
α5-GABAA Receptor Modulation Reverses Behavioral and Neurophysiological Correlates of Psychosis in Rats with Ventral Hippocampal Alzheimer’s Disease-like Pathology
 , , ,
, , ,  and
and 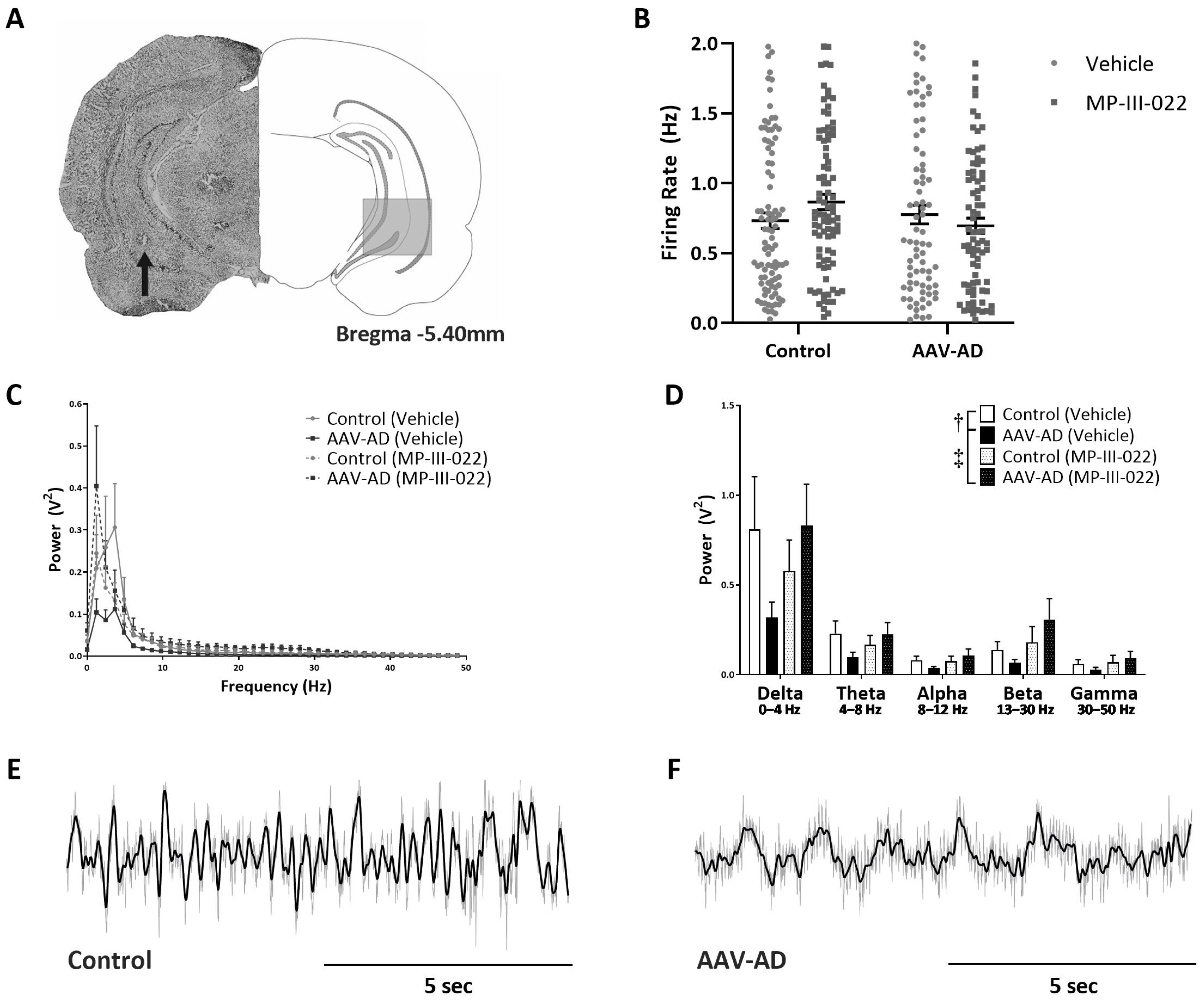{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. AAV-AD Rats Display Impairments in Hippocampal Activity That Are Reversed by Systemic Administration of the Selective α5-PAM, MP-III-022
2.2. AAV-AD Rats Exhibit Aberrant Dopamine System Function That Is Reversed by Systemic Administration of the Selective α5-PAM, MP-III-022
2.3. AAV-AD Rats Do Not Display Cognitive Deficits but Exhibit Specific Deficits in a dopamine-Dependent Behavior, Which Was Reversed By Systemic Administration of the Selective α5-PAM, MP-III-022
2.4. Parvalbumin Positive Interneurons are Decreased in AAV-AD Rats
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. In Vivo Extracellular Electrophysiology Recordings
4.3. Y-Maze Spontaneous Alternation Assay
4.4. Pre-Pulse Inhibition of Startle (PPI)
4.5. Stimulant-Induced Locomotor Activity
4.6. Immunohistochemistry
4.7. Histology
4.8. Analysis
4.9. Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rajan, K.B.; Weuve, J.; Barnes, L.L.; McAninch, E.A.; Wilson, R.S.; Evans, D.A. Population estimate of people with clinical Alzheimer’s disease and mild cognitive impairment in the United States (2020–2060). Alzheimer’s Dement. 2021, 17, 1966–1975. [Google Scholar] [CrossRef]
- Drevets, W.C.; Rubin, E.H. Psychotic symptoms and the longitudinal course of senile dementia of the Alzheimer type. Biol. Psychiatry 1989, 25, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Abi-Dargham, A. Do we still believe in the dopamine hypothesis? New data bring new evidence. Int. J. Neuropsychopharmacol. 2004, 7 (Suppl. S1), S1–S5. [Google Scholar] [CrossRef]
- Laruelle, M.; Abi-Dargham, A. Dopamine as the wind of the psychotic fire: New evidence from brain imaging studies. J. Psychopharmacol. 1999, 13, 358–371. [Google Scholar] [CrossRef]
- Palop, J.J.; Chin, J.; Roberson, E.D.; Wang, J.; Thwin, M.T.; Bien-Ly, N.; Yoo, J.; Ho, K.O.; Yu, G.Q.; Kreitzer, A.; et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 2007, 55, 697–711. [Google Scholar] [CrossRef] [Green Version]
- Schobel, S.A.; Kelly, M.A.; Corcoran, C.M.; Van Heertum, K.; Seckinger, R.; Goetz, R.; Harkavy-Friedman, J.; Malaspina, D. Anterior hippocampal and orbitofrontal cortical structural brain abnormalities in association with cognitive deficits in schizophrenia. Schizophr. Res. 2009, 114, 110–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodge, D.J.; Grace, A.A. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J. Neurosci. 2007, 27, 11424–11430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donegan, J.J.; Boley, A.M.; Yamaguchi, J.; Toney, G.M.; Lodge, D.J. Modulation of extrasynaptic GABAA alpha 5 receptors in the ventral hippocampus normalizes physiological and behavioral deficits in a circuit specific manner. Nat. Commun. 2019, 10, 2819. [Google Scholar] [CrossRef] [Green Version]
