
The Role of Macrophages in Connective Tissue Disease-Associated Interstitial Lung Disease: Focusing on Molecular Mechanisms and Potential Treatment Strategies

 ,
, 
Abstract
:1. Introduction
2. Roles of Signaling Pathways of Macrophages in ILD/PF in Animal Models

{kind=link}
{kind=link}
{kind=link}
| Signaling Pathways | Results | Reference |
|---|---|---|
| Caspase pathway | ||
| MCU ↑ CPT1A 🡪 ↑ Bcl-2 🡪 ↓ caspase-3 activity | ↓ macrophage apoptosis 🡪 ↑ fibrosis | [36] |
| CCR2 axis | ||
| CCR2 ↑ macrophage infiltration 🡪 ↑ MMP-2/MMP-9 | ↑ extracellular matrix deposition | [37] |
| CCR4 axis | ||
| CCR4 ↑ inflammation and tissue injury | ↑ fibrosis | [38] |
| CD204 axis | ||
| collagen type I monomers ↑ CD204 | ↑ CCL18 🡪 ↑ collagen production 🡪 ↑ CCL18 | [39,40] |
| Glycolysis | ||
| GLUT1 ↑ glycolysis | ↑ profibrotic macrophages | [42] |
| GSK pathway | ||
| TRIB3-GSK-3β interaction ↓ A20 activity and stabilizes C/EBPβ 🡪 macrophage activation | ↑ fibroblasts transform to myofibroblasts | [43] |
| HIF pathway | ||
| HIF1A ↑ ADORA2B 🡪 ↑ macrophage differentiation | ↑ profibrotic mediators | [44] |
| Itaconate axis | ||
| ACOD1 🡪 ↑ itaconate | ↓ fibroblast proliferation and profibrotic activity | [45] |
| Macrophage migration | ||
| PLXNC1 | ↓ macrophage migration | [46] |
| MAPK signaling | ||
| FOXM1 activates DUSP1 🡪 ↓ p38 MAPK signaling | ↓ fibrosis | [47] |
| STAT6 signaling | ||
| Cu,Zn-SOD ↑ H2O2 🡪 activates STAT6 | ↑ M2 polarization | [49] |
| Gab2 🡪 activates STAT6 | [50] | |
| SART1 🡪 ↑ activates STAT6 | [51] | |
| S1PR2 🡪 ↑ activates STAT6 | [52] | |
| TGF-β signaling | ||
| Akt ↑ ROS 🡪 ↑ mitophagy ↑ apoptosis resistance 🡪 ↑ TGF-β | ↑ fibroblast differentiation | [53] |
| CHOP ↓ SOCS1/SOCS3 🡪 ↑ STAT6/PPARγ signaling 🡪 ↑ TGF-β | [54] | |
| MBD2 ↓ SHIP 🡪 ↑ PI3K/Akt signaling 🡪 ↑ M2 polarization 🡪 ↑ TGF-β | [55] | |
| FBXW7 ↓ phagocyte recruitment ↑ c-Jun degradation 🡪 ↓ TGF-β | [56] | |
| TNF pathway | ||
| STAT1 🡪 ↑ ICAM-1 🡪 ↑ TNF | ↑ inflammatory cells infiltration 🡪 ↑ fibrosis | [58,59] |
| CD300c2 🡪 ↑ macrophage activation🡪 ↑ TNF | [60] |
3. Roles of Macrophage-Derived Secretory Proteins/microRNAs in ILD/PF in Animal Models
3.1. Macrophage-Derived Secretory Proteins/microRNAs That Aggravate ILD/PF in Animal Models
| Molecules | Characteristics | Signaling Pathway | Reference |
|---|---|---|---|
| Macrophage-derived secretory proteins/microRNAs that aggravate ILD/PF | |||
| ADAM17 | Protein | ↑ shedding of mIL-6Rα 🡪 ↑ IL-6 trans-signaling 🡪 ↑ fibroblast proliferation and ECM production | [61] |
| AT1R | Protein | AT1R taken up by fibroblasts 🡪 ↑ profibrotic ↑ TGF-β/Smad2/Smad3 pathway, ↑ α-collagen I production, ↑ Ang II secretion by fibroblasts 🡪 ↑ macrophage exosomes and AT1R in exosomes | [62] |
| CCL6 | Protein | ↑ fibrosis | [63] |
| Fibronectin | Protein | ↑ myofibroblasts 🡪 ↑ extracellular matrix deposition | [64,65] |
| Galectin-3 | Protein | ↑ TNF-α 🡪 ↑ galectin-3 🡪 ↑ inflammation, ↑ fibroblast proliferation | [66] |
| IGF-I | Protein | activates Akt 🡪 ↓ myofibroblast apoptosis | [67] |
| IL-1β | Protein | ↑ leukocyte influx, ↑ myofibroblasts 🡪 ↑ collagen deposition | [68,69] |
| MIP-1α | Protein | ↑ leukocyte accumulation 🡪 ↑ fibrosis | [37,70] |
| miR-328 | microRNA | ↓ FAM13A 🡪 ↑ fibroblast proliferation and ↑ collagen 1A, ↑ collagen 3A, ↑ α-SMA | [71] |
| NTN1 | Protein | interacts with DCC 🡪 impacts adrenergic nerve remodeling 🡪 ↑ fibrosis | [73] |
| PDGF | Protein | a mitogen for fibroblasts | [74] |
| S100A4 | Protein | ↑ fibroblast proliferation and activation | [75] |
| TGF-β | Protein | ↑ PAI-1 🡪 ↑ extracellular matrix | [77] |
| Wnt | Protein | ↑ differentiation of monocyte-macrophage 🡪 ↑ fibrosis | [79] |
| Macrophage-derived secretory proteins/microRNAs that ameliorate ILD/PF | |||
| Pentraxin-2 (serum amyloid P) [80] | Protein | ↓ M2 differentiation | [81] |
| miR-142-3p | microRNA | ↓ TGFβ-R1 🡪 ↓ profibrotic genes | [82] |
3.2. Macrophage-Derived Secretory Proteins/microRNAs That Ameliorate ILD/PF in Animal Models
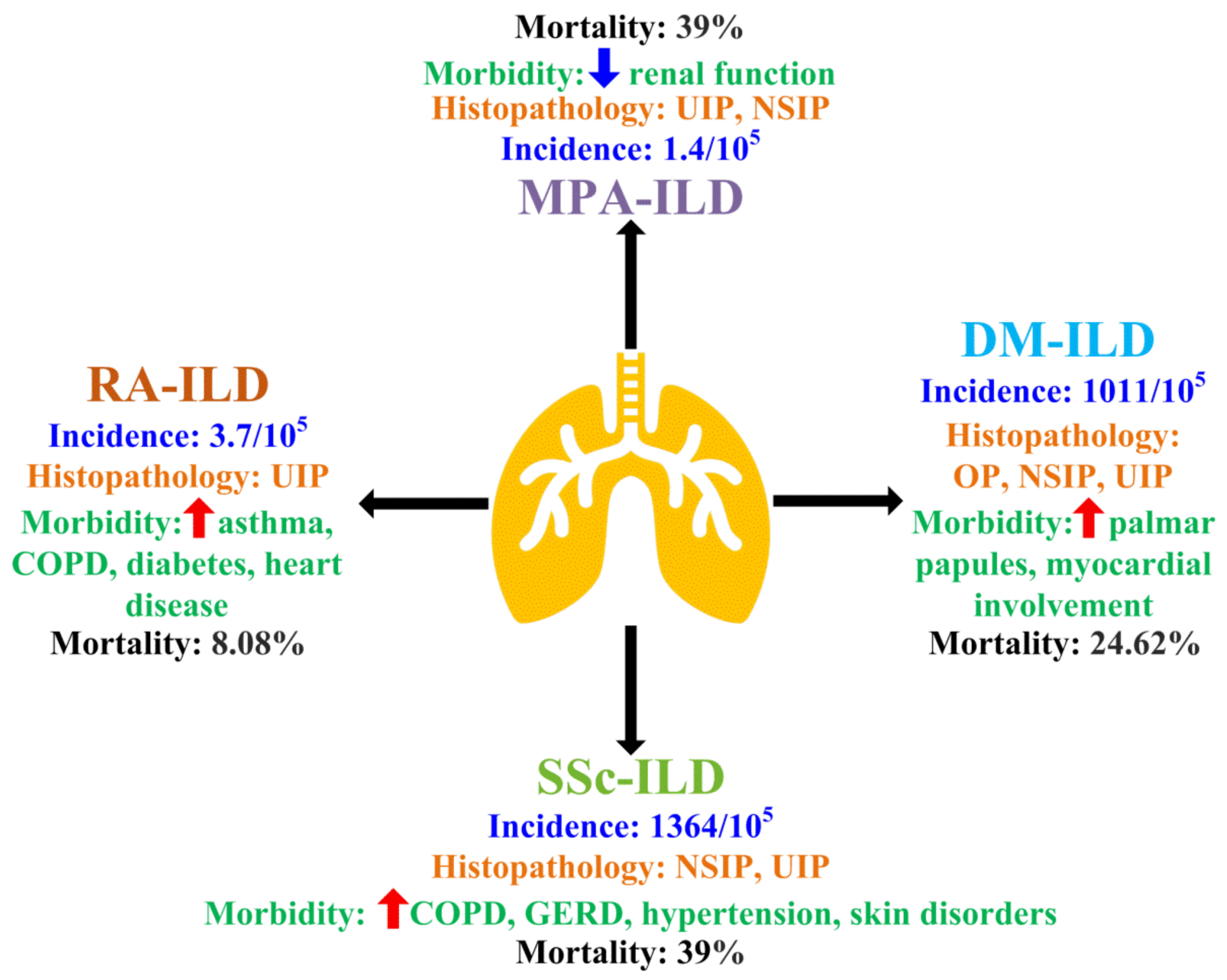
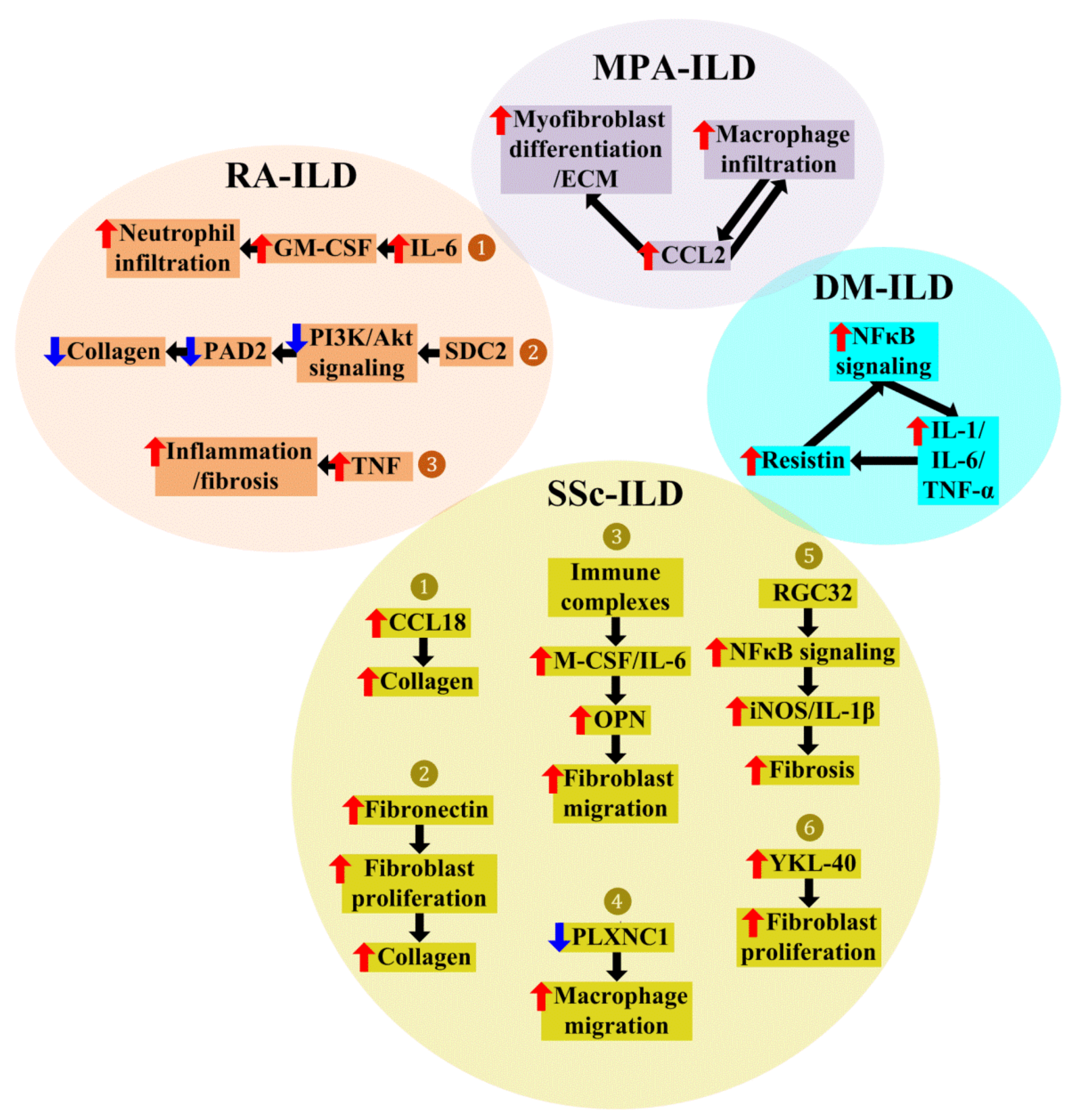
4. Roles of Macrophages in CTD-ILD
4.1. Macrophages in Dermatomyositis (DM)-Associated ILD (DM-ILD)
4.2. Macrophages in Microscopic Polyangiitis (MPA)-Associated ILD (MPA-ILD)
4.3. Macrophages in Rheumatoid Arthritis (RA)-Associated ILD (RA-ILD)
4.4. Macrophages in Systemic Sclerosis (SSc)-Associated ILD (SSc-ILD)
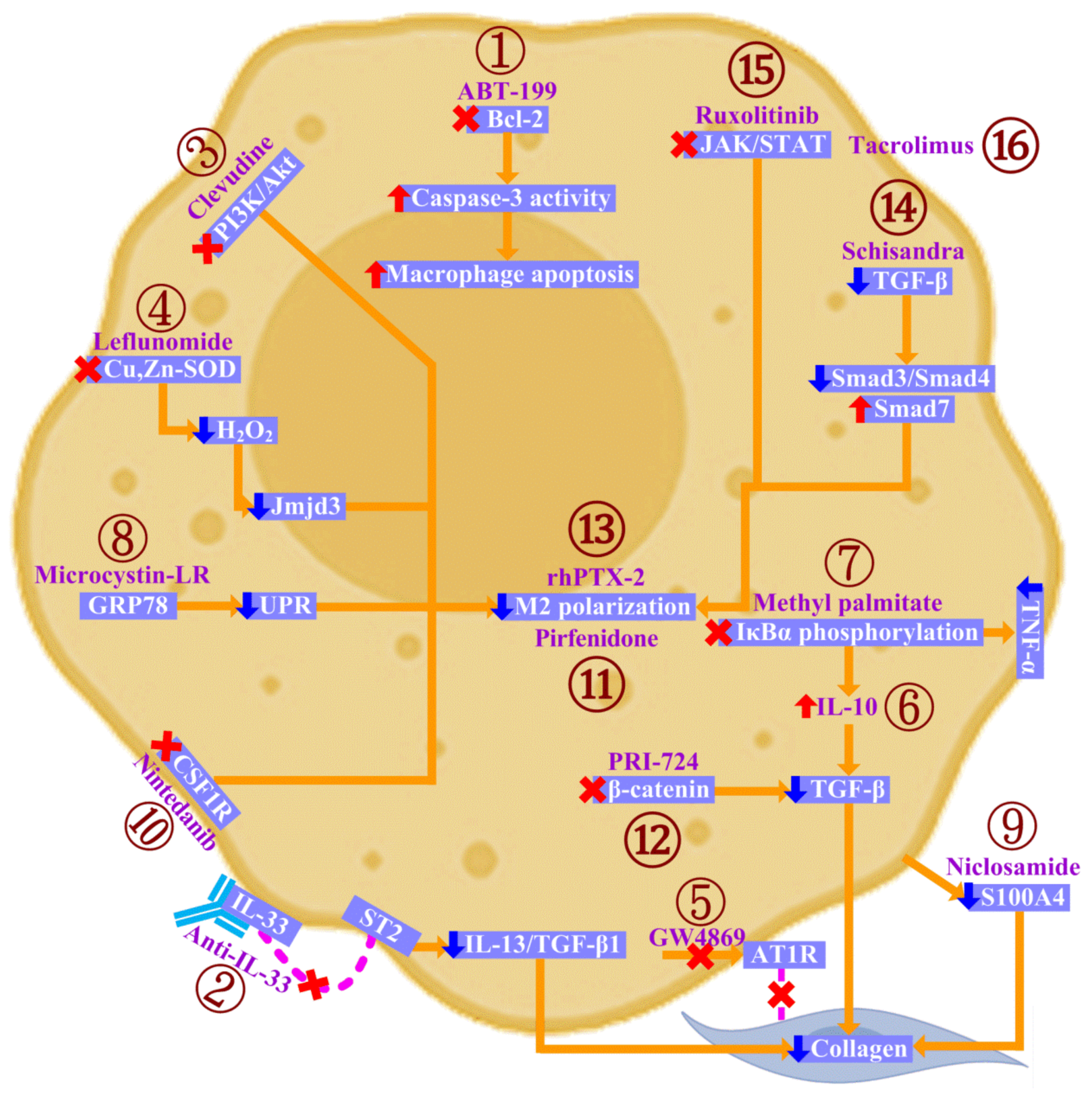
5. Targeting Macrophages in CTD-ILD
6. Investigational Agents in Preclinical and Clinical Trials of CTD-ILD
7. Concluding Remarks and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Spagnolo, P.; Distler, O.; Ryerson, C.J.; Tzouvelekis, A.; Lee, J.S.; Bonella, F.; Bouros, D.; Hoffmann-Vold, A.M.; Crestani, B.; Matteson, E.L. Mechanisms of progressive fibrosis in connective tissue disease (CTD)-associated interstitial lung diseases (ILDs). Ann. Rheum. Dis. 2021, 80, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.; Fernandes, A.; Pereira, A.R.; Madanelo, S.; Clemêncio, T.; Ferreira, P.G. Awareness towards the main ILD among primary care physicians. Multidiscip. Respir. Med. 2022, 17, 848. [Google Scholar] [CrossRef] [PubMed]
- Fisseler-Eckhoff, A.; Märker-Hermann, E. Interstitial lung disease associated with connective tissue disease. Pathologe 2021, 42, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.; Hino, T.; Han, J.; Franks, T.J.; Im, Y.; Hatabu, H.; Chung, M.P.; Lee, K.S. Connective tissue disease-related interstitial lung disease (CTD-ILD) and interstitial lung abnormality (ILA): Evolving concept of CT findings, pathology and management. Eur. J. Radiol. Open 2021, 8, 100311. [Google Scholar] [CrossRef]
- Ahmed, S.; Handa, R. Management of Connective Tissue Disease-related Interstitial Lung Disease. Curr. Pulmonol. Rep. 2022, 11, 86–98. [Google Scholar] [CrossRef]
