
Complement Drives Chronic Inflammation and Progressive Hydrocephalus in Murine Neonatal Germinal Matrix Hemorrhage
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
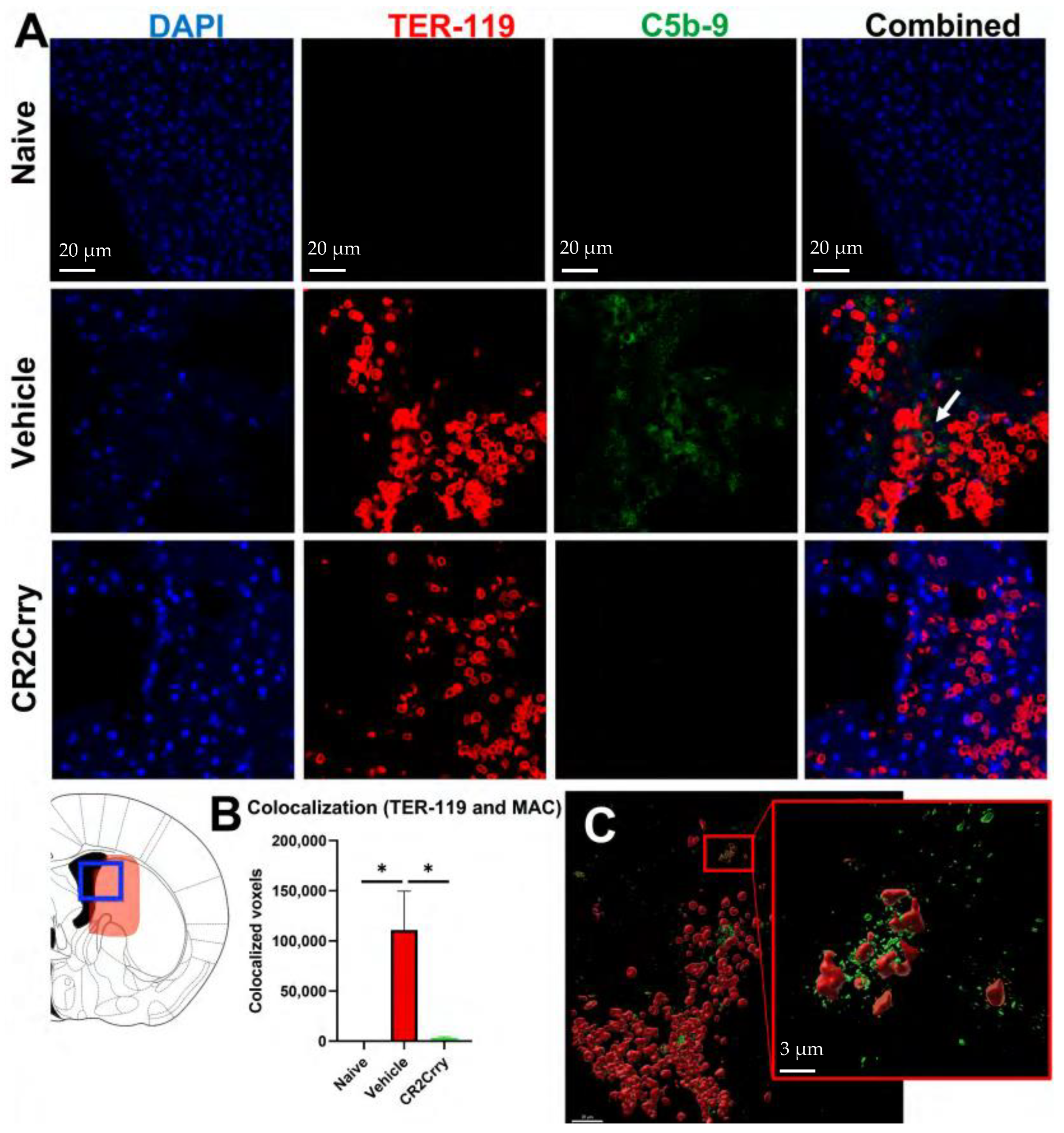
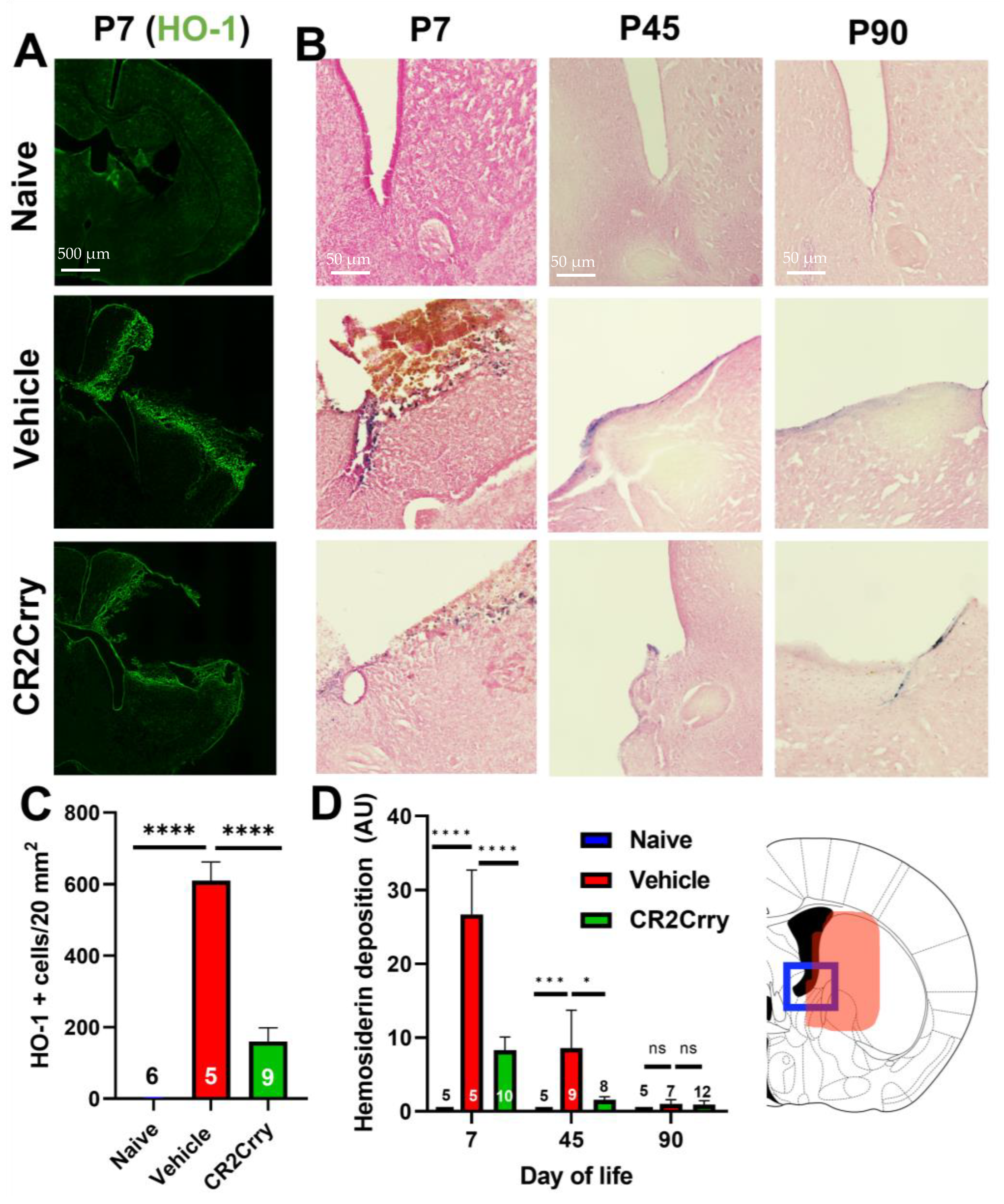
2.1. Germinal Matrix Hemorrhage Results in Complement-Dependent Heme Release and Iron-Dependent Inflammation
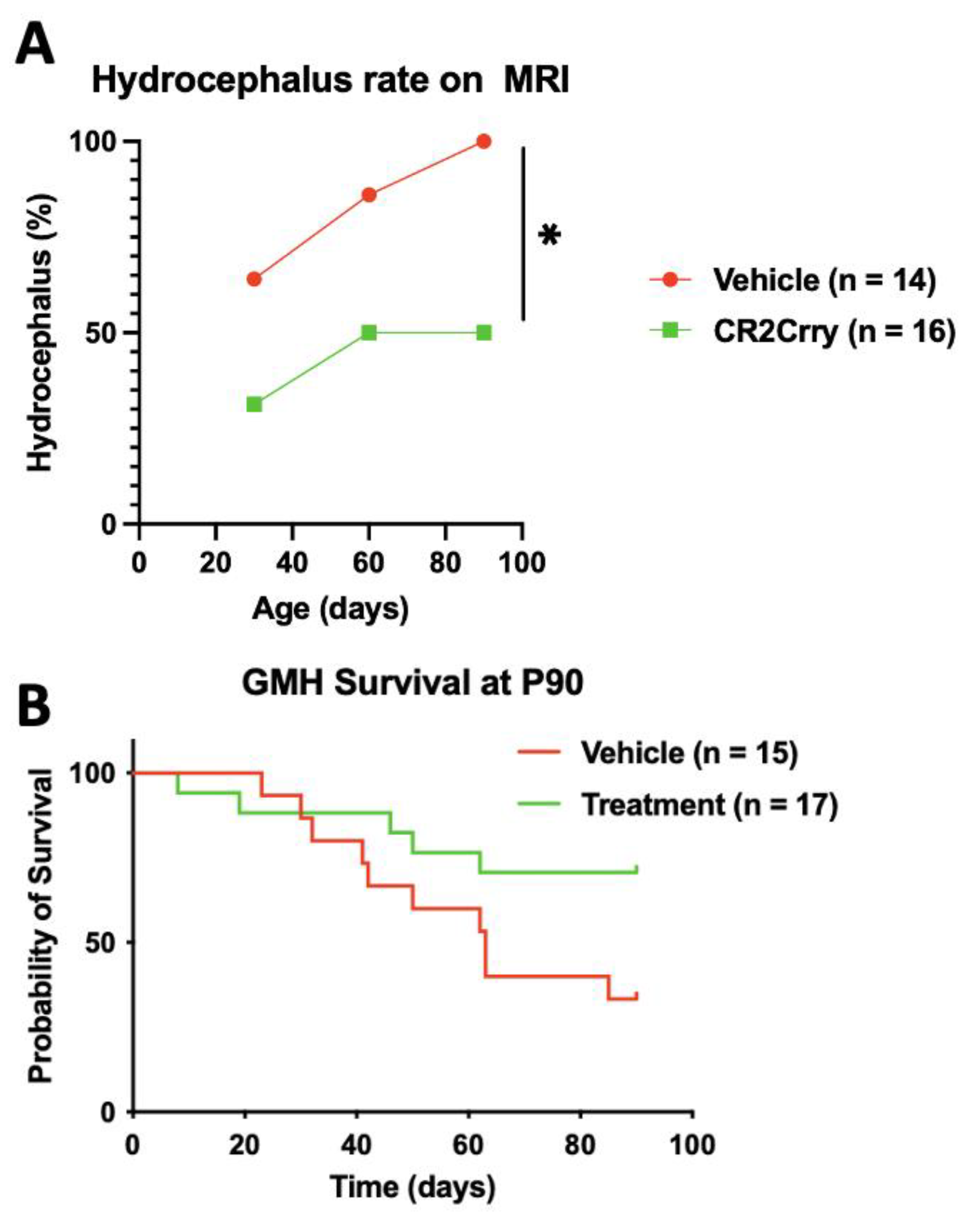
2.2. Complement Inhibition Reduces Chronic Progressive Hydrocephalus and Improves Survival in Chronic GMH Model
2.3. Chronic Inflammation Following GMH Leads to Radiographic Findings of Periventricular Leukomalacia
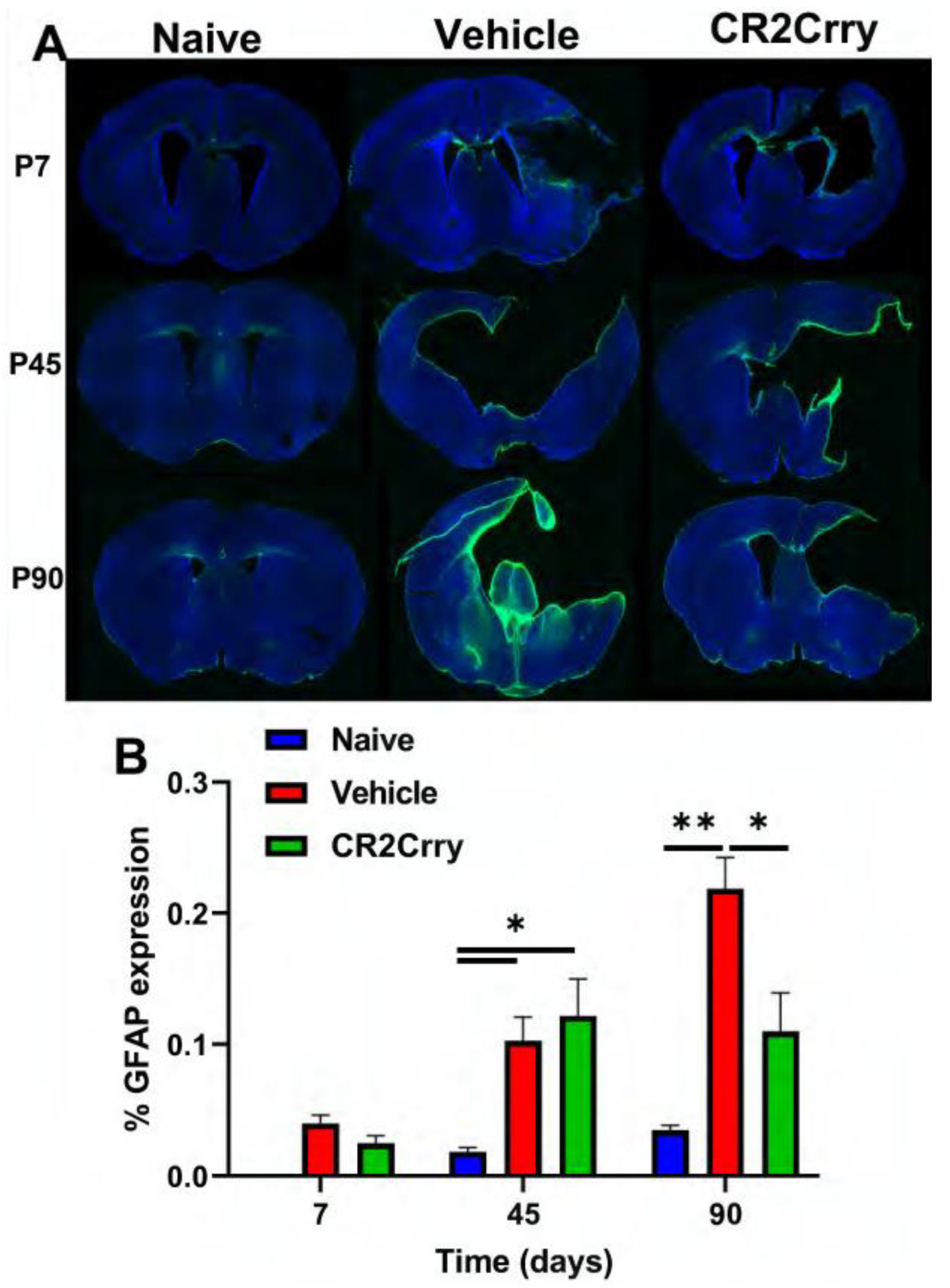
2.4. GMH Injury Leads to Increased Gliosis and Phagocytosis of White Matter Chronically after GMH
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Owens, R. Intraventricular hemorrhage in the premature neonate. Neonatal Netw. 2005, 24, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.P.; Inder, T.E.; Rooks, V.; Taylor, G.A.; Anderson, N.J.; Mogridge, N.; Horwood, L.J.; Volpe, J.J. Posthaemorrhagic ventricular dilatation in the premature infant: Natural history and predictors of outcome. Arch. Dis. Child. Fetal Neonatal. Ed. 2002, 87, F37–F41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klebe, D.; McBride, D.; Krafft, P.R.; Flores, J.J.; Tang, J.; Zhang, J.H. Posthemorrhagic hydrocephalus development after germinal matrix hemorrhage: Established mechanisms and proposed pathways. J. Neurosci. Res. 2020, 98, 105–120. [Google Scholar] [CrossRef] [Green Version]
- Bolisetty, S.; Dhawan, A.; Abdel-Latif, M.; Bajuk, B.; Stack, J.; Lui, K. New South Wales and Australian Capital Territory Neonatal Intensive Care Units’ Data Collection. Intraventricular Hemorrhage and Neurodevelopmental Outcomes in Extreme Preterm Infants. Pediatrics 2019, 144, e20192079. [Google Scholar]
- Radic, J.A.; Vincer, M.; McNeely, P.D. Outcomes of intraventricular hemorrhage and posthemorrhagic hydrocephalus in a population-based cohort of very preterm infants born to residents of Nova Scotia from 1993 to 2010. J. Neurosurg. Pediatr. 2015, 15, 580–588. [Google Scholar] [CrossRef] [Green Version]
