
Improving Soluble Expression of SARS-CoV-2 Spike Priming Protease TMPRSS2 with an Artificial Fusing Protein

Abstract
:1. Introduction
2. Results
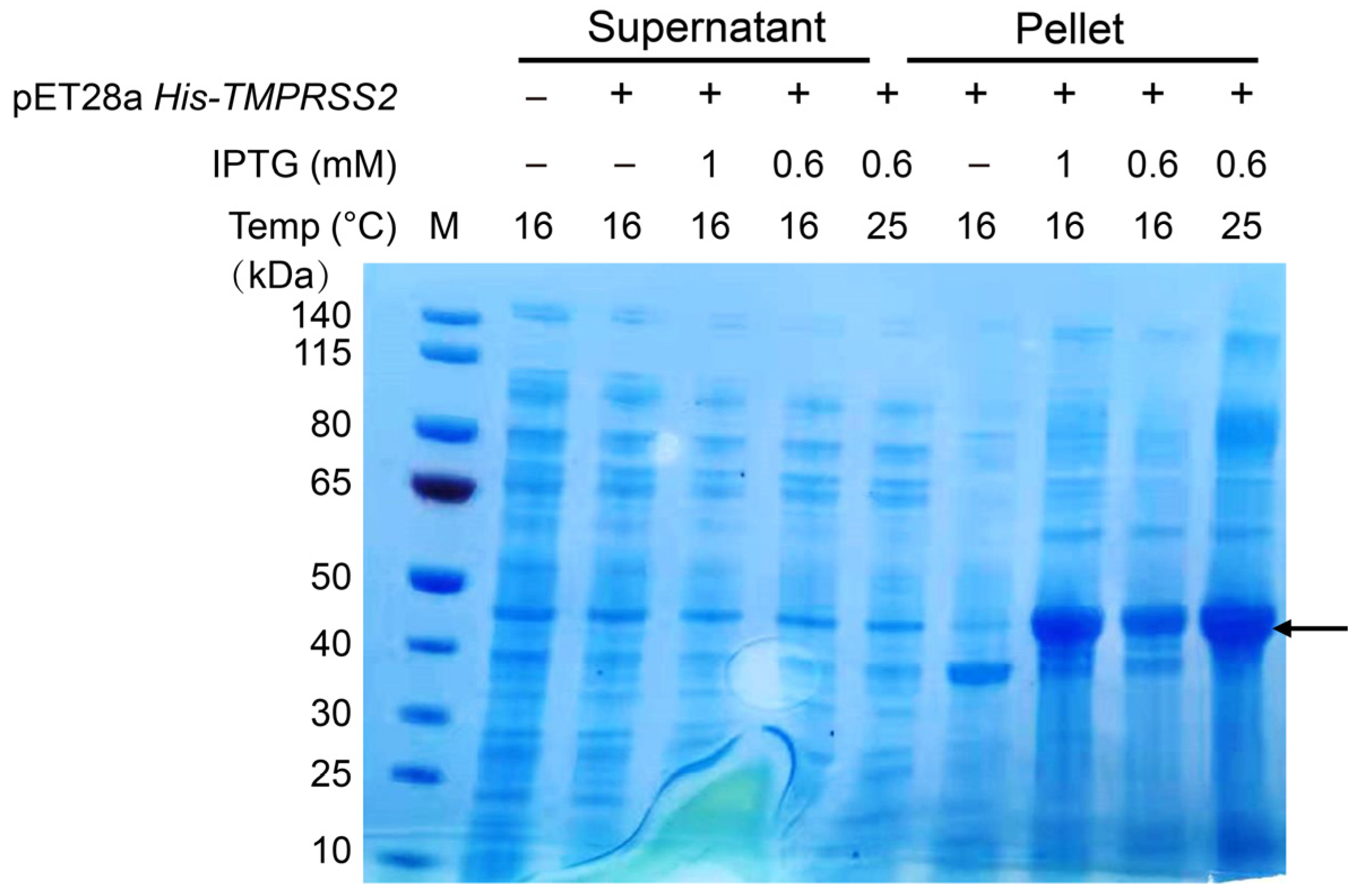
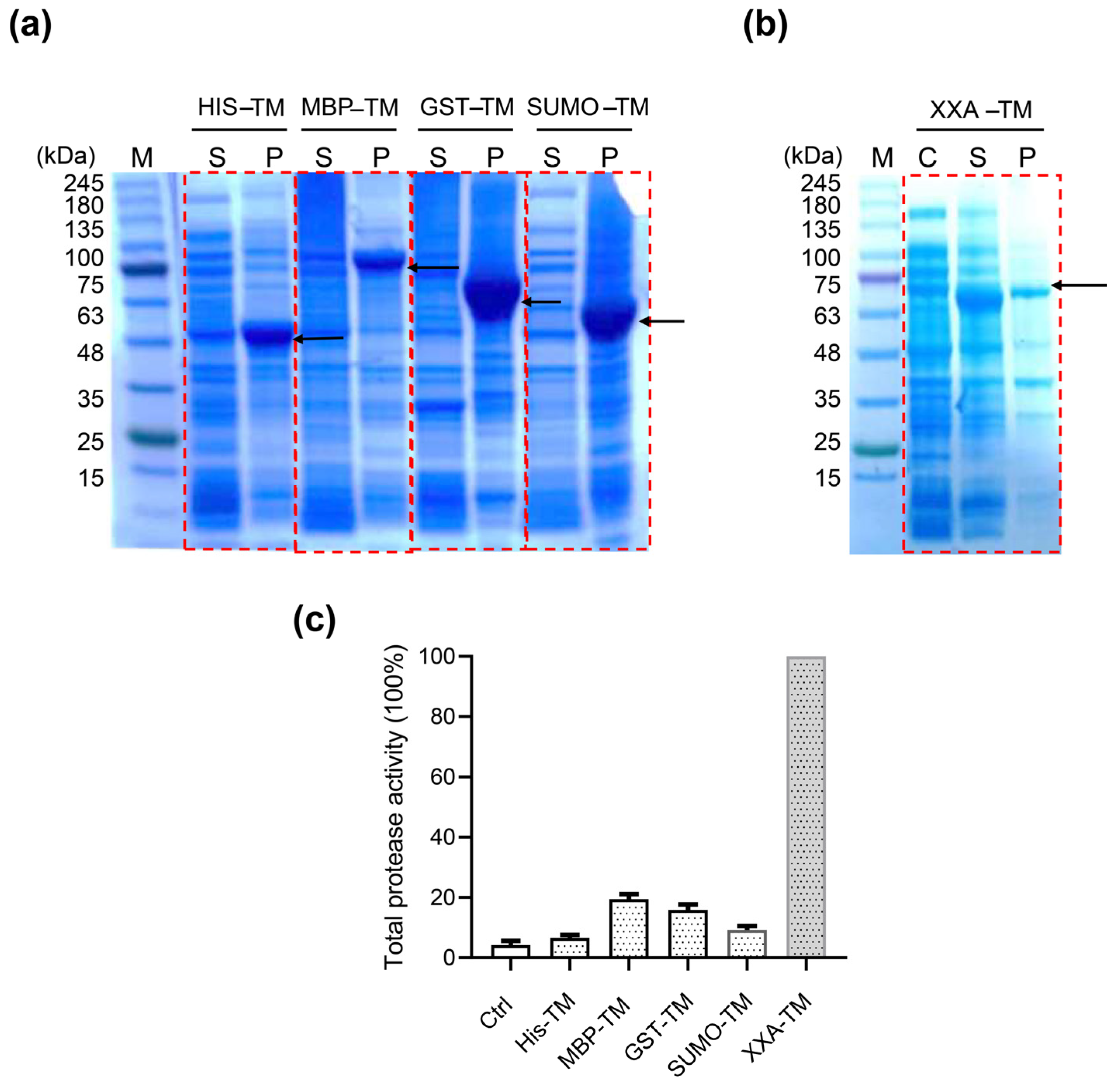
2.1. XXA Fusion Protein Increased the Expression of Soluble TMPRSS2 in E. coli
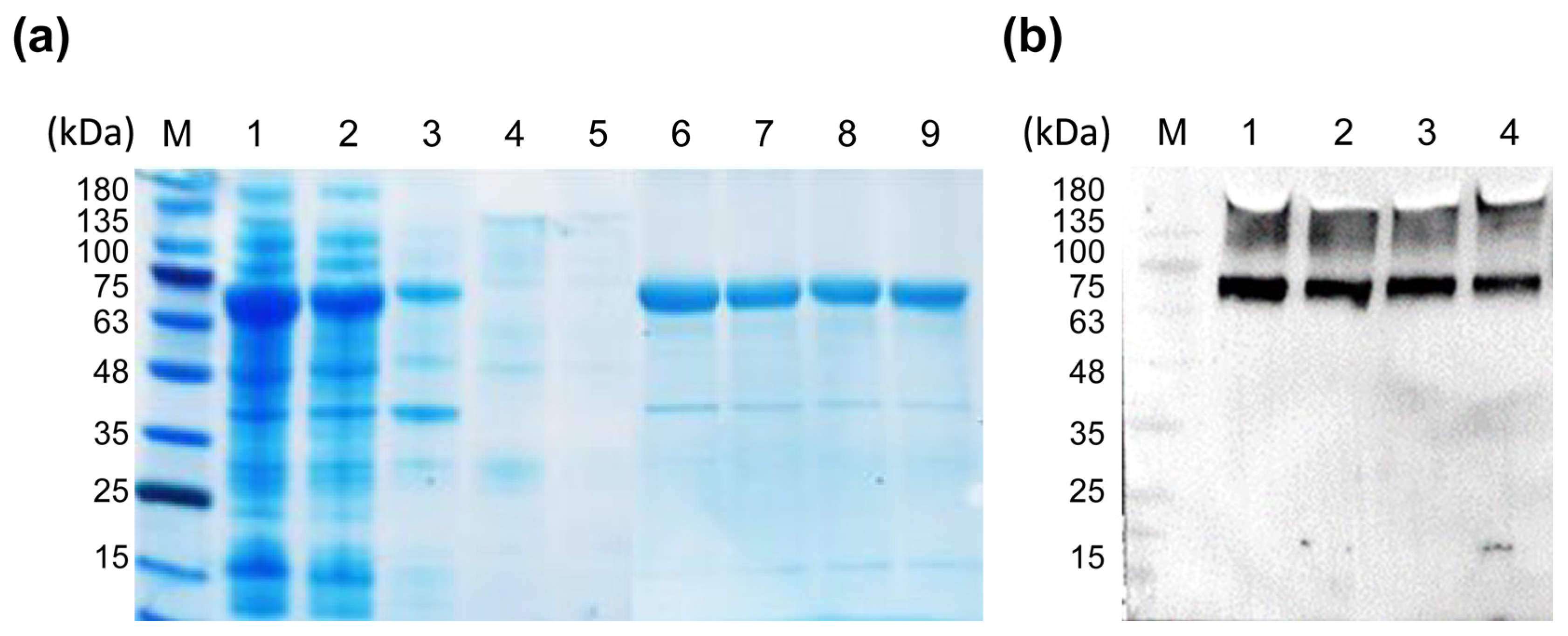
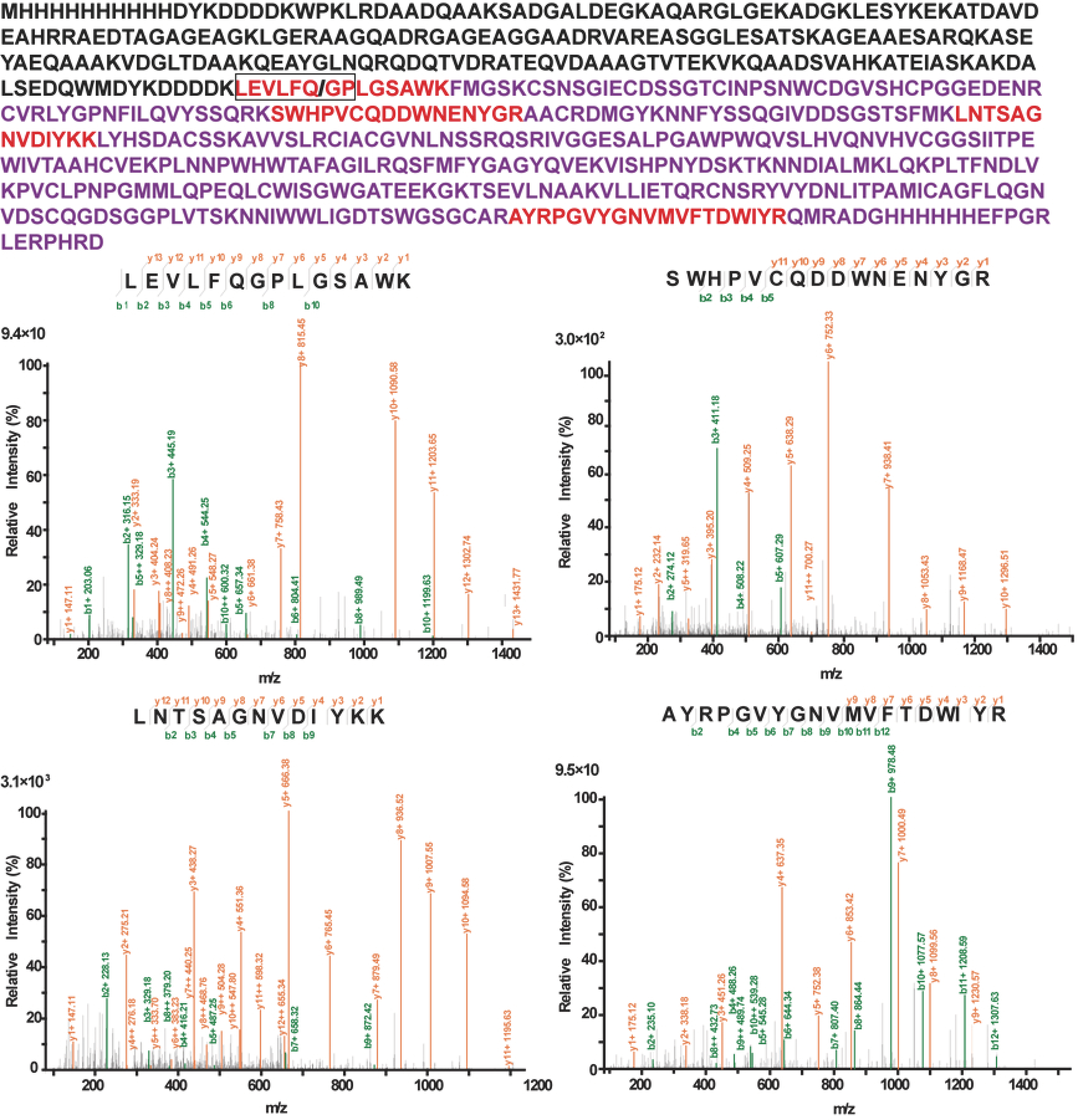
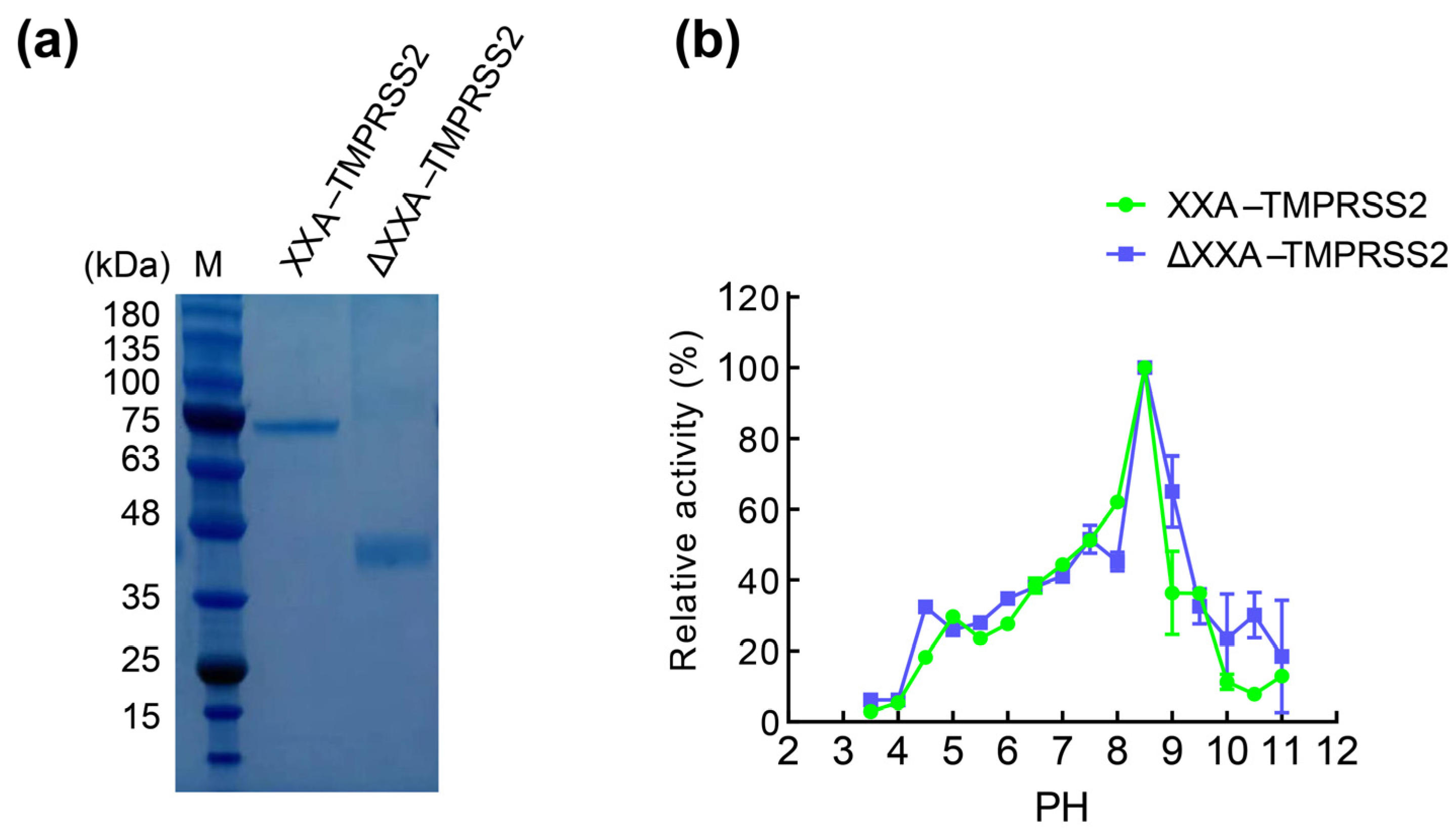
2.2. Purification and Identification of XXA–TMPRSS2
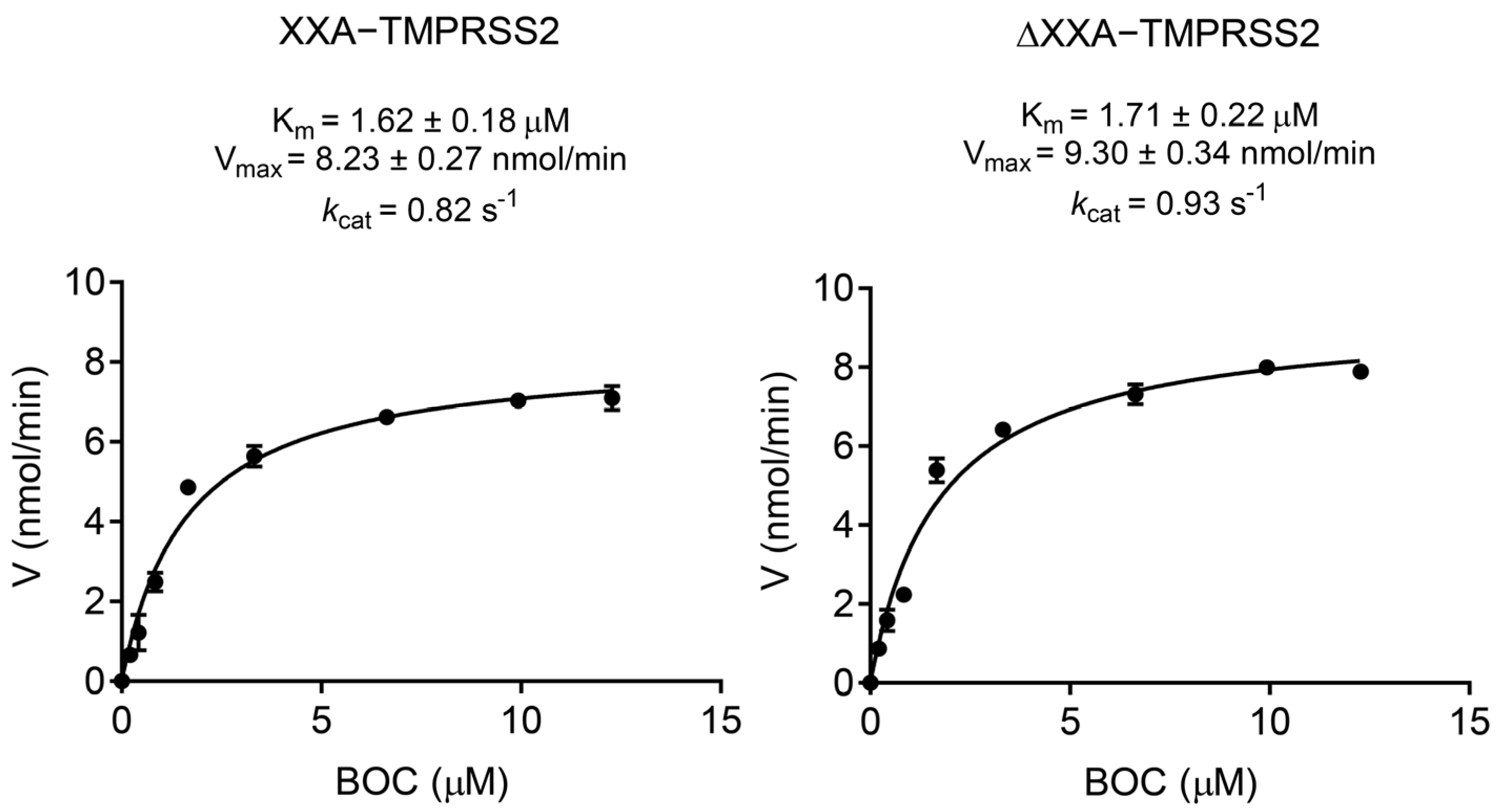
2.3. The Fusing Protein XXA Does Not Influence Catalytic Efficiency of TMPRSS2
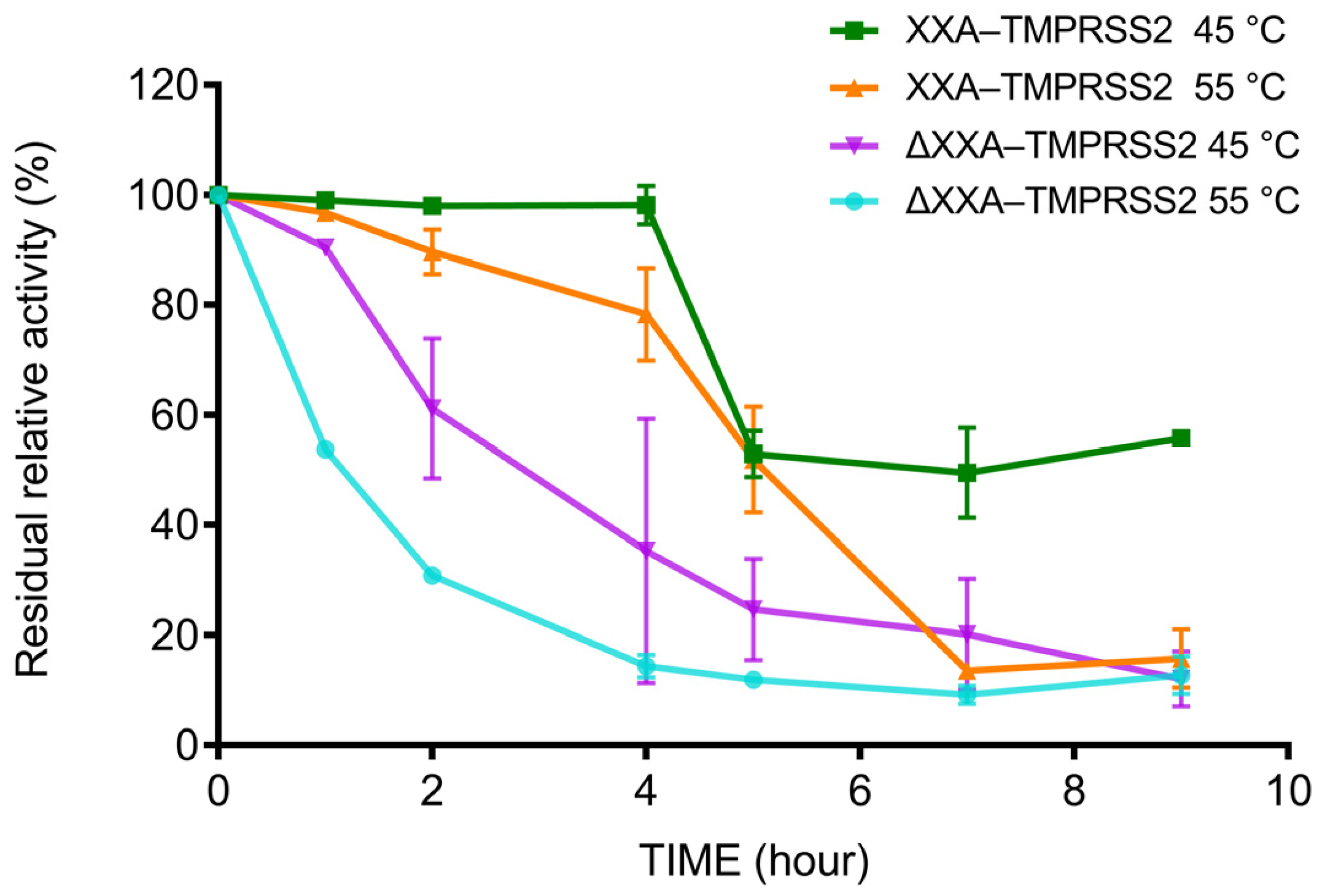
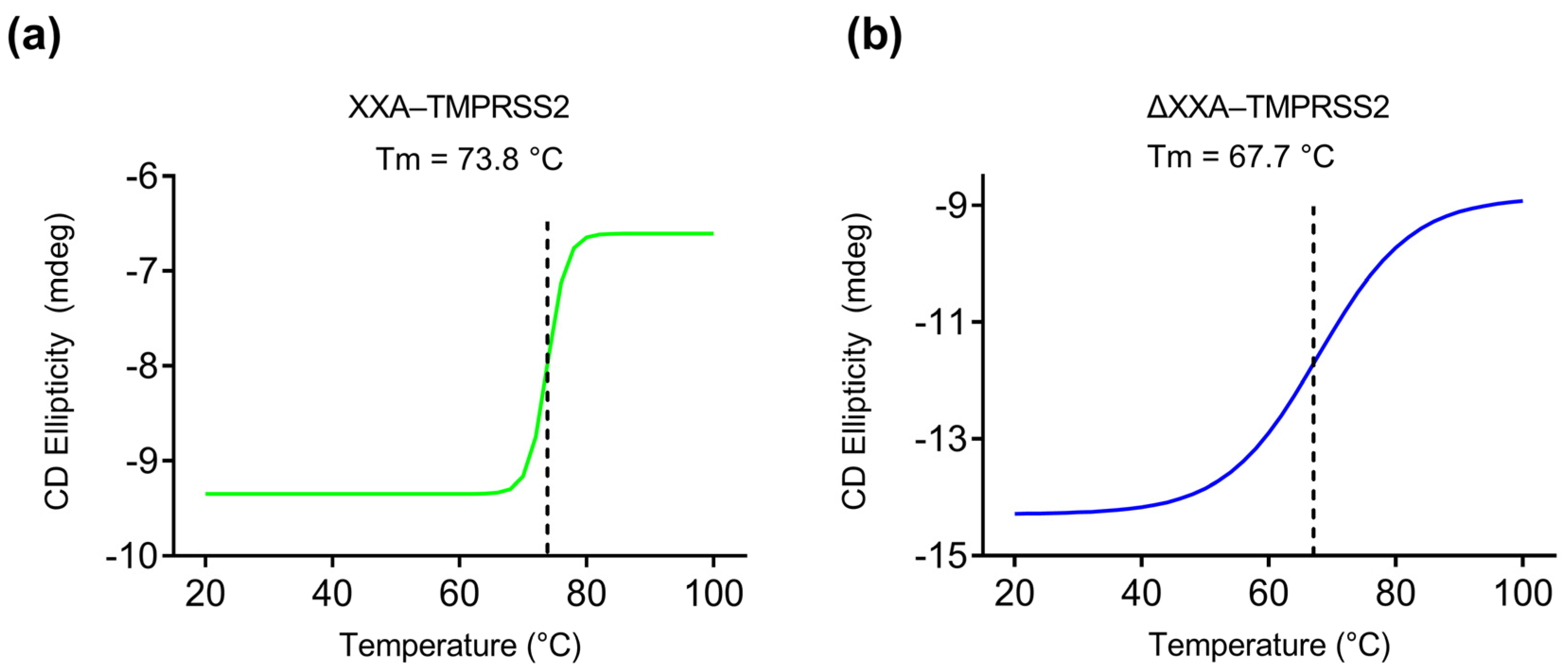
2.4. XXA Tag Improves the Thermostability of TMPRSS2
3. Discussion
4. Materials and Methods
4.1. E. coli Strains and Reagents
4.2. Amino Acid Sequence
4.3. Gene Cloning and Protein Expression
4.4. Protein Purification and XXA Tag Removal
4.5. SDS–PAGE and Western Blotting
4.6. Mass Spectrometry
4.7. Enzymatic Activity Assay
4.8. Enzyme Kinetic Studies
