
MCD Diet Modulates HuR and Oxidative Stress-Related HuR Targets in Rats
 , ,
, ,  , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Model Characterization
2.1.1. Enzyme Release after MCD Diet Administration
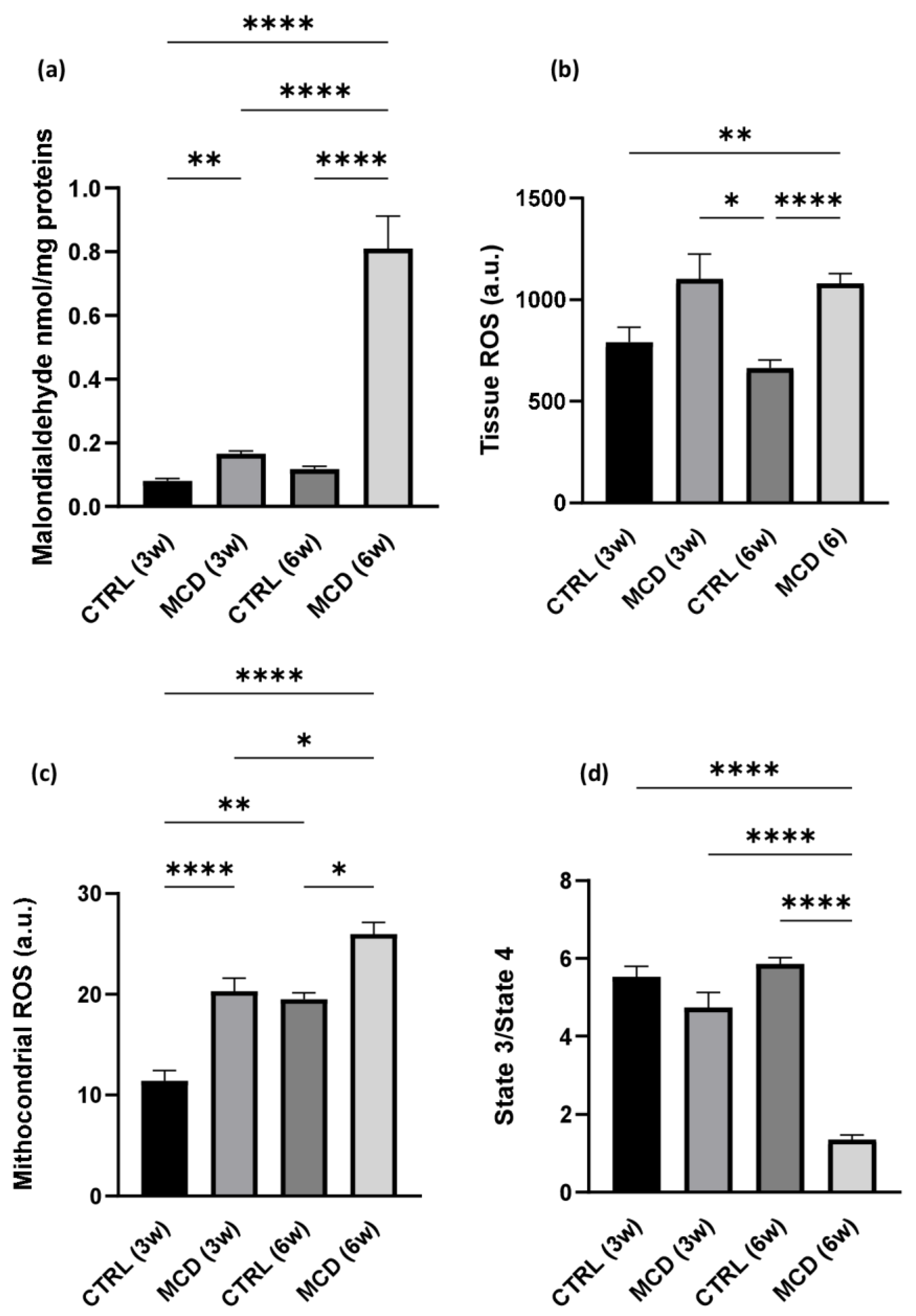
2.1.2. MCD Diet Administration Increases Oxidative Stress and Mitochondrial Dysfunction
2.1.3. MCD Diet Administration Promotes Hepatic Lipid Accumulation
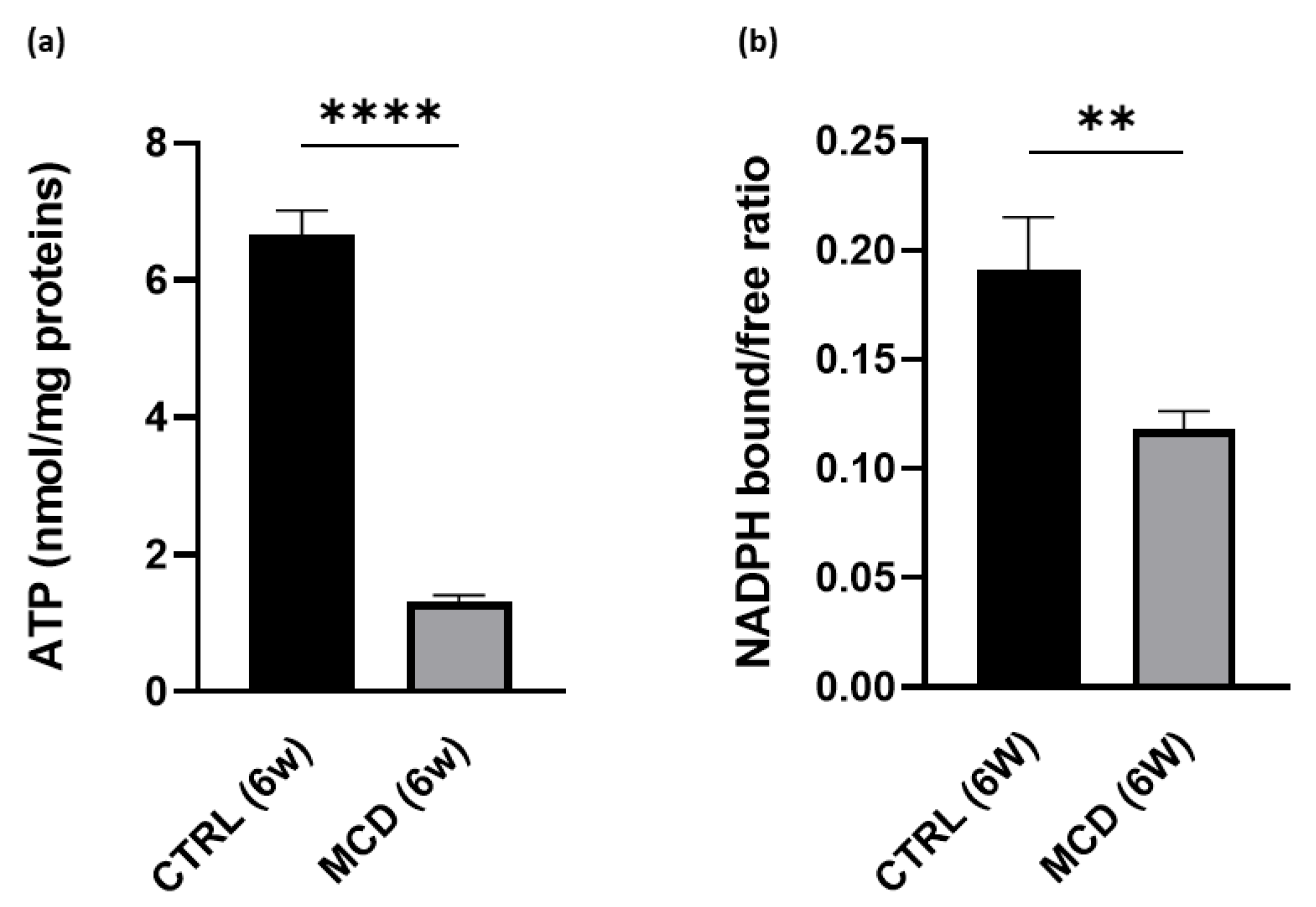
2.1.4. MCD Diet Administration Reduces Hepatic ATP Content and NADPH Bound/Free Ratio
2.2. HuR and Its Targets MnSOD and HO-1
2.2.1. Effect of the MCD Diet on HuR Protein Expression
2.2.2. Effect of the MCD Diet on MnSOD and HO-1 Protein Expression
2.2.3. Correlation between HuR, Oxidative Stress, and HuR Targets MnSOD and HO-1
3. Discussion
4. Materials and Methods
4.1. Animal Model and Experimental Procedures
4.2. Materials
4.3. Blood Sample Preparation and Enzyme Evaluation
4.4. Mitochondria Isolation
4.5. Respiratory Control Index Evaluation
4.6. Mitochondrial ROS Production
4.7. Oxidative Stress and ATP Content Evaluation
4.8. NADPH Bound/Free Ratio Evaluation
4.9. Tissue Histology and Staining
4.10. Hepatic Lipid Extraction and Quantification
4.11. Western Blot
4.12. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- LaBrecque, D.R.; Abbas, Z.; Anania, F.; Ferenci, P.; Khan, A.G.; Goh, K.-L.; Hamid, S.S.; Isakov, V.; Lizarzabal, M.; Peñaranda, M.M.; et al. World Gastroenterology Organisation Global Guidelines. J. Clin. Gastroenterol. 2014, 48, 467–473. [Google Scholar] [CrossRef]
- Berardo, C.; Di Pasqua, L.G.; Cagna, M.; Richelmi, P.; Vairetti, M.; Ferrigno, A. Nonalcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis: Current Issues and Future Perspectives in Preclinical and Clinical Research. Int. J. Mol. Sci. 2020, 21, 9646. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Tabas, I.; Pajvani, U.B. Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology 2020, 158, 1913–1928. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.A.; Friedman, S.L. Inflammatory and fibrotic mechanisms in NAFLD-Implications for new treatment strategies. J. Intern. Med. 2022, 291, 11–31. [Google Scholar] [CrossRef]
- Prasun, P.; Ginevic, I.; Oishi, K. Mitochondrial dysfunction in nonalcoholic fatty liver disease and alcohol related liver disease. Transl. Gastroenterol. Hepatol. 2021, 6, 4. [Google Scholar] [CrossRef]
