

Retinoic Acid Receptor β Loss in Hepatocytes Increases Steatosis and Elevates the Integrated Stress Response in Alcohol-Associated Liver Disease
 ,
,
Abstract
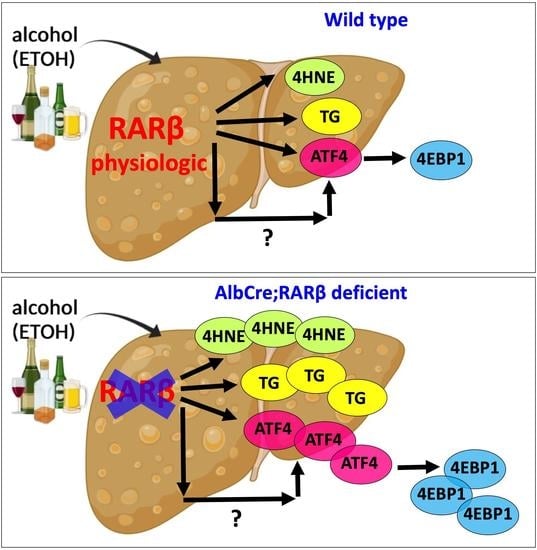
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. RARβ Protects Hepatocytes from Alcohol-Associated Steatosis
2.2. RARβ Limits the ETOH-Associated Integrated Stress Response (ISR) in Mice
2.3. RARβ Knockout-Cultured Hepatocytes also Show Increased Reactive Oxygen Species Generation
2.4. ATF4 Is Increased in the RARβ Knockout-Cultured Hepatocytes
2.5. RARβ Is a Negative Regulator of ATF4 but Not 4-EBP1 in Cultured Cells
3. Discussion
3.1. ETOH-Fed BKO Mice Develop Greater Hepatic Steatosis Than Wild Type Mice
3.2. Lack of RARβ Causes Greater Oxidative Stress and the Activation of the ATF4/Integrated Stress Response during ETOH Treatment
3.3. Cyp2E1 Is Not Required for the More Severe ALD and Increased ISR Network in the BKO Liver
4. Materials and Methods
4.1. Mice and Treatments
4.2. Triglyceride Measurements
4.3. Blood Alcohol Level Measurements
4.4. RNA Isolation and qRT-PCR
4.5. Southern Blotting and Immunostaining
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Primers 2018, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Moon, A.M.; Yang, J.Y.; Barritt, A.S.; Bataller, R.; Peery, A.F. Rising Mortality From Alcohol-Associated Liver Disease in the United States in the 21st Century. Am. J. Gastroenterol. 2020, 115, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wu, D.; Wang, X.; Ward, S.C.; Cederbaum, A.I. Chronic alcohol-induced liver injury and oxidant stress are decreased in cytochrome P4502E1 knockout mice and restored in humanized cytochrome P4502E1 knock-in mice. Free. Radic. Biol. Med. 2010, 49, 1406–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver Dis. 2012, 16, 667–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, C. New Insights into the Pathogenesis of Alcohol-Induced ER Stress and Liver Diseases. Int. J. Hepatol. 2014, 2014, 513787. [Google Scholar] [CrossRef] [Green Version]
- Hao, L.; Zhong, W.; Dong, H.; Guo, W.; Sun, X.; Zhang, W.; Yue, R.; Li, T.; Griffiths, A.; Ahmadi, A.R.; et al. ATF4 activation promotes hepatic mitochondrial dysfunction by repressing NRF1-TFAM signalling in alcoholic steatohepatitis. Gut 2021, 70, 1933–1945. [Google Scholar] [CrossRef]
- Miyata, T.; Nagy, L.E. Programmed cell death in alcohol-associated liver disease. Clin. Mol. Hepatol. 2020, 26, 618–625. [Google Scholar] [CrossRef]
- Leo, M.A.; Lieber, C.S. Hepatic vitamin A depletion in alcoholic liver injury. N. Engl. J. Med. 1982, 307, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Hepatic and metabolic effects of ethanol: Pathogenesis and prevention. Ann. Med. 1994, 26, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Blaner, W.S.; Li, Y.; Brun, P.J.; Yuen, J.J.; Lee, S.A.; Clugston, R.D. Vitamin A Absorption, Storage and Mobilization. Subcell. Biochem. 2016, 81, 95–125. [Google Scholar] [CrossRef]