4.9. Circular Dichroism (CD) Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- de Loyola, M.B.; Dos Reis, T.T.A.; de Oliveira, G.; da Fonseca Palmeira, J.; Arganaraz, G.A.; Arganaraz, E.R. Alpha–1–antitrypsin: A possible host protective factor against COVID-19. Rev. Med. Virol. 2021, 31, e2157. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine–Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Wang, M.Y.; Zhao, R.; Gao, L.J.; Gao, X.F.; Wang, D.P.; Cao, J.M. SARS-CoV-2: Structure, Biology, and Structure–Based Therapeutics Development. Front. Cell. Infect. Microbiol. 2020, 10, 587269. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 183, 1735. [Google Scholar] [CrossRef]
- Papa, G.; Mallery, D.L.; Albecka, A.; Welch, L.G.; Cattin–Ortola, J.; Luptak, J.; Paul, D.; McMahon, H.T.; Goodfellow, I.G.; Carter, A.; et al. Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell–cell fusion. PLoS Pathog. 2021, 17, e1009246. [Google Scholar] [CrossRef] [PubMed]
- Bertram, S.; Glowacka, I.; Blazejewska, P.; Soilleux, E.; Allen, P.; Danisch, S.; Steffen, I.; Choi, S.Y.; Park, Y.; Schneider, H.; et al. TMPRSS2 and TMPRSS4 facilitate trypsin–independent spread of influenza virus in Caco–2 cells. J. Virol. 2010, 84, 10016–10025. [Google Scholar] [CrossRef] [Green Version]
- Lam, D.K.; Dang, D.; Flynn, A.N.; Hardt, M.; Schmidt, B.L. TMPRSS2, a novel membrane–anchored mediator in cancer pain. Pain 2015, 156, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.S.; Heinlein, C.; Hackman, R.C.; Nelson, P.S. Phenotypic analysis of mice lacking the Tmprss2–encoded protease. Mol. Cell. Biol. 2006, 26, 965–975. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.W.; Mao, H.J.; Wu, Y.L.; Tanaka, Y.; Zhang, W. TMPRSS2: A potential target for treatment of influenza virus and coronavirus infections. Biochimie 2017, 142, 1–10. [Google Scholar] [CrossRef]
- Li, F.; Han, M.; Dai, P.; Xu, W.; He, J.; Tao, X.; Wu, Y.; Tong, X.; Xia, X.; Guo, W.; et al. Distinct mechanisms for TMPRSS2 expression explain organ–specific inhibition of SARS-CoV-2 infection by enzalutamide. Nat. Commun. 2021, 12, 866. [Google Scholar] [CrossRef]