- Mena-Vázquez, N.; Godoy-Navarrete, F.J.; Lisbona-Montañez, J.M.; Redondo-Rodriguez, R.; Manrique-Arija, S.; Rioja, J.; Mucientes, A.; Ruiz-Limón, P.; Garcia-Studer, A.; Ortiz-Márquez, F.; et al. Inflammatory Biomarkers in the Diagnosis and Prognosis of Rheumatoid Arthritis-Associated Interstitial Lung Disease. Int. J. Mol. Sci. 2023, 24, 6800. [Google Scholar] [CrossRef]
- Dsouza, K.G.; Alexander, A.S.; Watts, J.R.; Kulkarni, T. Management of interstitial lung disease in patients with autoimmune disease-related interstitial lung disease. Multidiscip. Respir. Med. 2023, 18, 890. [Google Scholar] [CrossRef]
- Ng, K.H.; Chen, D.Y.; Lin, C.H.; Chao, W.C.; Chen, Y.M.; Chen, Y.H.; Huang, W.N.; Hsieh, T.Y.; Lai, K.L.; Tang, K.T.; et al. Risk of interstitial lung disease in patients with newly diagnosed systemic autoimmune rheumatic disease: A nationwide, population-based cohort study. Semin. Arthritis Rheum. 2020, 50, 840–845. [Google Scholar] [CrossRef]
- Chen, F.; Wang, J.; Zhang, P.; Zuo, Y.; Ye, L.; Wang, G.; Shu, X. Interstitial Lung Disease in Dermatomyositis Without Myositis-Specific and Myositis-Associated Autoantibodies: Study of a Series of 72 Patients from a Single Cohort. Front. Immunol. 2022, 13, 879266. [Google Scholar] [CrossRef]
- Lu, J.; Liu, C.; Zhou, X.; Tang, J.; Liu, S.; Tang, M.; Li, M.; Zhu, L. Palmar erythema and palmar papules as predictors for dermatomyositis-related acute/subacute interstitial lung disease: A retrospective study. Rheumatology 2021, 61, 413–421. [Google Scholar] [CrossRef]
- Weng, C.; Ding, Z.; Zhou, Y.; Yang, Q.; Xue, L.; Zhang, L.; Wang, G.; Liu, Z. Clinical Characteristics of Dermatomyositis with Interstitial Lung Disease: A Retrospective Case-Control Study. Rheumatol. Ther. 2023, 10, 635–648. [Google Scholar] [CrossRef]
- Wang, Q.; Gao, C.; Zhang, C.; Yao, M.; Liang, W.; Sun, W.; Zheng, Z. Tumor markers are associated with rapidly progressive interstitial lung disease in adult-dermatomyositis. Clin. Rheumatol. 2022, 41, 1731–1739. [Google Scholar] [CrossRef]
- Pérez, N.; Gargiulo, M.L.Á.; Suarez, L.; Khoury, M.; Gómez, G. Clinical characteristics and prognostic factors in an Argentinian cohort with ANCA-associated vasculitis. Medicina 2021, 81, 198–207. [Google Scholar]
- Sweis, J.J.G.; Sweis, N.W.G.; Alnaimat, F.; Jansz, J.; Liao, T.E.; Alsakaty, A.; Azam, A.; Elmergawy, H.; Hanson, H.A.; Ascoli, C.; et al. Immune-mediated lung diseases: A narrative review. Front. Med. 2023, 10, 1160755. [Google Scholar] [CrossRef]
- Matsuda, S.; Kotani, T.; Suzuka, T.; Kiboshi, T.; Fukui, K.; Wakama, M.; Ishida, T.; Fujiki, Y.; Shiba, H.; Nagai, K.; et al. Evaluation of poor prognostic factors of respiratory related death in microscopic polyangiitis complicated by interstitial lung disease. Sci. Rep. 2021, 11, 1490. [Google Scholar] [CrossRef]
- Takakuwa, Y.; Yamasaki, Y.; Matsushita, H.; Kiyokawa, T.; Mizushima, M.; Tonooka, K.; Nagafuchi, H.; Matsuoka, S.; Ooka, S.; Kawahata, K. Long-term survival, causes of death, and prognostic factors for mortality in patients with microscopic polyangiitis and those with anti-neutrophil cytoplasmic antibody-positive interstitial lung disease: A single-center retrospective study. Int. J. Rheum. Dis. 2023, 26, 446–453. [Google Scholar] [CrossRef]
- Fidler, L.; Widdifield, J.; Fisher, J.H.; Shapera, S.; Gershon, A.S. Rheumatoid arthritis associated interstitial lung disease: Trends in epidemiology and mortality in Ontario from 2000 to 2018. Respir. Med. 2023, in press. [Google Scholar] [CrossRef]
- Ng, K.H.; Chen, D.Y.; Lin, C.H.; Chao, W.C.; Chen, H.H. Analysis of risk factors of mortality in rheumatoid arthritis patients with interstitial lung disease: A nationwide, population-based cohort study in Taiwan. RMD Open 2022, 8, e002343. [Google Scholar] [CrossRef]
- Sparks, J.A.; Jin, Y.; Cho, S.K.; Vine, S.; Desai, R.; Doyle, T.J.; Kim, S.C. Prevalence, incidence and cause-specific mortality of rheumatoid arthritis-associated interstitial lung disease among older rheumatoid arthritis patients. Rheumatology 2021, 60, 3689–3698. [Google Scholar] [CrossRef]
- Reid, P.; Guler, S.A. Mortality Trends in Rheumatoid Arthritis: Zooming in on Interstitial Lung Disease. Ann. Am. Thorac. Soc. 2021, 18, 1953–1954. [Google Scholar] [CrossRef]
- Panagopoulos, P.; Goules, A.; Hoffmann-Vold, A.M.; Matteson, E.L.; Tzioufas, A. Natural history and screening of interstitial lung disease in systemic autoimmune rheumatic disorders. Ther. Adv. Musculoskelet. Dis. 2021, 13, 1759720X211037519. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wallace, L.; Patnaik, P.; Alves, M.; Gahlemann, M.; Kohlbrenner, V.; Raabe, C.; Wang, J.R.; Garry, E.M. Disease frequency, patient characteristics, comorbidity outcomes and immunosuppressive therapy in systemic sclerosis and systemic sclerosis-associated interstitial lung disease: A US cohort study. Rheumatology 2021, 60, 1915–1925. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann-Vold, A.M.; Fretheim, H.; Halse, A.K.; Seip, M.; Bitter, H.; Wallenius, M.; Garen, T.; Salberg, A.; Brunborg, C.; Midtvedt, Ø.; et al. Tracking Impact of Interstitial Lung Disease in Systemic Sclerosis in a Complete Nationwide Cohort. Am. J. Respir. Crit. Care Med. 2019, 200, 1258–1266. [Google Scholar] [CrossRef]
- Hyldgaard, C.; Bendstrup, E.; Pedersen, A.B.; Pedersen, L.; Ellingsen, T. Interstitial Lung Disease in Connective Tissue Diseases: Survival Patterns in a Population-Based Cohort. J. Clin. Med. 2021, 10, 4830. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Saeed, A.F.U.H.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in immunoregulation and therapeutics. Signal Transduct. Target. Ther. 2023, 8, 207. [Google Scholar] [CrossRef]
- Moreira, A.C.; Silva, T.; Mesquita, G.; Gomes, A.C.; Bento, C.M.; Neves, J.V.; Rodrigues, D.F.; Rodrigues, P.N.; Almeida, A.A.; Santambrogio, P.; et al. H-Ferritin Produced by Myeloid Cells Is Released to the Circulation and Plays a Major Role in Liver Iron Distribution during Infection. Int. J. Mol. Sci. 2021, 23, 269. [Google Scholar] [CrossRef]
- Enomoto, N. Pathological Roles of Pulmonary Cells in Acute Lung Injury: Lessons from Clinical Practice. Int. J. Mol. Sci. 2022, 23, 15027. [Google Scholar] [CrossRef]