- Drake, J.M.; Kestle, J.R.; Tuli, S. CSF shunts 50 years on—past, present and future. Childs Nerv. Syst. 2000, 16, 800–804. [Google Scholar] [CrossRef]
- Strahle, J.; Garton, H.J.; Maher, C.O.; Muraszko, K.M.; Keep, R.F.; Xi, G. Mechanisms of hydrocephalus after neonatal and adult intraventricular hemorrhage. Transl. Stroke Res. 2012, 3 (Suppl. S1), 25–38. [Google Scholar] [CrossRef] [Green Version]
- Whitelaw, A.; Evans, D.; Carter, M.; Thoresen, M.; Wroblewska, J.; Mandera, M.; Swietlinski, J.; Simpson, J.; Hajivassiliou, C.; Hunt, L.P.; et al. Randomized clinical trial of prevention of hydrocephalus after intraventricular hemorrhage in preterm infants: Brain-washing versus tapping fluid. Pediatrics 2007, 119, e1071–e1078. [Google Scholar] [CrossRef]
- Whitelaw, A.; Jary, S.; Kmita, G.; Wroblewska, J.; Musialik-Swietlinska, E.; Mandera, M.; Hunt, L.; Carter, M.; Pople, I. Randomized trial of drainage, irrigation and fibrinolytic therapy for premature infants with posthemorrhagic ventricular dilatation: Developmental outcome at 2 years. Pediatrics 2010, 125, e852–e858. [Google Scholar] [CrossRef]
- Schulz, M.; Buhrer, C.; Pohl-Schickinger, A.; Haberl, H.; Thomale, U.W. Neuroendoscopic lavage for the treatment of intraventricular hemorrhage and hydrocephalus in neonates. J. Neurosurg. Pediatr. 2014, 13, 626–635. [Google Scholar] [CrossRef] [Green Version]
- Holste, K.G.; Xia, F.; Ye, F.; Keep, R.F.; Xi, G. Mechanisms of neuroinflammation in hydrocephalus after intraventricular hemorrhage: A review. Fluids Barriers CNS 2022, 19, 28. [Google Scholar] [CrossRef] [PubMed]
- Garton, T.; Hua, Y.; Xiang, J.; Xi, G.; Keep, R.F. Challenges for intraventricular hemorrhage research and emerging therapeutic targets. Expert Opin. Ther. Targets 2017, 21, 1111–1122. [Google Scholar] [CrossRef]