- Flessa, C.M.; Kyrou, I.; Nasiri-Ansari, N.; Kaltsas, G.; Papavassiliou, A.G.; Kassi, E.; Randeva, H.S. Endoplasmic Reticulum Stress and Autophagy in the Pathogenesis of Non-alcoholic Fatty Liver Disease (NAFLD): Current Evidence and Perspectives. Curr. Obes. Rep. 2021, 10, 134–161. [Google Scholar] [CrossRef] [PubMed]
- Han, A.L. Association between Non-Alcoholic Fatty Liver Disease and Dietary Habits, Stress, and Health-Related Quality of Life in Korean Adults. Nutrients 2020, 12, 1555. [Google Scholar] [CrossRef]
- Kitade, H.; Chen, G.; Ni, Y.; Ota, T. Nonalcoholic Fatty Liver Disease and Insulin Resistance: New Insights and Potential New Treatments. Nutrients 2017, 9, 387. [Google Scholar] [CrossRef] [Green Version]
- Hrncir, T.; Hrncirova, L.; Kverka, M.; Hromadka, R.; Machova, V.; Trckova, E.; Kostovcikova, K.; Kralickova, P.; Krejsek, J.; Tlaskalova-Hogenova, H. Gut Microbiota and NAFLD: Pathogenetic Mechanisms, Microbiota Signatures, and Therapeutic Interventions. Microorganisms 2021, 9, 957. [Google Scholar] [CrossRef]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Di Pasqua, L.G.; Cagna, M.; Berardo, C.; Vairetti, M.; Ferrigno, A. Detailed Molecular Mechanisms Involved in Drug-Induced Non-Alcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis: An Update. Biomedicines 2022, 10, 194. [Google Scholar] [CrossRef] [PubMed]
- Gabbia, D.; Cannella, L.; Martin, S. De The Role of Oxidative Stress in NAFLD–NASH–HCC Transition—Focus on NADPH Oxidases. Biomedicines 2021, 9, 687. [Google Scholar] [CrossRef]
- Delli Bovi, A.P.; Marciano, F.; Mandato, C.; Siano, M.A.; Savoia, M.; Vajro, P. Oxidative Stress in Non-alcoholic Fatty Liver Disease. An Updated Mini Review. Front. Med. 2021, 8, 595371. [Google Scholar] [CrossRef]
- Tariq, Z.; Green, C.J.; Hodson, L. Are oxidative stress mechanisms the common denominator in the progression from hepatic steatosis towards non-alcoholic steatohepatitis (NASH)? Liver Int. 2014, 34, e180–e190. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Haddad, A.; Osme, A.; Kim, C.; Borzou, A.; Ilchenko, S.; Allende, D.; Dasarathy, S.; McCullough, A.; Sadygov, R.G.; et al. Hepatic Mitochondrial Defects in a Nonalcoholic Fatty Liver Disease Mouse Model Are Associated with Increased Degradation of Oxidative Phosphorylation Subunits. Mol. Cell. Proteom. 2018, 17, 2371. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, H.; Fukura, M.; Tsuchishima, M.; Takase, S. Mutation of mitochondrial DNA in livers from patients with alcoholic hepatitis and nonalcoholic steatohepatitis. Alcohol. Clin. Exp. Res. 2007, 31, S54–S60. [Google Scholar] [CrossRef]