- Hofbauer, D.; Mougiakakos, D.; Broggini, L.; Zaiss, M.; Büttner-Herold, M.; Bach, C.; Spriewald, B.; Neumann, F.; Bisht, S.; Nolting, J.; et al. β2-microglobulin triggers NLRP3 inflammasome activation in tumor-associated macrophages to promote multiple myeloma progression. Immunity 2021, 54, 1772–1787.e9. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, R.; Xie, M.; Luan, C.; Li, X. Transcriptome Sequencing Identifies PLAUR as an Important Player in Patients with Dermatomyositis-Associated Interstitial Lung Disease. Front. Genet. 2021, 12, 784215. [Google Scholar] [CrossRef]
- Yang, Y.; Yin, G.; Hao, J.; Xie, Q.; Liu, Y. Serum Interleukin-18 Level is Associated with Disease Activity and Interstitial Lung Disease in Patients with Dermatomyositis. Arch. Rheumatol. 2017, 32, 181–188. [Google Scholar] [CrossRef]
- Byrne, A.J.; Maher, T.M.; Lloyd, C.M. Pulmonary Macrophages: A New Therapeutic Pathway in Fibrosing Lung Disease? Trends Mol. Med. 2016, 22, 303–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, B.S.; Park, K.Y.; Lee, H.J.; Kim, H.J.; Ahn, H.S.; Yim, S.Y.; Jun, J.B. Effect of combined pulmonary fibrosis and emphysema on patients with connective tissue diseases and systemic sclerosis: A systematic review and meta-analysis. Arthritis Res. Ther. 2021, 23, 100. [Google Scholar] [CrossRef] [PubMed]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef]
- Aegerter, H.; Lambrecht, B.N.; Jakubzick, C.V. Biology of lung macrophages in health and disease. Immunity 2022, 55, 1564–1580. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Surolia, R.; Larson-Casey, J.L.; He, C.; Davis, D.; Kang, J.; Antony, V.B.; Carter, A.B. Targeting Cpt1a-Bcl-2 interaction modulates apoptosis resistance and fibrotic remodeling. Cell Death Differ. 2022, 29, 118–132. [Google Scholar] [CrossRef]
- Ishida, Y.; Kuninaka, Y.; Mukaida, N.; Kondo, T. Immune Mechanisms of Pulmonary Fibrosis with Bleomycin. Int. J. Mol. Sci. 2023, 24, 3149. [Google Scholar] [CrossRef]
- Liu, S.; Liu, C.; Wang, Q.; Min, J. CC Chemokines in Idiopathic Pulmonary Fibrosis: Pathogenic Role and Therapeutic Potential. Biomolecules 2023, 13, 333. [Google Scholar] [CrossRef]
- Stahl, M.; Schupp, J.; Jäger, B.; Schmid, M.; Zissel, G.; Müller-Quernheim, J.; Prasse, A. Lung collagens perpetuate pulmonary fibrosis via CD204 and M2 macrophage activation. PLoS ONE 2013, 8, e81382. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, Y.; Wu, G.; Xiong, W.; Gu, W.; Wang, C.Y. Macrophages: Friend or foe in idiopathic pulmonary fibrosis? Respir. Res. 2018, 19, 170. [Google Scholar]
- Li, J.; Zhai, X.; Sun, X.; Cao, S.; Yuan, Q.; Wang, J. Metabolic reprogramming of pulmonary fibrosis. Front. Pharmacol. 2022, 13, 1031890. [Google Scholar] [CrossRef]
- Xie, N.; Cui, H.; Ge, J.; Banerjee, S.; Guo, S.; Dubey, S.; Abraham, E.; Liu, R.M.; Liu, G. Metabolic characterization and RNA profiling reveal glycolytic dependence of profibrotic phenotype of alveolar macrophages in lung fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L834–L844. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Lv, X.; Wei, X.; Liu, C.; Li, Q.; Min, J.; Hua, F.; Zhang, X.; Li, K.; Li, P.; et al. TRIB3–GSK-3β interaction promotes lung fibrosis and serves as a potential therapeutic target. Acta Pharm. Sin. B 2021, 11, 3105–3119. [Google Scholar] [CrossRef]
- Philip, K.; Mills, T.W.; Davies, J.; Chen, N.Y.; Karmouty-Quintana, H.; Luo, F.; Molina, J.G.; Amione-Guerra, J.; Sinha, N.; Guha, A.; et al. HIF1A up-regulates the ADORA2B receptor on alternatively activated macrophages and contributes to pulmonary fibrosis. FASEB J. 2017, 31, 4745–4758. [Google Scholar] [CrossRef] [Green Version]
- Ogger, P.P.; Albers, G.J.; Hewitt, R.J.; O’Sullivan, B.J.; Powell, J.E.; Calamita, E.; Ghai, P.; Walker, S.A.; McErlean, P.; Saunders, P.; et al. Itaconate controls the severity of pulmonary fibrosis. Sci. Immunol. 2020, 5, eabc1884. [Google Scholar] [CrossRef]
- Peng, X.; Moore, M.; Mathur, A.; Zhou, Y.; Sun, H.; Gan, Y.; Herazo-Maya, J.D.; Kaminski, N.; Hu, X.; Pan, H.; et al. Plexin C1 deficiency permits synaptotagmin 7-mediated macrophage migration and enhances mammalian lung fibrosis. FASEB J. 2016, 30, 4056–4070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goda, C.; Balli, D.; Black, M.; Milewski, D.; Le, T.; Ustiyan, V.; Ren, X.; Kalinichenko, V.V.; Kalin, T.V. Loss of FOXM1 in macrophages promotes pulmonary fibrosis by activating p38 MAPK signaling pathway. PLoS Genet. 2020, 16, e1008692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Ryan, A.J.; Murthy, S.; Carter, A.B. Accelerated development of pulmonary fibrosis via Cu,Zn-superoxide dismutase-induced alternative activation of macrophages. J. Biol. Chem. 2013, 288, 20745–20757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Larson-Casey, J.L.; Gu, L.; Ryan, A.J.; Murthy, S.; Carter, A.B. Cu,Zn-Superoxide Dismutase-Mediated Redox Regulation of Jumonji Domain Containing 3 Modulates Macrophage Polarization and Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2016, 55, 58–71. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Li, T.; Xu, Y.; Xu, X.; Zhu, Z.; Zhang, Y.; Xu, J.; Xu, K.; Cheng, H.; Zhang, X.; et al. Increased levels of Gab1 and Gab2 adaptor proteins skew interleukin-4 (IL-4) signaling toward M2 macrophage-driven pulmonary fibrosis in mice. J. Biol. Chem. 2017, 292, 14003–14015. [Google Scholar] [CrossRef] [Green Version]