- Strahle, J.M.; Garton, T.; Bazzi, A.A.; Kilaru, H.; Garton, H.J.; Maher, C.O.; Muraszko, K.M.; Keep, R.F.; Xi, G. Role of hemoglobin and iron in hydrocephalus after neonatal intraventricular hemorrhage. Neurosurgery 2014, 75, 696–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, F.; Liu, F.; Chen, Z.; Hua, Y.; Keep, R.F.; Xi, G. Hydrocephalus after intraventricular hemorrhage: The role of thrombin. J. Cereb. Blood Flow Metab. 2014, 34, 489–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayfrank, L.; Kissler, J.; Raoofi, R.; Delsing, P.; Weis, J.; Kuker, W.; Gilsbach, J.M. Ventricular dilatation in experimental intraventricular hemorrhage in pigs. Characterization of cerebrospinal fluid dynamics and the effects of fibrinolytic treatment. Stroke 1997, 28, 141–148. [Google Scholar] [CrossRef]
- Lodhia, K.R.; Shakui, P.; Keep, R.F. Hydrocephalus in a rat model of intraventricular hemorrhage. Acta Neurochir. Suppl. 2006, 96, 207–211. [Google Scholar]
- Chen, Z.; Gao, C.; Hua, Y.; Keep, R.F.; Muraszko, K.; Xi, G. Role of iron in brain injury after intraventricular hemorrhage. Stroke 2011, 42, 465–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaberel, T.; Montagne, A.; Lesept, F.; Gauberti, M.; Lemarchand, E.; Orset, C.; Goulay, R.; Bertrand, T.; Emery, E.; Vivien, D. Urokinase versus Alteplase for intraventricular hemorrhage fibrinolysis. Neuropharmacology 2014, 85, 158–165. [Google Scholar] [CrossRef]
- Chen, Q.; Tang, J.; Tan, L.; Guo, J.; Tao, Y.; Li, L.; Chen, Y.; Liu, X.; Zhang, J.H.; Chen, Z.; et al. Intracerebral Hematoma Contributes to Hydrocephalus After Intraventricular Hemorrhage via Aggravating Iron Accumulation. Stroke 2015, 46, 2902–2908. [Google Scholar] [CrossRef] [Green Version]
- Meoded, A.; Poretti, A.; Northington, F.J.; Tekes, A.; Intrapiromkul, J.; Huisman, T.A. Susceptibility weighted imaging of the neonatal brain. Clin. Radiol. 2012, 67, 793–801. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, U.; Meyer, S.; Gortner, L.; Korner, H.; Turkyilmaz, M.; Simgen, A.; Reith, W.; Muhl-Benninghaus, R. Superficial Siderosis after Germinal Matrix Hemorrhage. AJNR Am. J. Neuroradiol. 