- Brun, P.J.; Yang, K.J.; Lee, S.A.; Yuen, J.J.; Blaner, W.S. Retinoids: Potent regulators of metabolism. Biofactors 2013, 39, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Gudas, L.J. Emerging roles for retinoids in regeneration and differentiation in normal and disease states. Biochim. Biophys. Acta 2012, 1821, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, R.F.; Gudas, L.J. Retinoid regulated association of transcriptional co-regulators and the polycomb group protein SUZ12 with the retinoic acid response elements of Hoxa1, RARbeta(2), and Cyp26A1 in F9 embryonal carcinoma cells. J. Mol. Biol. 2007, 372, 298–316. [Google Scholar] [CrossRef] [Green Version]
- Clugston, R.D.; Blaner, W.S. The adverse effects of alcohol on vitamin A metabolism. Nutrients 2012, 4, 356–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, S.S.; Prathibha, P.; Rejitha, S.; Indira, M. Ethanol induced hepatic mitochondrial dysfunction is attenuated by all trans retinoic acid supplementation. Life Sci. 2015, 135, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Trasino, S.E.; Tang, X.H.; Jessurun, J.; Gudas, L.J. Retinoic acid receptor beta2 agonists restore glycaemic control in diabetes and reduce steatosis. Diabetes Obes. Metab. 2016, 18, 142–151. [Google Scholar] [CrossRef] [Green Version]
- Melis, M.; Tang, X.H.; Attarwala, N.; Chen, Q.; Prishker, C.; Qin, L.; Gross, S.S.; Gudas, L.J.; Trasino, S.E. A retinoic acid receptor β2 agonist protects against alcohol liver disease and modulates hepatic expression of canonical retinoid metabolism genes. Biofactors 2022, 48, 469–4801. [Google Scholar] [CrossRef] [PubMed]
- Melis, M.; Tang, X.H.; Trasino, S.E.; Patel, V.M.; Stummer, D.J.; Jessurun, J.; Gudas, L.J. Effects of AM80 compared to AC261066 in a high fat diet mouse model of liver disease. PLoS ONE 2019, 14, e0211071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.H.; Melis, M.; Lu, C.; Rappa, A.; Zhang, T.; Jessurun, J.; Gross, S.S.; Gudas, L.J. A Retinoic Acid Receptor β2 Agonist Attenuates Transcriptome and Metabolome Changes Underlying Non-Alcohol-Associated Fatty Liver Disease. J. Biol. Chem. 2021, 297, 101331. [Google Scholar] [CrossRef]
- Chapellier, B.; Mark, M.; Bastien, J.; Dierich, A.; LeMeur, M.; Chambon, P.; Ghyselinck, N.B. A conditional floxed (loxP-flanked) allele for the retinoic acid receptor beta (RARbeta) gene. Genesis 2002, 32, 91–94. [Google Scholar] [CrossRef]
- Kong, Y.; Zhao, C.; Huang, Y.; Liu, Y.; Liu, S.; Guo, Y.; Li, M.; Xu, T.; Zhao, B.; Wang, J. Angiopoietin-like protein 4 promotes very-low-density lipoprotein assembly and secretion in bovine hepatocytes in vitro. IUBMB Life 2020, 72, 2710–2721. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.Y.; He, H.; Nguyen, T.; Mennone, A.; Boyer, J.L. Retinoic acid represses CYP7A1 expression in human hepatocytes and HepG2 cells by FXR/RXR-dependent and independent mechanisms. J. Lipid Res. 2010, 51, 2265–2274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, Y. The biological significance of ω-oxidation of fatty acids. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2013, 89, 370–382. [Google Scholar] [CrossRef] [Green Version]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Lee, M.S. GDF15 as a central mediator for integrated stress response and a promising therapeutic molecule for metabolic disorders and NASH. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129834. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes. Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [PubMed]