- Pan, T.; Zhou, Q.; Miao, K.; Zhang, L.; Wu, G.; Yu, J.; Xu, Y.; Xiong, W.; Li, Y.; Wang, Y. Suppressing Sart1 to modulate macrophage polarization by siRNA-loaded liposomes: A promising therapeutic strategy for pulmonary fibrosis. Theranostics 2021, 11, 1192–1206. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Okamoto, Y.; Asano, Y.; Ishimaru, K.; Aki, S.; Yoshioka, K.; Takuwa, N.; Wada, T.; Inagaki, Y.; Takahashi, C.; et al. Sphingosine-1-phosphate receptor-2 facilitates pulmonary fibrosis through potentiating IL-13 pathway in macrophages. PLoS ONE 2018, 13, e0197604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson-Casey, J.L.; Deshane, J.S.; Ryan, A.J.; Thannickal, V.J.; Carter, A.B. Macrophage Akt1 Kinase-Mediated Mitophagy Modulates Apoptosis Resistance and Pulmonary Fibrosis. Immunity 2016, 44, 582–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Wang, Y.; Zhang, Z.; He, L.; Zhu, J.; Zhang, M.; He, X.; Cheng, Z.; Ao, Q.; Cao, Y.; et al. Chop Deficiency Protects Mice Against Bleomycin-induced Pulmonary Fibrosis by Attenuating M2 Macrophage Production. Mol. Ther. 2016, 24, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, L.; Wu, G.R.; Zhou, Q.; Yue, H.; Rao, L.Z.; Yuan, T.; Mo, B.; Wang, F.X.; Chen, L.M.; et al. MBD2 serves as a viable target against pulmonary fibrosis by inhibiting macrophage M2 program. Sci. Adv. 2021, 7, eabb6075. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Du, Y.; Li, G.; Xiao, P.; Sun, X.; Song, W.; Lai, L.; Xia, M.; Zhang, J.; Wang, Q. Myeloid Fbxw7 Prevents Pulmonary Fibrosis by Suppressing TGF-β Production. Front. Immunol. 2021, 12, 760138. [Google Scholar] [CrossRef] [PubMed]
- Huo, R.; Guo, Q.; Hu, J.; Li, N.; Gao, R.; Mi, L.; Zhang, Z.; Liu, H.; Guo, Z.; Zhao, H.; et al. Therapeutic Potential of Janus Kinase Inhibitors for the Management of Interstitial Lung Disease. Drug Des. Dev. Ther. 2022, 16, 991–998. [Google Scholar] [CrossRef]
- Fan, X.; Wang, Z. STAT1 antisense oligonucleotides attenuate the proinflammatory cytokine release of alveolar macrophages in bleomycin-induced fibrosis. Cell Mol. Immunol. 2005, 2, 211–217. [Google Scholar]
- Le, T.K.; Paris, C.; Khan, K.S.; Robson, F.; Ng, W.L.; Rocchi, P. Nucleic Acid-Based Technologies Targeting Coronaviruses. Trends Biochem. Sci. 2021, 46, 351–365. [Google Scholar] [CrossRef]
- Nakazawa, Y.; Ohtsuka, S.; Nakahashi-Oda, C.; Shibuya, A. Cutting Edge: Involvement of the Immunoreceptor CD300c2 on Alveolar Macrophages in Bleomycin-Induced Lung Fibrosis. J. Immunol. 2019, 203, 3107–3111. [Google Scholar] [CrossRef]
- Cardoneanu, A.; Burlui, A.M.; Macovei, L.A.; Bratoiu, I.; Richter, P.; Rezus, E. Targeting Systemic Sclerosis from Pathogenic Mechanisms to Clinical Manifestations: Why IL-6? Biomedicines 2022, 10, 318. [Google Scholar] [CrossRef]
- Sun, N.N.; Zhang, Y.; Huang, W.H.; Zheng, B.J.; Jin, S.Y.; Li, X.; Meng, Y. Macrophage exosomes transfer angiotensin II type 1 receptor to lung fibroblasts mediating bleomycin-induced pulmonary fibrosis. Chin. Med. J. 2021, 134, 2175–2185. [Google Scholar] [CrossRef]
- Chen, Y.; Hao, X.; Li, M.; Tian, Z.; Cheng, M. UGRP1-modulated MARCO+ alveolar macrophages contribute to age-related lung fibrosis. Immun. Ageing 2023, 20, 14. [Google Scholar] [CrossRef]
- Durairaj, P.; Venkatesan, S.; Narayanan, V.; Babu, M. Protective effects of curcumin on bleomycin-induced changes in lung glycoproteins. Mol. Cell Biochem. 2020, 469, 159–167. [Google Scholar] [CrossRef]
- Suka, M.; Kido, T.; Yoshioka, W.; Hachisuka, E.; Okoshi, H.; Yamauchi, T.; Hano, H.; Okano, T.; Yokoyama, M.; Yanagisawa, H. Single intratracheal administration of cross-linked water-soluble acrylic acid polymer causes acute alveolo-interstitial inflammation and the subsequent fibrotic formation possibly via the TGF-β1 pathway in the lung of rats. Toxicology 2021, 448, 152647. [Google Scholar] [CrossRef]
- Sciacchitano, S.; Lavra, L.; Morgante, A.; Ulivieri, A.; Magi, F.; De Francesco, G.P.; Bellotti, C.; Salehi, L.B.; Ricci, A. Galectin-3: One Molecule for an Alphabet of Diseases, from A to Z. Int. J. Mol. Sci. 2018, 19, 379. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Li, W.; Guo, Q.; Wang, Y.; Ma, L.; Zhang, X. Insulin-Like Growth Factor-1 Signaling in Lung Development and Inflammatory Lung Diseases. Biomed. Res. Int. 2018, 2018, 6057589. [Google Scholar] [CrossRef] [Green Version]
- Artlett, C.M. The Mechanism and Regulation of the NLRP3 Inflammasome during Fibrosis. Biomolecules 2022, 12, 634. [Google Scholar] [CrossRef]
- Ogawa, T.; Shichino, S.; Ueha, S.; Matsushima, K. Macrophages in lung fibrosis. Int. Immunol. 2021, 33, 665–671. [Google Scholar] [CrossRef]
- Liu, G.; Zhai, H.; Zhang, T.; Li, S.; Li, N.; Chen, J.; Gu, M.; Qin, Z.; Liu, X. New therapeutic strategies for IPF: Based on the “phagocytosis-secretion-immunization” network regulation mechanism of pulmonary macrophages. Biomed. Pharmacother. 2019, 118, 109230. [Google Scholar] [CrossRef]
- Ramírez-Hernández, A.A.; Velázquez-Enríquez, J.M.; Santos-Álvarez, J.C.; López-Martínez, A.; Reyes-Jiménez, E.; Carrasco-Torres, G.; González-García, K.; Vásquez-Garzón, V.R.; Baltierrez-Hoyos, R. The Role of Extracellular Vesicles in Idiopathic Pulmonary Fibrosis Progression: An Approach on Their Therapeutics Potential. Cells 2022, 11, 630. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.Y.; Zhang, W.H.; Ma, W.T.; Liu, Q.H.; Xing, L.H.; Zhao, G.F. microRNA-328 in exosomes derived from M2 macrophages exerts a promotive effect on the progression of pulmonary fibrosis via FAM13A in a rat model. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, R.; Peng, X.; Perry, C.; Sun, H.; Ntokou, A.; Ryu, C.; Gomez, J.L.; Reeves, B.C.; Walia, A.; Kaminski, N.; et al. Macrophage-derived netrin-1 drives adrenergic nerve-associated lung fibrosis. J. Clin. Investig. 2021, 131, e136542. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Lawrence, T.; Mohamed, R.; Liang, Y.; Yahaya, B.H. The emerging roles of interstitial macrophages in pulmonary fibrosis: A perspective from scRNA-seq analyses. Front. Immunol. 2022, 13, 923235. [Google Scholar] [CrossRef]
- Zhang, W.; Ohno, S.; Steer, B.; Klee, S.; Staab-Weijnitz, C.A.; Wagner, D.; Lehmann, M.; Stoeger, T.; Königshoff, M.; Adler, H. S100a4 Is Secreted by Alternatively Activated Alveolar Macrophages and Promotes Activation of Lung Fibroblasts in Pulmonary Fibrosis. Front. Immunol. 2018, 9, 1216. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Chakraborty, S.; Wong, S.W.; Hefner, N.A.; Stuart, A.; Qadir, A.S.; Mukhopadhyay, A.; Bachmaier, K.; Shin, J.W.; Rehman, J.; et al. Nanoparticle targeting of de novo profibrotic macrophages mitigates lung fibrosis. Proc. Natl. Acad. Sci. USA 2022, 119, e2121098119. [Google Scholar] [CrossRef]