2016, 37, 2389–2391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gram, M.; Sveinsdottir, S.; Ruscher, K.; Hansson, S.R.; Cinthio, M.; Akerstrom, B.; Ley, D. Hemoglobin induces inflammation after preterm intraventricular hemorrhage by methemoglobin formation. J. Neuroinflamm. 2013, 10, 100. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Du, H.; Hua, Y.; Keep, R.F.; Strahle, J.; Xi, G. Role of red blood cell lysis and iron in hydrocephalus after intraventricular hemorrhage. J. Cereb. Blood Flow Metab. 2014, 34, 1070–1075. [Google Scholar] [CrossRef] [Green Version]
- Flores, J.J.; Klebe, D.; Rolland, W.B.; Lekic, T.; Krafft, P.R.; Zhang, J.H. PPARgamma-induced upregulation of CD36 enhances hematoma resolution and attenuates long-term neurological deficits after germinal matrix hemorrhage in neonatal rats. Neurobiol. Dis. 2016, 87, 124–133. [Google Scholar] [CrossRef] [Green Version]
- Flores, J.J.; Klebe, D.; Tang, J.; Zhang, J.H. A comprehensive review of therapeutic targets that induce microglia/macrophage-mediated hematoma resolution after germinal matrix hemorrhage. J. Neurosci. Res. 2020, 98, 121–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducruet, A.F.; Zacharia, B.E.; Hickman, Z.L.; Grobelny, B.T.; Yeh, M.L.; Sosunov, S.A.; Connolly, E.S., Jr. The complement cascade as a therapeutic target in intracerebral hemorrhage. Exp. Neurol. 2009, 219, 398–403. [Google Scholar] [CrossRef] [Green Version]
- Galea, J.; Cruickshank, G.; Teeling, J.L.; Boche, D.; Garland, P.; Perry, V.H.; Galea, I. The intrathecal CD163-haptoglobin-hemoglobin scavenging system in subarachnoid hemorrhage. J. Neurochem. 2012, 121, 785–792. [Google Scholar] [CrossRef] [Green Version]
- Chavez-Bueno, S.; Beasley, J.A.; Goldbeck, J.M.; Bright, B.C.; Morton, D.J.; Whitby, P.W.; Stull, T.L. Haptoglobin concentrations in preterm and term newborns. J. Perinatol. 2011, 31, 500–503. [Google Scholar] [CrossRef] [Green Version]
- Alshareef, M.; Mallah, K.; Vasas, T.; Alawieh, A.; Borucki, D.; Couch, C.; Cutrone, J.; Shope, C.; Eskandari, R.; Tomlinson, S. A Role of Complement in the Pathogenic Sequelae of Mouse Neonatal Germinal Matrix Hemorrhage. Int. J. Mol. Sci. 2022, 23, 2943. [Google Scholar] [CrossRef]