- Chalasani, N.; Christopher Gorski, J.; Asghar, M.S.; Asghar, A.; Foresman, B.; Hall, S.D.; Crabb, D.W. Hepatic cytochrome P450 2E1 activity in nondiabetic patients with nonalcoholic steatohepatitis. Hepatology 2003, 37, 544–550. [Google Scholar] [CrossRef]
- Busch, C.J.L.; Hendrikx, T.; Weismann, D.; Jäckel, S.; Walenbergh, S.M.A.; Rendeiro, A.F.; Weißer, J.; Puhm, F.; Hladik, A.; Göderle, L.; et al. Malondialdehyde epitopes are sterile mediators of hepatic inflammation in hypercholesterolemic mice. Hepatology 2017, 65, 28970. [Google Scholar] [CrossRef] [Green Version]
- Papac-Milicevic, N.; Busch, C.J.L.; Binder, C.J. Malondialdehyde Epitopes as Targets of Immunity and the Implications for Atherosclerosis. Adv. Immunol. 2016, 131, 1–59. [Google Scholar]
- Thiele, G.M.; Duryee, M.J.; Willis, M.S.; Sorrell, M.F.; Freeman, T.L.; Tuma, D.J.; Klassen, L.W. Malondialdehyde-acetaldehyde (MAA) modified proteins induce pro-inflammatory and pro-fibrotic responses by liver endothelial cells. Comp. Hepatol. 2004, 3 (Suppl. 1), S25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascale, A.; Govoni, S. The complex world of post-transcriptional mechanisms: Is their deregulation a common link for diseases? Focus on ELAV-like RNA-binding proteins. Cell. Mol. Life Sci. 2012, 69, 501–517. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Xu, L. The RNA-binding protein HuR in human cancer: A friend or foe? Adv. Drug Deliv. Rev. 2022, 184, 114179. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, H.; Guo, C.; Yu, F.; Zhang, Y.; Qiao, L.; Zhang, H.; Zhang, C. Role of hsa_circ_0000280 in regulating vascular smooth muscle cell function and attenuating neointimal hyperplasia via ELAVL1. Cell. Mol. Life Sci. 2023, 80, 3. [Google Scholar] [CrossRef]
- Trivlidis, J.; Aloufi, N.; Al-Habeeb, F.; Nair, P.; Azuelos, I.; Eidelman, D.H.; Baglole, C.J. HuR drives lung fibroblast differentiation but not metabolic reprogramming in response to TGF-β and hypoxia. Respir. Res. 2021, 22, 323. [Google Scholar] [CrossRef] [PubMed]
- Pistono, C.; Monti, M.C.; Marchesi, N.; Boiocchi, C.; Campagnoli, L.I.M.; Morlotti, D.; Cuccia, M.; Govoni, S.; Montomoli, C.; Mallucci, G.; et al. Unraveling a new player in multiple sclerosis pathogenesis: The RNA-binding protein HuR. Mult. Scler. Relat. Disord. 2020, 41, 102048. [Google Scholar] [CrossRef]