- Han, S.; Lu, Q.; Liu, X. Advances in cellular senescence in idiopathic pulmonary fibrosis (Review). Exp. Ther. Med. 2023, 25, 145. [Google Scholar] [CrossRef]
- Hou, J.; Shi, J.; Chen, L.; Lv, Z.; Chen, X.; Cao, H.; Xiang, Z.; Han, X. M2 macrophages promote myofibroblast differentiation of LR-MSCs and are associated with pulmonary fibrogenesis. Cell Commun. Signal. 2018, 16, 89. [Google Scholar] [CrossRef] [Green Version]
- Sennello, J.A.; Misharin, A.V.; Flozak, A.S.; Berdnikovs, S.; Cheresh, P.; Varga, J.; Kamp, D.W.; Budinger, G.R.; Gottardi, C.J.; Lam, A.P. Lrp5/β-Catenin Signaling Controls Lung Macrophage Differentiation and Inhibits Resolution of Fibrosis. Am. J. Respir. Cell Mol. Biol. 2017, 56, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Glass, D.S.; Grossfeld, D.; Renna, H.A.; Agarwala, P.; Spiegler, P.; DeLeon, J.; Reiss, A.B. Idiopathic pulmonary fibrosis: Current and future treatment. Clin. Respir. J. 2022, 16, 84–96. [Google Scholar] [CrossRef]
- Maghsadi, Z.; Azadmehr, A.; Moghadamnia, A.A.; Feizi, F.; Hamidi, N. N-Acetylcysteine attenuated pulmonary fibrosis induced by bleomycin. Res. Pharm. Sci. 2023, 18, 177–184. [Google Scholar]
- Guiot, J.; Cambier, M.; Boeckx, A.; Henket, M.; Nivelles, O.; Gester, F.; Louis, E.; Malaise, M.; Dequiedt, F.; Louis, R.; et al. Macrophage-derived exosomes attenuate fibrosis in airway epithelial cells through delivery of antifibrotic miR-142-3p. Thorax 2020, 75, 870–881. [Google Scholar] [CrossRef]
- Enomoto, Y.; Suzuki, Y.; Hozumi, H.; Mori, K.; Kono, M.; Karayama, M.; Furuhashi, K.; Fujisawa, T.; Enomoto, N.; Nakamura, Y.; et al. Clinical significance of soluble CD163 in polymyositis-related or dermatomyositis-related interstitial lung disease. Arthritis Res. Ther. 2017, 19, 9. [Google Scholar] [CrossRef] [Green Version]
- Horiike, Y.; Suzuki, Y.; Fujisawa, T.; Yasui, H.; Karayama, M.; Hozumi, H.; Furuhashi, K.; Enomoto, N.; Nakamura, Y.; Inui, N.; et al. Successful classification of macrophage-mannose receptor CD206 in severity of anti-MDA5 antibody positive dermatomyositis associated ILD. Rheumatology 2019, 58, 2143–2152. [Google Scholar] [CrossRef]
- Ye, L.; Zuo, Y.; Chen, F.; Xu, Y.; Zhang, P.; Yang, H.; Lin, S.; Peng, Q.; Wang, G.; Shu, X. Resistin Expression Is Associated with Interstitial Lung Disease in Dermatomyositis. Front. Med. 2022, 9, 903887. [Google Scholar] [CrossRef]
- Matsuda, S.; Kotani, T.; Kuwabara, H.; Suzuka, T.; Kiboshi, T.; Wada, Y.; Ishida, T.; Fujiki, Y.; Shiba, H.; Hata, K.; et al. Association of M2 macrophages, Th2, and B cells with pathomechanism in microscopic polyangiitis complicated by interstitial lung disease. J. Rheumatol. 2022, 49, 913–921. [Google Scholar] [CrossRef]
- Matsuda, S.; Kotani, T.; Kuwabara, H.; Suzuka, T.; Kiboshi, T.; Fukui, K.; Ishida, T.; Fujiki, Y.; Shiba, H.; Hata, K.; et al. CCL2 produced by CD68+/CD163+ macrophages as a promising clinical biomarker of microscopic polyangiitis-interstitial lung disease. Rheumatology 2021, 60, 4643–4653. [Google Scholar] [CrossRef]
- Gaurav, R.; Mikuls, T.R.; Thiele, G.M.; Nelson, A.J.; Niu, M.; Guda, C.; Eudy, J.D.; Barry, A.E.; Wyatt, T.A.; Romberger, D.J.; et al. High-throughput analysis of lung immune cells in a combined murine model of agriculture dust-triggered airway inflammation with rheumatoid arthritis. PLoS ONE 2021, 16, e0240707. [Google Scholar] [CrossRef]
- Shiomi, A.; Usui, T.; Ishikawa, Y.; Shimizu, M.; Murakami, K.; Mimori, T. GM-CSF but not IL-17 is critical for the development of severe interstitial lung disease in SKG mice. J. Immunol. 2014, 193, 849–859. [Google Scholar] [CrossRef] [Green Version]
- Tsoyi, K.; Esposito, A.J.; Sun, B.; Bowen, R.G.; Xiong, K.; Poli, F.; Cardenas, R.; Chu, S.G.; Liang, X.; Ryter, S.W.; et al. Syndecan-2 regulates PAD2 to exert antifibrotic effects on RA-ILD fibroblasts. Sci. Rep. 2022, 12, 2847. [Google Scholar] [CrossRef]
- Xiong, L.; Ye, H.; Ma, W.L. Animal models of rheumatoid arthritis-associated interstitial lung disease. Immun. Inflamm. Dis. 2021, 9, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Wu, E.K.; Henkes, Z.I.; McGowan, B.; Bell, R.D.; Velez, M.J.; Livingstone, A.M.; Ritchlin, C.T.; Schwarz, E.M.; Rahimi, H. TNF-Induced Interstitial Lung Disease in a Murine Arthritis Model: Accumulation of Activated Monocytes, Conventional Dendritic Cells, and CD21+/CD23− B Cell Follicles Is Prevented with Anti-TNF Therapy. J. Immunol. 2019, 203, 2837–2849. [Google Scholar] [CrossRef] [PubMed]
- Rudnik, M.; Hukara, A.; Kocherova, I.; Jordan, S.; Schniering, J.; Milleret, V.; Ehrbar, M.; Klingel, K.; Feghali-Bostwick, C.; Distler, O.; et al. Elevated Fibronectin Levels in Profibrotic CD14+ Monocytes and CD14+ Macrophages in Systemic Sclerosis. Front. Immunol. 2021, 12, 642891. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Jia, G.; Guttman, A.; DePianto, D.J.; Morshead, K.B.; Sun, K.H.; Ramamoorthi, N.; Vander Heiden, J.A.; Modrusan, Z.; Wolters, P.J.; et al. Osteopontin Links Myeloid Activation and Disease Progression in Systemic Sclerosis. Cell Rep. Med. 2020, 1, 100140. [Google Scholar] [CrossRef]
- Sun, C.; Chen, S.Y. RGC32 Promotes Bleomycin-Induced Systemic Sclerosis in a Murine Disease Model by Modulating Classically Activated Macrophage Function. J. Immunol. 2018, 200, 2777–2785. [Google Scholar] [CrossRef] [Green Version]
- Bonhomme, O.; André, B.; Gester, F.; de Seny, D.; Moermans, C.; Struman, I.; Louis, R.; Malaise, M.; Guiot, J. Biomarkers in systemic sclerosis-associated interstitial lung disease: Review of the literature. Rheumatology 2019, 58, 1534–1546. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Guabiraba, R.; Besnard, A.G.; Komai-Koma, M.; Jabir, M.S.; Zhang, L.; Graham, G.J.; Kurowska-Stolarska, M.; Liew, F.Y.; McSharry, C.; et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J. Allergy Clin. Immunol. 2014, 134, 1422–1432.e11. [Google Scholar] [CrossRef]