- Fritschy, J.M.; Mohler, H. GABAA-receptor heterogeneity in the adult rat brain: Differential regional and cellular distribution of seven major subunits. J. Comp. Neurol. 1995, 359, 154–194. [Google Scholar] [CrossRef]
- Perez, S.M.; Boley, A.M.; McCoy, A.M.; Lodge, D.J. Aberrant dopamine system function in the ferrous amyloid buthionine (FAB) rat model for Alzheimer’s disease. Int. J. Mol. Sci. 2023, 24, 7196. [Google Scholar] [CrossRef]
- Stamenic, T.T.; Poe, M.M.; Rehman, S.; Santrac, A.; Divovic, B.; Scholze, P.; Ernst, M.; Cook, J.M.; Savic, M.M. Ester to amide substitution improves selectivity, efficacy and kinetic behavior of a benzodiazepine positive modulator of GABA(A) receptors containing the alpha5 subunit. Eur. J. Pharmacol. 2016, 791, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, S.M.; Lodge, D.J. Hippocampal interneuron transplants reverse aberrant dopamine system function and behavior in a rodent model of schizophrenia. Mol. Psychiatry 2013, 18, 1193–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boley, A.M.; Perez, S.M.; Lodge, D.J. A fundamental role for hippocampal parvalbumin in the dopamine hyperfunction associated with schizophrenia. Schizophr. Res. 2014, 157, 238–243. [Google Scholar] [CrossRef] [Green Version]
- Perez, S.M.; Boley, A.; Lodge, D.J. Region specific knockdown of Parvalbumin or Somatostatin produces neuronal and behavioral deficits consistent with those observed in schizophrenia. Transl. Psychiatry 2019, 9, 264. [Google Scholar] [CrossRef] [Green Version]
- Perez, S.M.; Aguilar, D.D.; Neary, J.L.; Carless, M.A.; Giuffrida, A.; Lodge, D.J. Schizophrenia-Like Phenotype Inherited by the F2 Generation of a Gestational Disruption Model of Schizophrenia. Neuropsychopharmacology 2016, 41, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Perez, S.M.; Chen, L.; Lodge, D.J. Alterations in dopamine system function across the estrous cycle of the MAM rodent model of schizophrenia. Psychoneuroendocrinology 2014, 47, 88–97. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, D.D.; Chen, L.; Lodge, D.J. Increasing Endocannabinoid Levels in the Ventral Pallidum Restores Aberrant Dopamine Neuron Activity in the Subchronic PCP Rodent Model of Schizophrenia. Int. J. Neuropsychopharmacol. 2015, 18, pyu035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, J.; Tabira, T.; Sano, M.; Nakayama, H.; Tateishi, J. Parvalbumin-immunoreactive neurons in the human central nervous system are decreased in Alzheimer’s disease. Acta Neuropathol. 1991, 81, 388–395. [Google Scholar] [CrossRef]
- Brady, D.R.; Mufson, E.J. Parvalbumin-immunoreactive neurons in the hippocampal formation of Alzheimer’s diseased brain. Neuroscience 1997, 80, 1113–1125. [Google Scholar] [CrossRef]
- Lodge, D.J.; Behrens, M.M.; Grace, A.A. A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J. Neurosci. 2009, 29, 2344–2354. [Google Scholar] [CrossRef] [Green Version]
- 2023 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2023, 19, 1598–1695. [CrossRef]