- Borgonetti, V.; Coppi, E.; Galeotti, N. Targeting the rna-binding protein hur as potential thera-peutic approach for neurological disorders: Focus on amyo-trophic lateral sclerosis (als), spinal muscle atrophy (sma) and multiple sclerosis. Int. J. Mol. Sci. 2021, 22, 10394. [Google Scholar] [CrossRef]
- Subramanian, P.; Gargani, S.; Palladini, A.; Chatzimike, M.; Grzybek, M.; Peitzsch, M.; Papanastasiou, A.D.; Pyrina, I.; Ntafis, V.; Gercken, B.; et al. The RNA binding protein human antigen R is a gatekeeper of liver homeostasis. Hepatology 2022, 75, 881–897. [Google Scholar] [CrossRef]
- Li, J.; Gong, L.; Liu, S.; Zhang, Y.; Zhang, C.; Tian, M.; Lu, H.; Bu, P.; Yang, J.; Ouyang, C.; et al. Adipose HuR protects against diet-induced obesity and insulin resistance. Nat. Commun. 2019, 10, 2375. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tai, Y.L.; Way, G.; Zeng, J.; Zhao, D.; Su, L.; Jiang, X.; Jackson, K.G.; Wang, X.; Gurley, E.C.; et al. RNA binding protein HuR protects against NAFLD by suppressing long noncoding RNA H19 expression. Cell Biosci. 2022, 12, 172. [Google Scholar] [CrossRef]
- Lebedeva, S.; Jens, M.; Theil, K.; Schwanhäusser, B.; Selbach, M.; Landthaler, M.; Rajewsky, N. Transcriptome-wide Analysis of Regulatory Interactions of the RNA-Binding Protein HuR. Mol. Cell 2011, 43, 340–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.S.; Tang, P.W.; Welles, J.E.; Pan, W.; Javed, Z.; Elhaw, A.T.; Mythreye, K.; Kimball, S.R.; Hempel, N. HuR-dependent SOD2 protein synthesis is an early adaptation to anchorage-independence. Redox Biol. 2022, 53, 102329. [Google Scholar] [CrossRef] [PubMed]
- Millien, G.; Wang, H.; Zhang, Z.; Alkon, D.L.; Hongpaisan, J. PKCε Activation Restores Loss of PKCε, Manganese Superoxide Dismutase, Vascular Endothelial Growth Factor, and Microvessels in Aged and Alzheimer’s Disease Hippocampus. Front. Aging Neurosci. 2022, 14, 836634. [Google Scholar] [CrossRef] [PubMed]
- Mallucci, G.; Marchesi, N.; Campagnoli, L.I.M.; Boschi, F.; Fahmideh, F.; Fusco, S.; Tavazzi, E.; Govoni, S.; Bergamaschi, R.; Pascale, A. Evidence for novel cell defense mechanisms sustained by dimethyl fumarate in multiple sclerosis patients: The HuR/SOD2 cascade. Mult. Scler. Relat. Disord. 2022, 68, 104197. [Google Scholar] [CrossRef]
- Amadio, M.; Scapagnini, G.; Davinelli, S.; Calabrese, V.; Govoni, S.; Pascale, A. Involvement of ELAV RNA-binding proteins in the post-transcriptional regulation of HO-1. Front. Cell. Neurosci. 2015, 8, 459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, C.; Stewart, A.G.; Woodman, O.L.; Ritchie, R.H.; Qin, C.X. Non-Alcoholic Steatohepatitis: A Review of Its Mechanism, Models and Medical Treatments. Front. Pharmacol. 2020, 11, 603926. [Google Scholar] [CrossRef]