- Li, S.; Gao, S.; Jiang, Q.; Liang, Q.; Luan, J.; Zhang, R.; Zhang, F.; Ruan, H.; Li, X.; Zhou, H.; et al. Clevudine attenuates bleomycin-induced early pulmonary fibrosis via regulating M2 macrophage polarization. Int. Immunopharmacol. 2021, 101, 108271. [Google Scholar] [CrossRef]
- Nakagome, K.; Dohi, M.; Okunishi, K.; Tanaka, R.; Miyazaki, J.; Yamamoto, K. In Vivo IL-10 gene delivery attenuates bleomycin induced pulmonary fibrosis by inhibiting the production and activation of TGF-beta in the lung. Thorax 2006, 61, 886–894. [Google Scholar] [CrossRef] [Green Version]
- El-Demerdash, E. Anti-inflammatory and antifibrotic effects of methyl palmitate. Toxicol. Appl. Pharmacol. 2011, 254, 238–244. [Google Scholar] [CrossRef]
- Wang, J.; Xu, L.; Xiang, Z.; Ren, Y.; Zheng, X.; Zhao, Q.; Zhou, Q.; Zhou, Y.; Wang, Y. Microcystin-LR ameliorates pulmonary fibrosis via modulating CD206+ M2-like macrophage polarization. Cell Death Dis. 2020, 11, 136. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Maier, C.; Zhang, Y.; Soare, A.; Dees, C.; Beyer, C.; Harre, U.; Chen, C.W.; Distler, O.; Schett, G.; et al. Nintedanib inhibits macrophage activation and ameliorates vascular and fibrotic manifestations in the Fra2 mouse model of systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 1941–1948. [Google Scholar] [CrossRef]
- Toda, M.; Mizuguchi, S.; Minamiyama, Y.; Yamamoto-Oka, H.; Aota, T.; Kubo, S.; Nishiyama, N.; Shibata, T.; Takemura, S. Pirfenidone suppresses polarization to M2 phenotype macrophages and the fibrogenic activity of rat lung fibroblasts. J. Clin. Biochem. Nutr. 2018, 63, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, H.; Sato, S.; Koyama, K.; Morizumi, S.; Abe, S.; Azuma, M.; Chen, Y.; Goto, H.; Aono, Y.; Ogawa, H.; et al. The novel inhibitor PRI-724 for Wnt/β-catenin/CBP signaling ameliorates bleomycin-induced pulmonary fibrosis in mice. Exp. Lung Res. 2019, 45, 188–199. [Google Scholar] [CrossRef]
- Raghu, G.; Hamblin, M.J.; Brown, A.W.; Golden, J.A.; Ho, L.A.; Wijsenbeek, M.S.; Vasakova, M.; Pesci, A.; Antin-Ozerkis, D.E.; Meyer, K.C.; et al. Long-term evaluation of the safety and efficacy of recombinant human pentraxin-2 (rhPTX-2) in patients with idiopathic pulmonary fibrosis (IPF): An open-label extension study. Respir. Res. 2022, 23, 129. [Google Scholar] [CrossRef]
- Guo, Z.; Li, S.; Zhang, N.; Kang, Q.; Zhai, H. Schisandra Inhibit Bleomycin-Induced Idiopathic Pulmonary Fibrosis in Rats via Suppressing M2 Macrophage Polarization. Biomed. Res. Int. 2020, 2020, 5137349. [Google Scholar] [CrossRef]
- Lescoat, A.; Lelong, M.; Jeljeli, M.; Piquet-Pellorce, C.; Morzadec, C.; Ballerie, A.; Jouneau, S.; Jego, P.; Vernhet, L.; Batteux, F.; et al. Combined anti-fibrotic and anti-inflammatory properties of JAK-inhibitors on macrophages in vitro and in vivo: Perspectives for scleroderma-associated interstitial lung disease. Biochem. Pharmacol. 2020, 178, 114103. [Google Scholar] [CrossRef]
- Liu, B.; Jiang, Q.; Chen, R.; Gao, S.; Xia, Q.; Zhu, J.; Zhang, F.; Shao, C.; Liu, X.; Li, X.; et al. Tacrolimus ameliorates bleomycin-induced pulmonary fibrosis by inhibiting M2 macrophage polarization via JAK2/STAT3 signaling. Int. Immunopharmacol. 2022, 113, 109424. [Google Scholar] [CrossRef]
- Boutel, M.; Boutou, A.; Pitsiou, G.; Garyfallos, A.; Dimitroulas, T. Efficacy and Safety of Nintedanib in Patients with Connective Tissue Disease-Interstitial Lung Disease (CTD-ILD): A Real-World Single Center Experience. Diagnostics 2023, 13, 1221. [Google Scholar] [CrossRef]
- Kamenova, A.; Tzouvelekis, A.; Margaritopoulos, G.A. Recent advances in the treatment of systemic sclerosis associated interstitial lung disease. Front. Med. 2023, 10, 1155771. [Google Scholar] [CrossRef]
- Tardella, M.; Di Carlo, M.; Carotti, M.; Giovagnoni, A.; Salaffi, F. Abatacept in rheumatoid arthritis-associated interstitial lung disease: Short-term outcomes and predictors of progression. Clin. Rheumatol. 2021, 40, 4861–4867. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Tashkin, D.P.; Wells, A.U.; Seibold, J.R.; Wax, S.; Vazquez-Mateo, C.; Fleuranceau-Morel, P.; Damian, D.; Denton, C.P. STRATUS: A Phase II Study of Abituzumab in Patients With Systemic Sclerosis-associated Interstitial Lung Disease. J. Rheumatol. 2021, 48, 1295–1298. [Google Scholar] [CrossRef]
- Ll Wilkinson, M.G.; Deakin, C.T.; Papadopoulou, C.; Eleftheriou, D.; Wedderburn, L.R. JAK inhibitors: A potential treatment for JDM in the context of the role of interferon-driven pathology. Pediatr. Rheumatol. Online J. 2021, 19, 146. [Google Scholar] [CrossRef] [PubMed]
- Mwangi, J.; Litteken, C.; Gorthi, R.; Attoti, Y.; Atluri, R. Belimumab in the Treatment of Connective Tissue Disease-Associated Interstitial Lung Disease: Case Report and Literature Review. Cureus 2021, 13, e19218. [Google Scholar] [CrossRef] [PubMed]
- Penke, L.R.K.; Speth, J.; Wettlaufer, S.; Draijer, C.; Peters-Golden, M. Bortezomib Inhibits Lung Fibrosis and Fibroblast Activation without Proteasome Inhibition. Am. J. Respir. Cell Mol. Biol. 2022, 66, 23–37. [Google Scholar] [CrossRef]
- Giacomelli, R.; Liakouli, V.; Berardicurti, O.; Ruscitti, P.; Di Benedetto, P.; Carubbi, F.; Guggino, G.; Di Bartolomeo, S.; Ciccia, F.; Triolo, G.; et al. Interstitial lung disease in systemic sclerosis: Current and future treatment. Rheumatol. Int. 2017, 37, 853–863. [Google Scholar] [CrossRef]
- Harari, S.; Wells, A.U.; Wuyts, W.A.; Nathan, S.D.; Kirchgaessler, K.U.; Bengus, M.; Behr, J. The 6-min walk test as a primary end-point in interstitial lung disease. Eur. Respir. Rev. 2022, 31, 220087. [Google Scholar] [CrossRef]
- Vonk, M.C. Is there still a role for cyclophosphamide in the treatment of systemic sclerosis? J. Scleroderma Relat. Disord. 2021, 6, 117–122. [Google Scholar] [CrossRef]
- Kim, G.H.J.; Tashkin, D.P.; Lo, P.; Brown, M.S.; Volkmann, E.R.; Gjertson, D.W.; Khanna, D.; Elashoff, R.M.; Tseng, C.H.; Roth, M.D.; et al. Using Transitional Changes on High-Resolution Computed Tomography to Monitor the Impact of Cyclophosphamide or Mycophenolate Mofetil on Systemic Sclerosis-Related Interstitial Lung Disease. Arthritis Rheumatol. 2020, 72, 316–325. [Google Scholar] [CrossRef]