- Murray, P.S.; Kumar, S.; Demichele-Sweet, M.A.; Sweet, R.A. Psychosis in Alzheimer’s disease. Biol. Psychiatry 2014, 75, 542–552. [Google Scholar] [CrossRef] [Green Version]
- Kales, H.C.; Kim, H.M.; Zivin, K.; Valenstein, M.; Seyfried, L.S.; Chiang, C.; Cunningham, F.; Schneider, L.S.; Blow, F.C. Risk of mortality among individual antipsychotics in patients with dementia. Am. J. Psychiatry 2012, 169, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Marchani, E.E.; Bird, T.D.; Steinbart, E.J.; Rosenthal, E.; Yu, C.E.; Schellenberg, G.D.; Wijsman, E.M. Evidence for three loci modifying age-at-onset of Alzheimer’s disease in early-onset PSEN2 families. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2010, 153B, 1031–1041. [Google Scholar]
- Pedersen, W.A.; McCullers, D.; Culmsee, C.; Haughey, N.J.; Herman, J.P.; Mattson, M.P. Corticotropin-releasing hormone protects neurons against insults relevant to the pathogenesis of Alzheimer’s disease. Neurobiol. Dis. 2001, 8, 492–503. [Google Scholar] [CrossRef] [Green Version]
- Cohen, R.M.; Rezai-Zadeh, K.; Weitz, T.M.; Rentsendorj, A.; Gate, D.; Spivak, I.; Bholat, Y.; Vasilevko, V.; Glabe, C.G.; Breunig, J.J.; et al. A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric abeta, and frank neuronal loss. J. Neurosci. 2013, 33, 6245–6256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jawhar, S.; Trawicka, A.; Jenneckens, C.; Bayer, T.A.; Wirths, O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Abeta aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 196.e29–196.e40. [Google Scholar] [CrossRef] [PubMed]
- Gordon, B.A.; Blazey, T.M.; Su, Y.; Hari-Raj, A.; Dincer, A.; Flores, S.; Christensen, J.; McDade, E.; Wang, G.; Xiong, C.; et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: A longitudinal study. Lancet Neurol. 2018, 17, 241–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palop, J.J.; Mucke, L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2016, 17, 777–792. [Google Scholar] [CrossRef] [PubMed]
- Giesers, N.K.; Wirths, O. Loss of Hippocampal Calretinin and Parvalbumin Interneurons in the 5XFAD Mouse Model of Alzheimer’s Disease. ASN Neuro 2020, 12, 1759091420925356. [Google Scholar] [CrossRef]
- Sosulina, L.; Mittag, M.; Geis, H.R.; Hoffmann, K.; Klyubin, I.; Qi, Y.; Steffen, J.; Friedrichs, D.; Henneberg, N.; Fuhrmann, F.; et al. Hippocampal hyperactivity in a rat model of Alzheimer’s disease. J. Neurochem. 2021, 157, 2128–2144. [Google Scholar] [CrossRef]
- Janowsky, D.S.; el-Yousel, M.K.; Davis, J.M.; Sekerke, H.J. Provocation of schizophrenic symptoms by intravenous administration of methylphenidate. Arch. Gen. Psychiatry 1973, 28, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.; Gendelman, D.S.; Tischler, M.D.; Gendelman, P.M. A primary acoustic startle circuit: Lesion and stimulation studies. J. Neurosci. 1982, 2, 791–805. [Google Scholar] [CrossRef] [PubMed]