- Shrimp, J.H.; Kales, S.C.; Sanderson, P.E.; Simeonov, A.; Shen, M.; Hall, M.D. An Enzymatic TMPRSS2 Assay for Assessment of Clinical Candidates and Discovery of Inhibitors as Potential Treatment of COVID-19. ACS Pharmacol. Transl. Sci. 2020, 3, 997–1007. [Google Scholar] [CrossRef]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Van Lam van, T.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef]
- Makrides, S.C. Strategies for achieving high–level expression of genes in Escherichia coli. Microbiol. Rev. 1996, 60, 512–538. [Google Scholar] [CrossRef]
- Meyer, D.; Sielaff, F.; Hammami, M.; Bottcher–Friebertshauser, E.; Garten, W.; Steinmetzer, T. Identification of the first synthetic inhibitors of the type II transmembrane serine protease TMPRSS2 suitable for inhibition of influenza virus activation. Biochem. J. 2013, 452, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Wu, P.; Huang, X.; Bai, W.; Li, B.; Shi, N. Retro–Protein XXA is a remarkable solubilizing fusion tag for inclusion bodies. Microb. Cell Fact. 2022, 21, 51. [Google Scholar] [CrossRef]
- Fraser, B.J.; Beldar, S.; Seitova, A.; Hutchinson, A.; Mannar, D.; Li, Y.; Kwon, D.; Tan, R.; Wilson, R.P.; Leopold, K.; et al. Structure, activity and inhibition of human TMPRSS2, a protease implicated in SARS-CoV-2 activation. bioRxiv 2021. bioRxiv 2021.06.23.449282. [Google Scholar]
- Paoloni–Giacobino, A.; Chen, H.; Peitsch, M.C.; Rossier, C.; Antonarakis, S.E. Cloning of the TMPRSS2 gene, which encodes a novel serine protease with transmembrane, LDLRA, and SRCR domains and maps to 21q22.3. Genomics 1997, 44, 309–320. [Google Scholar] [CrossRef]
- Peiffer, A.L.; Garlick, J.M.; Wu, Y.; Soellner, M.B.; Brooks, C.L.; Mapp, A.K. TMPRSS2 inhibitor discovery facilitated through an in silico and biochemical screening platform. bioRxiv 2021. [Google Scholar] [CrossRef]
- Peciak, K.; Tommasi, R.; Choi, J.W.; Brocchini, S.; Laurine, E. Expression of soluble and active interferon consensus in SUMO fusion expression system in E. coli. Protein Expr. Purif. 2014, 99, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.D.; Kapust, R.B.; Waugh, D.S. Single amino acid substitutions on the surface of Escherichia coli maltose–binding protein can have a profound impact on the solubility of fusion proteins. Protein Sci. 2001, 10, 622–630. [Google Scholar] [CrossRef] [Green Version]