- Tsikas, D. Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: Analytical and biological challenges. Anal. Biochem. 2017, 524, 13–30. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Vance, D.E. Phosphatidylcholine and choline homeostasis. J. Lipid Res. 2008, 49, 1187–1194. [Google Scholar] [CrossRef] [Green Version]
- Marí, M.; Colell, A.; Morales, A.; Von Montfort, C.; Garcia-Ruiz, C.; Fernández-Checa, J.C. Redox control of liver function in health and disease. Antioxid. Redox Signal. 2010, 12, 1295–1331. [Google Scholar] [CrossRef] [Green Version]
- Palladini, G.; Di Pasqua, L.G.; Cagna, M.; Croce, A.C.; Perlini, S.; Mannucci, B.; Profumo, A.; Ferrigno, A.; Vairetti, M. MCD Diet Rat Model Induces Alterations in Zinc and Iron during NAFLD Progression from Steatosis to Steatohepatitis. Int. J. Mol. Sci. 2022, 23, 6817. [Google Scholar] [CrossRef] [PubMed]
- Ferrigno, A.; Cagna, M.; Bosco, O.; Trucchi, M.; Berardo, C.; Nicoletti, F.; Vairetti, M.; Di Pasqua, L.G. MPEP Attenuates Intrahepatic Fat Accumulation in Obese Mice. Int. J. Mol. Sci. 2023, 24, 6076. [Google Scholar] [CrossRef] [PubMed]
- Spooner, H.C.; Derrick, S.A.; Maj, M.; Manjarín, R.; Hernandez, G.V.; Tailor, D.S.; Bastani, P.S.; Fanter, R.K.; Fiorotto, M.L.; Burrin, D.G.; et al. High-Fructose, High-Fat Diet Alters Muscle Composition and Fuel Utilization in a Juvenile Iberian Pig Model of Non-Alcoholic Fatty Liver Disease. Nutrients 2021, 13, 4195. [Google Scholar] [CrossRef]
- Bayliak, M.M.; Vatashchuk, M.V.; Gospodaryov, D.V.; Hurza, V.V.; Demianchuk, O.I.; Ivanochko, M.V.; Burdyliuk, N.I.; Storey, K.B.; Lushchak, O.; Lushchak, V.I. High fat high fructose diet induces mild oxidative stress and reorganizes intermediary metabolism in male mouse liver: Alpha-ketoglutarate effects. Biochim. Biophys. Acta Gen. Subj. 2022, 1866, 130226. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Gambino, R. Non-alcoholic steatohepatitis: Emerging molecular targets and therapeutic strategies. Nat. Rev. Drug Discov. 2016, 15, 249–274. [Google Scholar] [CrossRef]
- Tian, M.; Wang, J.; Liu, S.; Li, X.; Li, J.; Yang, J.; Zhang, C.; Zhang, W. Hepatic HuR protects against the pathogenesis of non-alcoholic fatty liver disease by targeting PTEN. Cell Death Dis. 2021, 12, 236. [Google Scholar] [CrossRef]