- Krivinko, J.M.; Koppel, J.; Savonenko, A.; Sweet, R.A. Animal Models of Psychosis in Alzheimer Disease. Am. J. Geriatr. Psychiatry 2020, 28, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.M.; McCoy, A.M.; Prevot, T.D.; Mian, M.Y.; Carreno, F.R.; Frazer, A.; Cook, J.M.; Sibille, E.; Lodge, D.J. Hippocampal alpha5-GABA(A) Receptors Modulate Dopamine Neuron Activity in the Rat Ventral Tegmental Area. Biol. Psychiatry Glob. Open Sci. 2023, 3, 78–86. [Google Scholar] [CrossRef]
- Santrac, A.; Batinic, B.; Stamenic, T.T.; Arandelovic, J.; Sharmin, D.; Knutson, D.E.; Cook, J.M.; Savic, M.M. Positive modulation of alpha5GABAA receptors leads to dichotomous effects in rats on memory pattern and GABRA5 expression in prefrontal cortex and hippocampus. Behav. Brain Res. 2022, 416, 113578. [Google Scholar] [CrossRef]
- Hyland, B.I.; Reynolds, J.N.; Hay, J.; Perk, C.G.; Miller, R. Firing modes of midbrain dopamine cells in the freely moving rat. Neuroscience 2002, 114, 475–492. [Google Scholar] [CrossRef]
- Shah, A.; Lodge, D.J. A loss of hippocampal perineuronal nets produces deficits in dopamine system function: Relevance to the positive symptoms of schizophrenia. Transl. Psychiatry 2013, 3, e215. [Google Scholar] [CrossRef] [Green Version]
- Grace, A.A.; Bunney, B.S. Intracellular and extracellular electrophysiology of nigral dopaminergic neurons--1. Identification and characterization. Neuroscience 1983, 10, 301–315. [Google Scholar] [CrossRef]
- Ungless, M.A.; Grace, A.A. Are you or aren’t you? Challenges associated with physiologically identifying dopamine neurons. Trends Neurosci. 2012, 35, 422–430. [Google Scholar] [CrossRef] [Green Version]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 2nd ed.; Academic Press: Sydney, Australia, 1986. [Google Scholar]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eassa, N.E.; Perez, S.M.; Boley, A.M.; Elam, H.B.; Sharmin, D.; Cook, J.M.; Lodge, D.J. α5-GABAA Receptor Modulation Reverses Behavioral and Neurophysiological Correlates of Psychosis in Rats with Ventral Hippocampal Alzheimer’s Disease-like Pathology. Int. J. Mol. Sci. 2023, 24, 11788. https://doi.org/10.3390/ijms241411788
Eassa NE, Perez SM, Boley AM, Elam HB, Sharmin D, Cook JM, Lodge DJ. α5-GABAA Receptor Modulation Reverses Behavioral and Neurophysiological Correlates of Psychosis in Rats with Ventral Hippocampal Alzheimer’s Disease-like Pathology. International Journal of Molecular Sciences. 2023; 24(14):11788. https://doi.org/10.3390/ijms241411788
Chicago/Turabian StyleEassa, Nicole E., Stephanie M. Perez, Angela M. Boley, Hannah B. Elam, Dishary Sharmin, James M. Cook, and Daniel J. Lodge. 2023. "α5-GABAA Receptor Modulation Reverses Behavioral and Neurophysiological Correlates of Psychosis in Rats with Ventral Hippocampal Alzheimer’s Disease-like Pathology" International Journal of Molecular Sciences 24, no. 14: 11788. https://doi.org/10.3390/ijms241411788
APA StyleEassa, N. E., Perez, S. M., Boley, A. M., Elam, H. B., Sharmin, D., Cook, J. M., & Lodge, D. J. (2023). α5-GABAA Receptor Modulation Reverses Behavioral and Neurophysiological Correlates of Psychosis in Rats with Ventral Hippocampal Alzheimer’s Disease-like Pathology. International Journal of Molecular Sciences, 24(14), 11788. https://doi.org/10.3390/ijms241411788