- Kapust, R.B.; Waugh, D.S. Escherichia coli maltose–binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 1999, 8, 1668–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.B.; Johnson, K.S. Single–step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S–transferase. Gene 1988, 67, 31–40. [Google Scholar] [CrossRef]
- Bottcher–Friebertshauser, E.; Klenk, H.D.; Garten, W. Activation of influenza viruses by proteases from host cells and bacteria in the human airway epithelium. Pathog. Dis. 2013, 69, 87–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahoney, M.; Damalanka, V.C.; Tartell, M.A.; Chung, D.H.; Lourenco, A.L.; Pwee, D.; Mayer Bridwell, A.E.; Hoffmann, M.; Voss, J.; Karmakar, P.; et al. A novel class of TMPRSS2 inhibitors potently block SARS-CoV-2 and MERS–CoV viral entry and protect human epithelial lung cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2108728118. [Google Scholar] [CrossRef]
- de Marco, A. Strategies for successful recombinant expression of disulfide bond–dependent proteins in Escherichia coli. Microb. Cell Fact. 2009, 8, 26. [Google Scholar] [CrossRef] [Green Version]
- Schlegel, S.; Rujas, E.; Ytterberg, A.J.; Zubarev, R.A.; Luirink, J.; de Gier, J.W. Optimizing heterologous protein production in the periplasm of E. coli by regulating gene expression levels. Microb. Cell Fact. 2013, 12, 24. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid | Inserted Gene | Primer F | Primer R |
|---|---|---|---|
| pET−His−TMPRSS2 | His−TMPRSS2 | TGGGTCGCGGATCCGCCTGGAAATTTATGGGC | GTGCGGCCGCAAGCTTGCCATCCGCGCGCATC |
| pET−MBP−TMPRSS2 | MBP−TMPRSS2 | AACAACCTCGGGGAATTCTGGAAATTTATGGGC | GGTGGTGGTGCTCGAGTTAGCCATCCGCGCGCATCTGAC |
| pET−SUMO−TMPRSS2 | SUMO−TMPRSS2 | CCAGGGGCCCGGATCCTGGAAATTTATGGGC | CGGCCGCAAGCTTGTTAGCCATCCGCGCGCA |
| pGEX−GST−TMPRSS2 | GST−TMPRSS2 | GGGGCCCCTGGGATCCTGGAAATTTATGGGC | AATTCTTAATGATGATGATGATGATGGCCATCCGCGCGC |