- Horie, Y.; Suzuki, A.; Kataoka, E.; Sasaki, T.; Hamada, K.; Sasaki, J.; Mizuno, K.; Hasegawa, G.; Kishimoto, H.; Iizuka, M.; et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J. Clin. Investig. 2004, 113, 1774–1783. [Google Scholar] [CrossRef] [Green Version]
- Lan, Q.; Ren, Z.; Chen, Y.; Cui, G.; Choi, I.C.; Ung, C.O.L.; Yu, H.H.; Lee, S.M.Y. Hepatoprotective effect of Qushihuayu formula on non-alcoholic steatohepatitis induced by MCD diet in rat. Chin. Med. 2021, 16, 27. [Google Scholar] [CrossRef]
- Stephenson, K.; Kennedy, L.; Hargrove, L.; Demieville, J.; Thomson, J.; Alpini, G.; Francis, H. Updates on Dietary Models of Nonalcoholic Fatty Liver Disease: Current Studies and Insights. Gene Expr. 2018, 18, 5. [Google Scholar] [CrossRef]
- Caballero, F.; Fernández, A.; Matías, N.; Martínez, L.; Fucho, R.; Elena, M.; Caballeria, J.; Morales, A.; Fernández-Checa, J.C.; García-Ruiz, C. Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: Impact on mitochondrial S-adenosyl-L-methionine and glutathione. J. Biol. Chem. 2010, 285, 18528–18536. [Google Scholar] [CrossRef] [Green Version]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.H.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Croce, A.C.; Ferrigno, A.; Di Pasqua, L.G.; Berardo, C.; Bottiroli, G.; Vairetti, M. NAD(P)H and Flavin Autofluorescence Correlation with ATP in Rat Livers with Different Metabolic Steady-State Conditions. Photochem. Photobiol. 2017, 93, 1519–1524. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zong, C.; Jiang, M.; Hu, H.; Cheng, X.; Ni, J.; Yi, X.; Jiang, B.; Tian, F.; Chang, M.W.; et al. Hepatic HuR modulates lipid homeostasis in response to high-fat diet. Nat. Commun. 2020, 11, 3067. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tang, T.; Wang, G.D.; Liu, B. LncRNA-H19 promotes hepatic lipogenesis by directly regulating miR-130a/PPARγ axis in non-alcoholic fatty liver disease. Biosci. Rep. 2019, 39, 20181722. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.S.; Chang, T.E.; Perng, C.L.; Huang, Y.H. Genetic variations of three important antioxidative enzymes SOD2, CAT, and GPX1 in nonalcoholic steatohepatitis. J. Chin. Med. Assoc. 2021, 84, 14–18. [Google Scholar] [CrossRef]