- Huang, Y.; Qiao, F.; Atkinson, C.; Holers, V.M.; Tomlinson, S. A novel targeted inhibitor of the alternative pathway of complement and its therapeutic application in ischemia/reperfusion injury. J. Immunol. 2008, 181, 8068–8076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alawieh, A.; Langley, E.F.; Weber, S.; Adkins, D.; Tomlinson, S. Identifying the Role of Complement in Triggering Neuroinflammation after Traumatic Brain Injury. J. Neurosci. 2018, 38, 2519–2532. [Google Scholar] [CrossRef] [Green Version]
- Alawieh, A.; Tomlinson, S. Injury site-specific targeting of complement inhibitors for treating stroke. Immunol. Rev. 2016, 274, 270–280. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Luo, Y.; Zeng, H.; Reis, C.; Chen, S. Research Advances of Germinal Matrix Hemorrhage: An Update Review. Cell. Mol. Neurobiol. 2019, 39, 1–10. [Google Scholar] [CrossRef]
- Christian, E.A.; Melamed, E.F.; Peck, E.; Krieger, M.D.; McComb, J.G. Surgical management of hydrocephalus secondary to intraventricular hemorrhage in the preterm infant. J. Neurosurg. Pediatr. 2016, 17, 278–284. [Google Scholar] [CrossRef] [Green Version]
- Lan, X.; Han, X.; Li, Q.; Yang, Q.W.; Wang, J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat. Rev. Neurol. 2017, 13, 420–433. [Google Scholar] [CrossRef] [Green Version]
- Jing, C.; Bian, L.; Wang, M.; Keep, R.F.; Xi, G.; Hua, Y. Enhancement of Hematoma Clearance With CD47 Blocking Antibody in Experimental Intracerebral Hemorrhage. Stroke 2019, 50, 1539–1547. [Google Scholar] [CrossRef]
- Mevorach, D.; Mascarenhas, J.O.; Gershov, D.; Elkon, K.B. Complement-dependent clearance of apoptotic cells by human macrophages. J. Exp. Med. 1998, 188, 2313–2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Schmidt, C.Q.; Koutsogiannaki, S.; Ricci, P.; Risitano, A.M.; Lambris, J.D.; Ricklin, D. Complement C3dg-mediated erythrophagocytosis: Implications for paroxysmal nocturnal hemoglobinuria. Blood 2015, 126, 891–894. [Google Scholar] [CrossRef] [Green Version]