| pET−XXA–TMPRSS2 | XXA–TMPRSS2 | CAGGGGCCCGAATTCTGGAAATTTATGGGC | GGTGGTGGTGCTCGAGTTAGCCATCCGCGCGC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, X.; Ling, X.; Wu, M.; Bai, G.; Yuan, M.; Rao, L. Improving Soluble Expression of SARS-CoV-2 Spike Priming Protease TMPRSS2 with an Artificial Fusing Protein. Int. J. Mol. Sci. 2023, 24, 10475. https://doi.org/10.3390/ijms241310475
Ye X, Ling X, Wu M, Bai G, Yuan M, Rao L. Improving Soluble Expression of SARS-CoV-2 Spike Priming Protease TMPRSS2 with an Artificial Fusing Protein. International Journal of Molecular Sciences. 2023; 24(13):10475. https://doi.org/10.3390/ijms241310475
Chicago/Turabian StyleYe, Xiao, Xue Ling, Min Wu, Guijie Bai, Meng Yuan, and Lang Rao. 2023. "Improving Soluble Expression of SARS-CoV-2 Spike Priming Protease TMPRSS2 with an Artificial Fusing Protein" International Journal of Molecular Sciences 24, no. 13: 10475. https://doi.org/10.3390/ijms241310475
APA StyleYe, X., Ling, X., Wu, M., Bai, G., Yuan, M., & Rao, L. (2023). Improving Soluble Expression of SARS-CoV-2 Spike Priming Protease TMPRSS2 with an Artificial Fusing Protein. International Journal of Molecular Sciences, 24(13), 10475. https://doi.org/10.3390/ijms241310475