- He, J.; Hu, B.; Shi, X.; Weidert, E.R.; Lu, P.; Xu, M.; Huang, M.; Kelley, E.E.; Xie, W. Activation of the Aryl Hydrocarbon Receptor Sensitizes Mice to Nonalcoholic Steatohepatitis by Deactivating Mitochondrial Sirtuin Deacetylase Sirt3. Mol. Cell. Biol. 2013, 33, 2047–2055. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Ren, W.; Zhang, Q.; Fu, N.; Han, F.; Cui, P.; Li, W.; Kong, L.; Zhao, S.; Wang, R.; et al. Heme Oxygenase-1 Suppresses Wnt Signaling Pathway in Nonalcoholic Steatohepatitis-Related Liver Fibrosis. BioMed Res. Int. 2020, 2020, 4910601. [Google Scholar] [CrossRef]
- Li, D.; Zhao, D.; Du, J.; Dong, S.; Aldhamin, Z.; Yuan, X.; Li, W.; Du, H.; Zhao, W.; Cui, L.; et al. Heme oxygenase-1 alleviated non-alcoholic fatty liver disease via suppressing ROS-dependent endoplasmic reticulum stress. Life Sci. 2020, 253, 117678. [Google Scholar] [CrossRef]
- Li, S.; Qiu, B.; Lu, H.; Lai, Y.; Liu, J.; Luo, J.; Zhu, F.; Hu, Z.; Zhou, M.; Tian, J.; et al. Hyperhomocysteinemia Accelerates Acute Kidney Injury to Chronic Kidney Disease Progression by Downregulating Heme Oxygenase-1 Expression. Antioxid. Redox Signal. 2019, 30, 1635–1650. [Google Scholar] [CrossRef]
- Dery, K.J.; Nakamura, K.; Kadono, K.; Hirao, H.; Kageyama, S.; Ito, T.; Kojima, H.; Kaldas, F.M.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Human Antigen R (HuR): A Regulator of Heme Oxygenase-1 Cytoprotection in Mouse and Human Liver Transplant Injury. Hepatology 2020, 72, 31093. [Google Scholar] [CrossRef]
- Di Pasqua, L.G.; Berardo, C.; Cagna, M.; Mannucci, B.; Milanesi, G.; Croce, A.C.; Ferrigno, A.; Vairetti, M. Long-term cold storage preservation does not affect fatty livers from rats fed with a methionine and choline deficient diet. Lipids Health Dis. 2021, 20, 78. [Google Scholar] [CrossRef]
- Lehninger, A.L.; Vercesi, A.; Bababunmi, E.A. Regulation of Ca2+ release from mitochondria by the oxidation-reduction state of pyridine nucleotides. Proc. Natl. Acad. Sci. USA 1978, 75, 1690–1694. [Google Scholar] [CrossRef] [Green Version]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Ferrigno, A.; Berardo, C.; Di Pasqua, L.G.; Siciliano, V.; Richelmi, P.; Nicoletti, F.; Vairetti, M. Selective blockade of the metabotropic glutamate receptor mGluR5 protects mouse livers in in vitro and ex vivo models of ischemia reperfusion injury. Int. J. Mol. Sci. 2018, 19, 314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esterbauer, H.; Cheeseman, K.H. Determination of aldehydic lipid peroxidation products: Malonaldehyde and 4-hydroxynonenal. Methods Enzymol. 1990, 186, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Croce, A.C.A.C.; Ferrigno, A.; Di Pasqua, L.G.L.G.; Berardo, C.; Piccolini, V.M.V.M.; Bertone, V.; Bottiroli, G.; Vairetti, M. Autofluorescence discrimination of metabolic fingerprint in nutritional and genetic fatty liver models. J. Photochem. Photobiol. B Biol. 2016, 164, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, D.W. An Algorithm for Least-Squares Estimation of Nonlinear Parameters. J. Soc. Ind. Appl. Math. 1963, 11, 431–441. [Google Scholar] [CrossRef]