- Lutz, H.U. Naturally occurring autoantibodies in mediating clearance of senescent red blood cells. Adv. Exp. Med. Biol. 2012, 750, 76–90. [Google Scholar] [PubMed]
- Cao, S.; Zheng, M.; Hua, Y.; Chen, G.; Keep, R.F.; Xi, G. Hematoma Changes During Clot Resolution After Experimental Intracerebral Hemorrhage. Stroke 2016, 47, 1626–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.C.; Tang, S.C.; Lee, J.E.; Lai, D.M.; Huang, S.J.; Hsieh, S.T.; Jeng, J.S.; Tu, Y.K. Prognostic value of intrathecal heme oxygenase-1 concentration in patients with Fisher Grade III aneurysmal subarachnoid hemorrhage. J. Neurosurg. 2014, 121, 1388–1393. [Google Scholar] [CrossRef] [Green Version]
- Okubo, S.; Xi, G.; Keep, R.F.; Muraszko, K.M.; Hua, Y. Cerebral hemorrhage, brain edema, and heme oxygenase-1 expression after experimental traumatic brain injury. Acta Neurochir. Suppl. 2013, 118, 83–87. [Google Scholar]
- Imaizumi, T.; Chiba, M.; Honma, T.; Niwa, J. Detection of hemosiderin deposition by T2*-weighted MRI after subarachnoid hemorrhage. Stroke 2003, 34, 1693–1698. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Pleasure, J.; Pleasure, D. Progress in periventricular leukomalacia. Arch. Neurol. 2008, 65, 1291–1295. [Google Scholar] [CrossRef] [Green Version]
- Zaghloul, N.; Patel, H.; Ahmed, M.N. A model of Periventricular Leukomalacia (PVL) in neonate mice with histopathological and neurodevelopmental outcomes mimicking human PVL in neonates. PLoS ONE 2017, 12, e0175438. [Google Scholar] [CrossRef] [Green Version]
- Pierson, C.R.; Folkerth, R.D.; Billiards, S.S.; Trachtenberg, F.L.; Drinkwater, M.E.; Volpe, J.J.; Kinney, H.C. Gray matter injury associated with periventricular leukomalacia in the premature infant. Acta Neuropathol. 2007, 114, 619–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinney, H.C.; Panigrahy, A.; Newburger, J.W.; Jonas, R.A.; Sleeper, L.A. Hypoxic-ischemic brain injury in infants with congenital heart disease dying after cardiac surgery. Acta Neuropathol. 2005, 110, 563–578. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, E.B.; Lucas, A.; Chong, W.K.; Wood, S.J.; Johnson, C.L.; Marshall, C.; Vargha-Khadem, F.; Gadian, D.G. Hippocampal volume and everyday memory in children of very low birth weight. Pediatr. Res. 2000, 47, 713–720. [Google Scholar] [CrossRef]