- Eapen, M.S.; Gaikwad, A.V.; Thompson, I.E.; Lu, W.; Myers, S.; Sharma, P.; Sohal, S.S. The effectiveness of immunosuppressive cyclosporin in attenuating the progression of interstitial lung diseases. J. Thorac. Dis. 2019, 11, S1139–S1142. [Google Scholar] [CrossRef]
- Chen, I.C.; Liu, Y.C.; Wu, Y.H.; Lo, S.H.; Dai, Z.K.; Hsu, J.H.; Tseng, Y.H. Evaluation of Proteasome Inhibitors in the Treatment of Idiopathic Pulmonary Fibrosis. Cells 2022, 11, 1543. [Google Scholar] [CrossRef]
- Assassi, S.; Volkmann, E.R.; Zheng, W.J.; Wang, X.; Wilhalme, H.; Lyons, M.A.; Roth, M.D.; Tashkin, D.P. Peripheral blood gene expression profiling shows predictive significance for response to mycophenolate in systemic sclerosis-related interstitial lung disease. Ann. Rheum. Dis. 2022, 81, 854–860. [Google Scholar] [CrossRef]
- Naidu, G.S.R.S.; Sharma, S.K.; Adarsh, M.B.; Dhir, V.; Sinha, A.; Dhooria, S.; Jain, S. Effect of mycophenolate mofetil (MMF) on systemic sclerosis-related interstitial lung disease with mildly impaired lung function: A double-blind, placebo-controlled, randomized trial. Rheumatol. Int. 2020, 40, 207–216. [Google Scholar] [CrossRef]
- Kulshrestha, R.; Pandey, A.; Jaggi, A.; Bansal, S. Beneficial effects of N-acetylcysteine on protease-antiprotease balance in attenuating bleomycin-induced pulmonary fibrosis in rats. Iran. J. Basic. Med. Sci. 2020, 23, 396–405. [Google Scholar]
- Denton, C.P.; Goh, N.S.; Humphries, S.M.; Maher, T.M.; Spiera, R.; Devaraj, A.; Ho, L.; Stock, C.; Erhardt, E.; Alves, M.; et al. Extent of fibrosis and lung function decline in patients with systemic sclerosis and interstitial lung disease: Data from the SENSCIS trial. Rheumatology 2023, 62, 1870–1876. [Google Scholar] [CrossRef] [PubMed]
- Solomon, J.J.; Danoff, S.K.; Woodhead, F.A.; Hurwitz, S.; Maurer, R.; Glaspole, I.; Dellaripa, P.F.; Gooptu, B.; Vassallo, R.; Cox, P.G.; et al. Safety, tolerability, and efficacy of pirfenidone in patients with rheumatoid arthritis-associated interstitial lung disease: A randomised, double-blind, placebo-controlled, phase 2 study. Lancet Respir. Med. 2023, 11, 87–96. [Google Scholar] [CrossRef]
- Hsu, V.M.; Denton, C.P.; Domsic, R.T.; Furst, D.E.; Rischmueller, M.; Stanislav, M.; Steen, V.D.; Distler, J.H.W.; Korish, S.; Cooper, A.; et al. Pomalidomide in Patients with Interstitial Lung Disease due to Systemic Sclerosis: A Phase II, Multicenter, Randomized, Double-blind, Placebo-controlled, Parallel-group Study. J. Rheumatol. 2018, 45, 405–410. [Google Scholar] [CrossRef]
- Available online: https://www.prometheusbiosciences.com/pipeline/pra023/tl1a/ (accessed on 30 March 2023).
- Maher, T.M.; Tudor, V.A.; Saunders, P.; Gibbons, M.A.; Fletcher, S.V.; Denton, C.P.; Hoyles, R.K.; Parfrey, H.; Renzoni, E.A.; Kokosi, M.; et al. Rituximab versus intravenous cyclophosphamide in patients with connective tissue disease-associated interstitial lung disease in the UK (RECITAL): A double-blind, double-dummy, randomised, controlled, phase 2b trial. Lancet Respir. Med. 2023, 11, 45–54. [Google Scholar] [CrossRef]
- Takada, K.; Katada, Y.; Ito, S.; Hayashi, T.; Kishi, J.; Itoh, K.; Yamashita, H.; Hirakata, M.; Kawahata, K.; Kawakami, A.; et al. Impact of adding tacrolimus to initial treatment of interstitial pneumonitis in polymyositis/dermatomyositis: A single-arm clinical trial. Rheumatology 2020, 59, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Kuwana, M.; Wakasugi, N.; Furuya, T.; Uno, S.; Suda, T. Tacrolimus in Patients With Interstitial Pneumonia Associated With Polymyositis or Dermatomyositis: Interim Report of Postmarketing Surveillance in Japan. J. Rheumatol. 2022, 49, 707–718. [Google Scholar] [CrossRef]
- Mansour, S.M.; El-Abhar, H.S.; Soubh, A.A. MiR-200a inversely correlates with Hedgehog and TGF-β canonical/non-canonical trajectories to orchestrate the anti-fibrotic effect of Tadalafil in a bleomycin-induced pulmonary fibrosis model. Inflammopharmacology 2021, 29, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Parida, J.; Nath, A.; Neyaz, Z.; Agarwal, V. A double blind randomized control trial of oral tadalafil in interstitial lung disease of scleroderma [abstract]. Arthritis Rheum. 2014, 66, S739. [Google Scholar]
- Kalyoncu, U.; Bilgin, E.; Erden, A.; Satış, H.; Tufan, A.; Tekgöz, E.; Ateş, A.; Coşkun, B.N.; Yağız, B.; Küçükşahin, O.; et al. Efficacy and safety of tofacitinib in rheumatoid arthritis-associated interstitial lung disease: TReasure real-life data. Clin. Exp. Rheumatol. 2022, 40, 2071–2077. [Google Scholar] [PubMed]
- Harrison, C. Itch receptor OSMR attracts industry. Nat. Biotechnol. 2022, 40, 1306. [Google Scholar] [CrossRef]
- Yaseen, B.; Lopez, H.; Taki, Z.; Zafar, S.; Rosario, H.; Abdi, B.A.; Vigneswaran, S.; Xing, F.; Arumalla, N.; Black, S.; et al. Interleukin-31 promotes pathogenic mechanisms underlying skin and lung fibrosis in scleroderma. Rheumatology 2020, 59, 2625–2636. [Google Scholar] [CrossRef]
- Erre, G.L.; Sebastiani, M.; Manfredi, A.; Gerratana, E.; Atzeni, F.; Passiu, G.; Mangoni, A.A. Antifibrotic drugs in connective tissue disease-related interstitial lung disease (CTD-ILD): From mechanistic insights to therapeutic applications. Drugs Context 2021, 10, 2020-8-6. [Google Scholar] [CrossRef]
- Faust, A.; Bäumer, N.; Schlütermann, A.; Becht, M.; Greune, L.; Geyer, C.; Rüter, C.; Margeta, R.; Wittmann, L.; Dersch, P.; et al. Tumor-Cell-Specific Targeting of Ibrutinib: Introducing Electrostatic Antibody-Inhibitor Conjugates (AiCs). Angew. Chem. Int. Ed. Engl. 2022, 61, e202109769. [Google Scholar] [CrossRef]
- Gearty, S.V.; Dündar, F.; Zumbo, P.; Espinosa-Carrasco, G.; Shakiba, M.; Sanchez-Rivera, F.J.; Socci, N.D.; Trivedi, P.; Lowe, S.W.; Lauer, P.; et al. An autoimmune stem-like CD8 T cell population drives type 1 diabetes. Nature 2022, 602, 156–161. [Google Scholar] [CrossRef]