- van der Mijn, J.C.; Chen, Q.; Laursen, K.B.; Khani, F.; Wang, X.; Dorsaint, P.; Sboner, A.; Gross, S.S.; Nanus, D.M.; Gudas, L.J. Transcriptional and metabolic remodeling in clear cell renal cell carcinoma caused by ATF4 activation and the integrated stress response (ISR). Mol. Carcinog. 2022, 61, 851–864. [Google Scholar] [CrossRef]
- Tameire, F.; Verginadis, I.I.; Leli, N.M.; Polte, C.; Conn, C.S.; Ojha, R.; Salas Salinas, C.; Chinga, F.; Monroy, A.M.; Fu, W.; et al. ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat. Cell Biol. 2019, 21, 889–899. [Google Scholar] [CrossRef]
- Sommer, T.; Jarosch, E. BiP binding keeps ATF6 at bay. Dev. Cell 2002, 3, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Benedetti, A.; Comporti, M.; Esterbauer, H. Identification of 4-hydroxynonenal as a cytotoxic product originating from the peroxidation of liver microsomal lipids. Biochim. Biophys. Acta 1980, 620, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Zou, J.; Liu, C.; Khan, M.; Xie, T.; Huang, X.; Zhang, K.; Yuan, Y.; Wang, B. A Multi-Omics Pan-Cancer Analysis of 4-EBP1 in Cancer Prognosis and Cancer-Associated Fibroblasts Infiltration. Front. Genet. 2022, 13, 845751. [Google Scholar] [CrossRef] [PubMed]
- Bertola, A.; Mathews, S.; Ki, S.H.; Wang, H.; Gao, B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 2013, 8, 627–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, R.M.; Dhir, R.; Yin, X.; Agarwal, B.; Ahima, R.S. Temporal effects of ethanol consumption on energy homeostasis, hepatic steatosis, and insulin sensitivity in mice. Alcohol. Clin. Exp. Res. 2013, 37, 1091–1099. [Google Scholar] [CrossRef] [Green Version]
- Bideyan, L.; Fan, W.; Kaczor-Urbanowicz, K.E.; Priest, C.; Casero, D.; Tontonoz, P. Integrative analysis reveals multiple modes of LXR transcriptional regulation in liver. Proc. Natl. Acad. Sci. USA 2022, 119, e2122683119. [Google Scholar] [CrossRef]
- Fondevila, M.F.; Fernandez, U.; Heras, V.; Parracho, T.; Gonzalez-Rellan, M.J.; Novoa, E.; Porteiro, B.; Alonso, C.; Mayo, R.; da Silva Lima, N.; et al. Inhibition of carnitine palmitoyltransferase 1A in hepatic stellate cells protects against fibrosis. J. Hepatol. 2022, 77, 15–28. [Google Scholar] [CrossRef]
- Li, Y.; Wong, K.; Walsh, K.; Gao, B.; Zang, M. Retinoic acid receptor β stimulates hepatic induction of fibroblast growth factor 21 to promote fatty acid oxidation and control whole-body energy homeostasis in mice. J. Biol. Chem. 2013, 288, 10490–10504. [Google Scholar] [CrossRef] [Green Version]
- Yanagitani, A.; Yamada, S.; Yasui, S.; Shimomura, T.; Murai, R.; Murawaki, Y.; Hashiguchi, K.; Kanbe, T.; Saeki, T.; Ichiba, M.; et al. Retinoic acid receptor alpha dominant negative form causes steatohepatitis and liver tumors in transgenic mice. Hepatology 2004, 40, 366–375. [Google Scholar] [CrossRef]
- Cassim Bawa, F.N.; Xu, Y.; Gopoju, R.; Plonski, N.M.; Shiyab, A.; Hu, S.; Chen, S.; Zhu, Y.; Jadhav, K.; Kasumov, T.; et al. Hepatic retinoic acid receptor alpha mediates all-trans retinoic acid’s effect on diet-induced hepatosteatosis. Hepatol. Commun. 2022, 6, 2665–2675. [Google Scholar] [CrossRef]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005, 24, 1243–1255. [Google Scholar] [CrossRef]
