each scaffold for 24 h. Then, cells were repeatedly washed with cold PBS (3 times per 5 min) and fixed with 0.4% paraformaldehyde (PFA) at RT for 1 h. Then, cells were washed again with PBS (3 times per 5 min) and permeabilized using 0.1% Tryton X-100 in PBS for 20 min. Finally, cells were stained with 5 μL of Phalloidin-488, which binds to cell actin, in PBS supplemented with 1% Bovine Serum Albumin (BSA) and 0.01% Tryton X-100 for 1 h.
Confocal images were used to quantify the percent of nonlinear and linear actin filaments within the cell cytoskeleton for all the configurations. After selecting the channel of interest via actin staining, all the images were firstly binarized; then, a threshold was generated using the ‘Default’ threshold method in ImageJ. These steps converted the images into binary image masks that defined groups of pixels as objects. Using ‘Analyze Particle’ function of ImageJ, the analysis parameters were set for size (pixel2): 0-infinity; circularity: 0–1. Based on this selection, the entire collection of objects was quantified and classified as concerns the aspect ratio, which was defined as the ratio among the major axis and minor axis of the object of interest. Finally, we arbitrarily established linear actin as the objects with an aspect ratio larger than 5 and nonlinear actin as the objects with an aspect ratio smaller than 5.
4.8. Secretome Analysis for hADSC
The study was performed in a model of inflamed microenvironment to observe any paracrine activity of hADSC depending on the surrounding microenvironment and the types of scaffolds used. A total of 5 × 105 hADSC were cultured under controlled conditions (37 °C in 5% CO2), suspended in culture medium, and seeded on top of different substrates. After an 8 h incubation period, all the cell substrates were moved to a new well plate that already contained 10 mL of DMEM supplemented with 2% FBS, 1% L-Glutamine, 1% Penicillum–Streptavicin, and TNF-α (10 ng/mL), which from now on, is referred to as the pro-inflammatory culture medium. Supernatants were collected at 48 h after seeding, and cytokines, chemokines, and growth factors released by the hADSC were analyzed via cytokine/antibody array (Human Cytokine Antibody Array, Membrane, 80 Targets, ab133998) according to the manufacturer’s instructions. The arrays could assess 80 different factors simultaneously. Briefly, supernatants were first characterized in terms of total protein via a BCA assay (Bicinchoninic Protein Assay kit, Euroclone) so that we used the same amount of protein for every condition. Then, each supernatant was added to a corresponding membrane provided in the kit and kept overnight at 4 °C. The day after, following several washes with Wash Buffer I and Wash Buffer II, biotin-conjugated anti-cytokines antibody was added to all the membranes at RT for 2 h. This was followed by incubation with HRP-Conjugated Streptavidin at RT for 2 h. Finally, all the membranes were analyzed using the chemiluminescent method (ChemiDoc MP Imaging System by Bio-Rad). By using ImageJ, the luminescence associated with each spot on the membrane was quantified: higher luminescence levels are associated with higher amounts of the cytokine, chemokine, or growth factor of interest. The baseline, which is derived as the average value of the negative control spots, was subtracted from all the measured quantities. Finally, all the obtained values were normalized according to the average value of the positive control spots.
4.9. Statistics Analysis
All the statistical tests were performed using Graph Pad Prism 8.0. Values are presented as mean ± standard deviation (SD). Each experiment was repeated multiple times independently (≥3). Statistically significant differences among experimental groups were evaluated with an ordinary one-way ANOVA test using a Tukey’s Multiple Comparison Test as post hoc test.
5. Conclusions
Taking advantage of the accurate control of the size, shape, surface proprieties, and mechanical stiffness that could be obtained by using the top-down fabrication technique, three different PLGA fabric configurations, namely 5 μm, 10 μm, and 20 μm, were realized. All the three fabric configurations successfully established an intimate interaction with stem cells favoring their organization in 3D spheroidal-like structures, without affecting their viability. The investigations of the cell cytoskeleton architecture demonstrated that the fabric configuration influenced the arrangement of actin fibers favoring nonlinear organization. In addition, it was shown that the 5, 10, and 20 μm fabrics can enhance the secretion of several factors involved in key biological processes favoring injury resolution, including angiogenesis, ECM remodeling, and stem cell homing. Finally, these results highlight the possibility of modulating stem cells biological activities (i.e., angiogenesis) via using only the geometrical features of the scaffold and without using any type of active molecule. Therefore, although additional biological characterizations will be needed in future studies to assess the actual biocompatibility of the proposed PLGA microstructured fabrics, this study could pave the way for the realization of a novel, bio-functional, and tunable scaffold in regenerative medicine.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