- Nie, Y.; Zhao, L.; Zhang, X. B Cell Aberrance in Lupus: The Ringleader and the Solution. Clin. Rev. Allergy Immunol. 2022, 62, 301–323. [Google Scholar] [CrossRef]
- Bohdziewicz, A.; Pawlik, K.K.; Maciejewska, M.; Sikora, M.; Alda-Malicka, R.; Czuwara, J.; Rudnicka, L. Future Treatment Options in Systemic Sclerosis-Potential Targets and Ongoing Clinical Trials. J. Clin. Med. 2022, 11, 1310. [Google Scholar] [CrossRef]
- Radziszewska, A.; Moulder, Z.; Jury, E.C.; Ciurtin, C. CD8+ T Cell Phenotype and Function in Childhood and Adult-Onset Connective Tissue Disease. Int. J. Mol. Sci. 2022, 23, 11431. [Google Scholar] [CrossRef]
- Xu, L.; Wang, F.; Luo, F. Rituximab for the treatment of connective tissue disease-associated interstitial lung disease: A systematic review and meta-analysis. Front. Pharmacol. 2022, 13, 1019915. [Google Scholar] [CrossRef]
- Bellan, M.; Patrucco, F.; Barone-Adesi, F.; Gavelli, F.; Castello, L.M.; Nerviani, A.; Andreoli, L.; Cavagna, L.; Pirisi, M.; Sainaghi, P.P. Targeting CD20 in the treatment of interstitial lung diseases related to connective tissue diseases: A systematic review. Autoimmun. Rev. 2020, 19, 102453. [Google Scholar] [CrossRef]



| Agents | Targeted Cells | Diseases | Study | Outcome a |
|---|---|---|---|---|
| Abatacept | T cells [111] | Antisynthetase syndrome-associated ILD | NCT03215927 | Active |
| Abatacept | T cells [111] | RA-ILD | NCT03084419 | Unknown |
| Abituzumab | Epithelial cells [112] | SSc-ILD | NCT02745145 | Terminated due to slow enrollment [112] |
| Basiliximab | Lymphocytes [113] | DM-ILD | NCT03192657 | Unknown |
| Belimumab | B cells [114] | SSc-ILD | NCT05878717 | Not yet recruiting |
| Bortezomib | Fibroblasts [115] | SSc-ILD | NCT02370693 | Completed |
| Bosentan | Fibroblasts [116] | SSc-ILD | NCT00070590 | Ineffective [117] |
| Bosentan | Fibroblasts [116] | SSc-ILD | NCT00319033 | Completed |
| Cyclophosphamide | Lymphocytes [118] | Antisynthetase syndrome-associated ILD | NCT03770663 | Recruiting |
| Cyclophosphamide | Lymphocytes [118] | SSc-ILD | NCT00883129 | Improve [119] |
| Cyclophosphamide | Lymphocytes [118] | SSc-ILD | NCT01570764 | Completed |
| Cyclosporin A | T cells [120] | Sjogren’s syndrome-associated ILD | NCT02370550 | Unknown |
| Ixazomib | Fibroblasts [121] | SSc-ILD | NCT04837131 | Recruiting |
| Mycophenolate mofetil | Lymphocytes [122] | Myositis-associated ILD | NCT05129410 | Recruiting |
| Mycophenolate mofetil | Lymphocytes [122] | SSc-ILD | NCT00883129 | Improve [119] |
| Mycophenolate mofetil | Lymphocytes [122] | SSc-ILD | NCT02896205 | Ineffective [123] |
| Mycophenolate mofetil | Lymphocytes [122] | SSc-ILD | NCT05785065 | Not yet recruiting |
| N-acetylcysteine | Macrophages [124] | CTD-ILD | NCT01424033 | Terminated due to departure of principal investigator. |
| Nintedanib | Macrophages | Myositis-associated ILD | NCT05335278 | Recruiting |
| Nintedanib | Macrophages | Myositis-associated ILD | NCT05799755 | Not yet recruiting |
| Nintedanib | Macrophages | SSc-ILD | NCT02597933 | Effective [125] |
| Nintedanib | Macrophages | SSc-ILD | NCT03313180 | Completed |
| Pirfenidone | Macrophages | CTD-ILD | NCT04928586 | Recruiting |
| Pirfenidone | Macrophages | CTD-ILD | NCT05505409 | Recruiting |
| Pirfenidone | Macrophages | DM-ILD | NCT02821689 | Unknown |
| Pirfenidone | Macrophages | DM-ILD | NCT03857854 | Unknown |
| Pirfenidone | Macrophages | RA-ILD | NCT02808871 | Slow FVC decline [126] |
| Pirfenidone | Macrophages | SSc-ILD | NCT03221257 | Completed |
| Pirfenidone | Macrophages | SSc-ILD | NCT03856853 | Unknown |
| Pomalidomide | T cells [127] | SSc-ILD | NCT01559129 | Discontinued [127] |
| PRA023 | Fibroblasts [128] | SSc-ILD | NCT05270668 | Recruiting |
| Rituximab | B cells [129] | CTD-ILD | NCT01862926 | Not superior to cyclophosphamide [129] |
| Rituximab | B cells [129] | RA-ILD | NCT00578565 | Inconclusive |
| Tacrolimus | Macrophages | Myositis-associated ILD | NCT00504348 | Improve [130] |
| Tacrolimus | Macrophages | Myositis-associated ILD | NCT02159651 | Improve [131] |
| Tadalafil | Fibroblasts [132] | SSc-ILD | NCT01553981 | Improve [133] |
| Tofacitinib | Myeloid derived suppressor cells [134] | RA-ILD | NCT04311567 | Recruiting |
| Tofacitinib | Myeloid derived suppressor cells [134] | RA-ILD | NCT05246293 | Recruiting |
| Vixarelimab | Fibroblasts [135,136] | SSc-ILD | NCT05785624 | Recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tseng, C.-C.; Sung, Y.-W.; Chen, K.-Y.; Wang, P.-Y.; Yen, C.-Y.; Sung, W.-Y.; Wu, C.-C.; Ou, T.-T.; Tsai, W.-C.; Liao, W.-T.; et al. The Role of Macrophages in Connective Tissue Disease-Associated Interstitial Lung Disease: Focusing on Molecular Mechanisms and Potential Treatment Strategies. Int. J. Mol. Sci. 2023, 24, 11995. https://doi.org/10.3390/ijms241511995
Tseng C-C, Sung Y-W, Chen K-Y, Wang P-Y, Yen C-Y, Sung W-Y, Wu C-C, Ou T-T, Tsai W-C, Liao W-T, et al. The Role of Macrophages in Connective Tissue Disease-Associated Interstitial Lung Disease: Focusing on Molecular Mechanisms and Potential Treatment Strategies. International Journal of Molecular Sciences. 2023; 24(15):11995. https://doi.org/10.3390/ijms241511995
Chicago/Turabian StyleTseng, Chia-Chun, Ya-Wen Sung, Kuan-Yu Chen, Pin-Yi Wang, Chang-Yi Yen, Wan-Yu Sung, Cheng-Chin Wu, Tsan-Teng Ou, Wen-Chan Tsai, Wei-Ting Liao, and et al. 2023. "The Role of Macrophages in Connective Tissue Disease-Associated Interstitial Lung Disease: Focusing on Molecular Mechanisms and Potential Treatment Strategies" International Journal of Molecular Sciences 24, no. 15: 11995. https://doi.org/10.3390/ijms241511995
APA StyleTseng, C. -C., Sung, Y. -W., Chen, K. -Y., Wang, P. -Y., Yen, C. -Y., Sung, W. -Y., Wu, C. -C., Ou, T. -T., Tsai, W. -C., Liao, W. -T., Chen, C. -J., Lee, S. -C., Chang, S. -J., & Yen, J. -H. (2023). The Role of Macrophages in Connective Tissue Disease-Associated Interstitial Lung Disease: Focusing on Molecular Mechanisms and Potential Treatment Strategies. International Journal of Molecular Sciences, 24(15), 11995. https://doi.org/10.3390/ijms241511995