- Li, K.; Xiao, Y.; Yu, J.; Xia, T.; Liu, B.; Guo, Y.; Deng, J.; Chen, S.; Wang, C.; Guo, F. Liver-specific Gene Inactivation of the Transcription Factor ATF4 Alleviates Alcoholic Liver Steatosis in Mice. J. Biol. Chem. 2016, 291, 18536–18546. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.S.; Das, S.S.; Nair, R.P.; Indira, M. Supplementation of all trans retinoic acid ameliorates ethanol-induced endoplasmic reticulum stress. Arch. Physiol. Biochem. 2018, 124, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, A.A.; Driedzic, W.R.; Campos, D.; Heinrichs-Caldas, W.; Almeida-Val, V.M.F.; Val, A.L.; Lamarre, S.G. Protein synthesis is lowered by 4-EBP1 and eIF2-α signaling while protein degradation may be maintained in fasting, hypoxic Amazonian cichlids. J. Exp. Biol. 2018, 221, jeb167601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magne, L.; Blanc, E.; Legrand, B.; Lucas, D.; Barouki, R.; Rouach, H.; Garlatti, M. ATF4 and the integrated stress response are induced by ethanol and cytochrome P450 2E1 in human hepatocytes. J. Hepatol. 2011, 54, 729–737. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Kleiner, D.E.; Hosler, B.A.; Rogers, M.B.; Kozak, C.A.; Gudas, L.J. An octamer motif contributes to the expression of the retinoic acid-regulated zinc finger gene Rex-1 (Zfp-42) in F9 teratocarcinoma cells. Mol. Cell Biol. 1993, 13, 2919–2928. [Google Scholar]
- Wilson, L.A.; Belt, P.; Neuschwander-Tetri, B.A. (CRN) NCRN. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: Distinct clinicopathologic meanings. Hepatology 2011, 53, 810–820. [Google Scholar]
- Trasino, S.E.; Tang, X.H.; Jessurun, J.; Gudas, L.J. A retinoic acid receptor β2 agonist reduces hepatic stellate cell activation in nonalcoholic fatty liver disease. J. Mol. Med. 2016, 94, 1143–1151. [Google Scholar] [PubMed] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melis, M.; Trasino, S.E.; Tang, X.-H.; Rappa, A.; Zhang, T.; Qin, L.; Gudas, L.J. Retinoic Acid Receptor β Loss in Hepatocytes Increases Steatosis and Elevates the Integrated Stress Response in Alcohol-Associated Liver Disease. Int. J. Mol. Sci. 2023, 24, 12035. https://doi.org/10.3390/ijms241512035
Melis M, Trasino SE, Tang X-H, Rappa A, Zhang T, Qin L, Gudas LJ. Retinoic Acid Receptor β Loss in Hepatocytes Increases Steatosis and Elevates the Integrated Stress Response in Alcohol-Associated Liver Disease. International Journal of Molecular Sciences. 2023; 24(15):12035. https://doi.org/10.3390/ijms241512035
Chicago/Turabian StyleMelis, Marta, Steven E. Trasino, Xiao-Han Tang, Andrew Rappa, Tuo Zhang, Lihui Qin, and Lorraine J. Gudas. 2023. "Retinoic Acid Receptor β Loss in Hepatocytes Increases Steatosis and Elevates the Integrated Stress Response in Alcohol-Associated Liver Disease" International Journal of Molecular Sciences 24, no. 15: 12035. https://doi.org/10.3390/ijms241512035
APA StyleMelis, M., Trasino, S. E., Tang, X. -H., Rappa, A., Zhang, T., Qin, L., & Gudas, L. J. (2023). Retinoic Acid Receptor β Loss in Hepatocytes Increases Steatosis and Elevates the Integrated Stress Response in Alcohol-Associated Liver Disease. International Journal of Molecular Sciences, 24(15), 12035. https://doi.org/10.3390/ijms241512035
{kind=link}