- Inder, T.E.; Wells, S.J.; Mogridge, N.B.; Spencer, C.; Volpe, J.J. Defining the nature of the cerebral abnormalities in the premature infant: A qualitative magnetic resonance imaging study. J. Pediatr. 2003, 143, 171–179. [Google Scholar] [CrossRef]
- Inder, T.E.; Warfield, S.K.; Wang, H.; Huppi, P.S.; Volpe, J.J. Abnormal cerebral structure is present at term in premature infants. Pediatrics 2005, 115, 286–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosun, D.; Dabbs, K.; Caplan, R.; Siddarth, P.; Toga, A.; Seidenberg, M.; Hermann, B. Deformation-based morphometry of prospective neurodevelopmental changes in new onset paediatric epilepsy. Brain 2011, 134, 1003–1014. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, A.; Okoshi, Y.; Hachiya, Y.; Ozawa, Y.; Ito, M.; Kida, Y.; Imai, Y.; Kohsaka, S.; Takashima, S. Early immunohistochemical detection of axonal damage and glial activation in extremely immature brains with periventricular leukomalacia. Clin. Neuropathol. 2001, 20, 87–91. [Google Scholar]
- Billiards, S.S.; Haynes, R.L.; Folkerth, R.D.; Borenstein, N.S.; Trachtenberg, F.L.; Rowitch, D.H.; Ligon, K.L.; Volpe, J.J.; Kinney, H.C. Myelin abnormalities without oligodendrocyte loss in periventricular leukomalacia. Brain Pathol. 2008, 18, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, C.; Song, H.; Lu, B.; Qiao, F.; Burns, T.A.; Holers, V.M.; Tsokos, G.C.; Tomlinson, S. Targeted complement inhibition by C3d recognition ameliorates tissue injury without apparent increase in susceptibility to infection. J. Clin. Investig. 2005, 115, 2444–2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallah, K.; Couch, C.; Alshareef, M.; Borucki, D.; Yang, X.; Alawieh, A.; Tomlinson, S. Complement mediates neuroinflammation and cognitive decline at extended chronic time points after traumatic brain injury. Acta Neuropathol. Commun. 2021, 9, 72. [Google Scholar] [CrossRef] [PubMed]