- Lyn-Cook, L.E.; Lawton, M.; Tong, M.; Silbermann, E.; Longato, L.; Jiao, P.; Mark, P.; Wands, J.R.; Xu, H.; de la Monte, S.M. Hepatic ceramide may mediate brain insulin resistance and neurodegeneration in type 2 diabetes and non-alcoholic steatohepatitis. J. Alzheimer’s Dis. 2009, 16, 715–729. [Google Scholar] [CrossRef] [Green Version]
- Ferrigno, A.; Di Pasqua, L.; Berardo, C.; Siciliano, V.; Rizzo, V.; Mannucci, B.; Richelmi, P.; Croce, A.; Vairetti, M. Liver Graft Susceptibility during Static Cold Storage and Dynamic Machine Perfusion: DCD versus Fatty Livers. Int. J. Mol. Sci. 2017, 19, 109. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3w | 6w | |||
|---|---|---|---|---|
| CTRL | MCD | CTRL | MCD | |
| AST (mU/mL) | 84.5 ± 1.7 | 109.5 ± 7.5 * | 73.6 ± 13.7 | 144 ± 17.4 * |
| ALT (mU/mL) | 21.5 ± 2 | 81.5 ± 15 * | 37.3 ± 7.6 | 168.6 ± 9.8 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrigno, A.; Campagnoli, L.I.M.; Barbieri, A.; Marchesi, N.; Pascale, A.; Croce, A.C.; Vairetti, M.; Di Pasqua, L.G. MCD Diet Modulates HuR and Oxidative Stress-Related HuR Targets in Rats. Int. J. Mol. Sci. 2023, 24, 9808. https://doi.org/10.3390/ijms24129808
Ferrigno A, Campagnoli LIM, Barbieri A, Marchesi N, Pascale A, Croce AC, Vairetti M, Di Pasqua LG. MCD Diet Modulates HuR and Oxidative Stress-Related HuR Targets in Rats. International Journal of Molecular Sciences. 2023; 24(12):9808. https://doi.org/10.3390/ijms24129808
Chicago/Turabian StyleFerrigno, Andrea, Lucrezia Irene Maria Campagnoli, Annalisa Barbieri, Nicoletta Marchesi, Alessia Pascale, Anna Cleta Croce, Mariapia Vairetti, and Laura Giuseppina Di Pasqua. 2023. "MCD Diet Modulates HuR and Oxidative Stress-Related HuR Targets in Rats" International Journal of Molecular Sciences 24, no. 12: 9808. https://doi.org/10.3390/ijms24129808
APA StyleFerrigno, A., Campagnoli, L. I. M., Barbieri, A., Marchesi, N., Pascale, A., Croce, A. C., Vairetti, M., & Di Pasqua, L. G. (2023). MCD Diet Modulates HuR and Oxidative Stress-Related HuR Targets in Rats. International Journal of Molecular Sciences, 24(12), 9808. https://doi.org/10.3390/ijms24129808