- Van Duijn, S.; Nabuurs, R.J.; van Duinen, S.G.; Natte, R. Comparison of histological techniques to visualize iron in paraffin-embedded brain tissue of patients with Alzheimer’s disease. J. Histochem. Cytochem. 2013, 61, 785–792. [Google Scholar] [CrossRef] [Green Version]
- Alawieh, A.; Langley, E.F.; Tomlinson, S. Targeted complement inhibition salvages stressed neurons and inhibits neuroinflammation after stroke in mice. Sci. Transl. Med. 2018, 10, eaao6459. [Google Scholar] [CrossRef] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshareef, M.; Hatchell, D.; Vasas, T.; Mallah, K.; Shingala, A.; Cutrone, J.; Alawieh, A.; Guo, C.; Tomlinson, S.; Eskandari, R. Complement Drives Chronic Inflammation and Progressive Hydrocephalus in Murine Neonatal Germinal Matrix Hemorrhage. Int. J. Mol. Sci. 2023, 24, 10171. https://doi.org/10.3390/ijms241210171
Alshareef M, Hatchell D, Vasas T, Mallah K, Shingala A, Cutrone J, Alawieh A, Guo C, Tomlinson S, Eskandari R. Complement Drives Chronic Inflammation and Progressive Hydrocephalus in Murine Neonatal Germinal Matrix Hemorrhage. International Journal of Molecular Sciences. 2023; 24(12):10171. https://doi.org/10.3390/ijms241210171
Chicago/Turabian StyleAlshareef, Mohammed, Devin Hatchell, Tyler Vasas, Khalil Mallah, Aakash Shingala, Jonathan Cutrone, Ali Alawieh, Chunfang Guo, Stephen Tomlinson, and Ramin Eskandari. 2023. "Complement Drives Chronic Inflammation and Progressive Hydrocephalus in Murine Neonatal Germinal Matrix Hemorrhage" International Journal of Molecular Sciences 24, no. 12: 10171. https://doi.org/10.3390/ijms241210171
APA StyleAlshareef, M., Hatchell, D., Vasas, T., Mallah, K., Shingala, A., Cutrone, J., Alawieh, A., Guo, C., Tomlinson, S., & Eskandari, R. (2023). Complement Drives Chronic Inflammation and Progressive Hydrocephalus in Murine Neonatal Germinal Matrix Hemorrhage. International Journal of Molecular Sciences, 24(12), 10171. https://doi.org/10.3390/ijms241210171