
Comprehensive Evaluation and Transcriptome Analysis Reveal the Salt Tolerance Mechanism in Semi-Wild Cotton (Gossypium purpurascens)

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
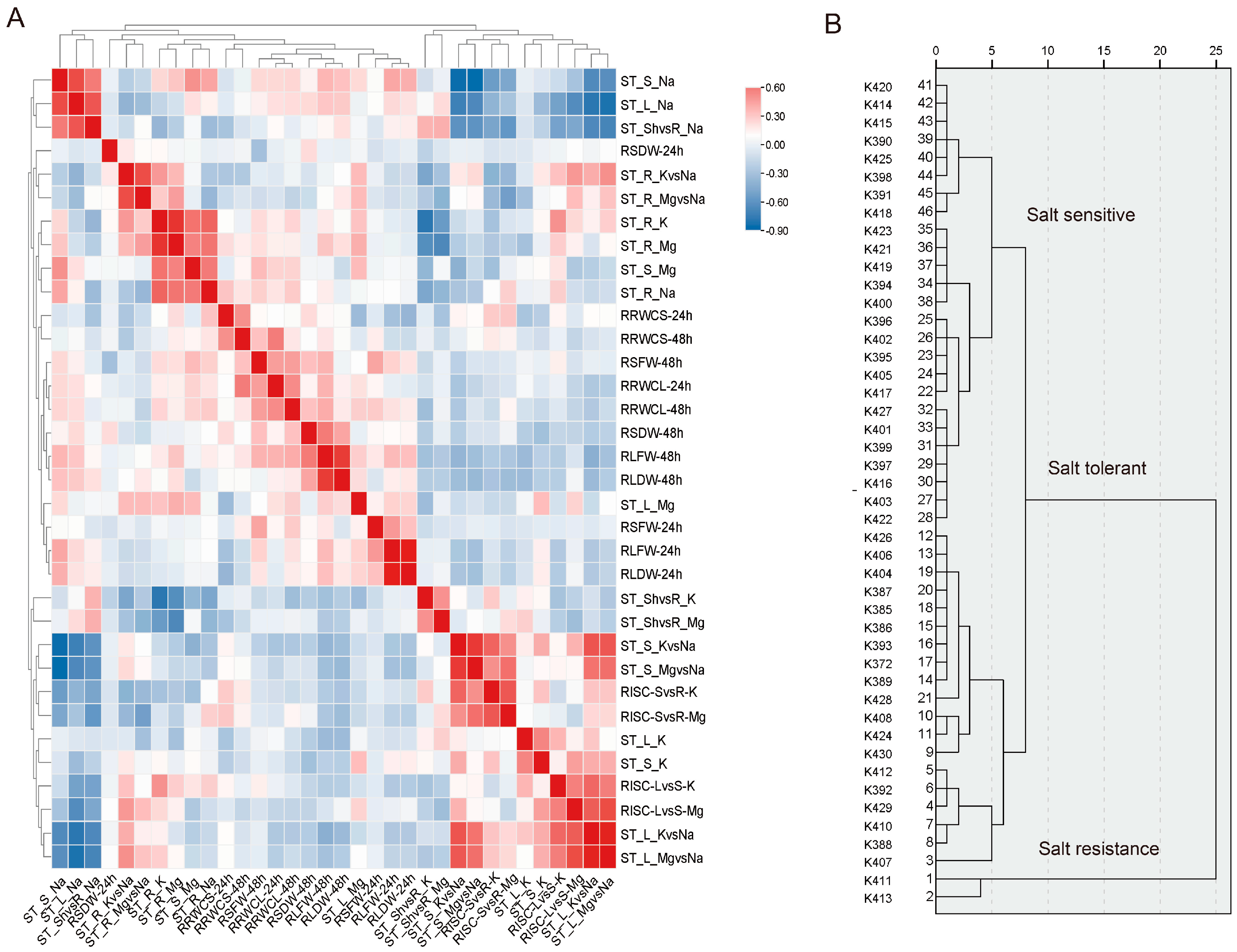
2.1. Comprehensive Evaluation of Salt Tolerance of 45 G. purpurascens at the Seedling Stage
2.2. The Phenome of Two Genotypes with Contrasting Differences in Salt Tolerance Showed Significant Differences under Salinity Stress
2.3. RNA-Seq Analysis
2.4. Identification of DEGs Using RNA-Seq
2.5. Identification of DE Salt Tolerance-Related Genes and Key KEGG Pathways
2.6. Analysis of DEGs Based on the Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG)
2.7. TF, RLK, CAZy, and Other Key Genes Involved in Salinity Tolerance
2.8. Gene Co-Expression Network Analysis Identifies Specific Modules Related to Salt Stress in G. purpurascens
2.9. Identification of Highly Connected and Central Hub Genes
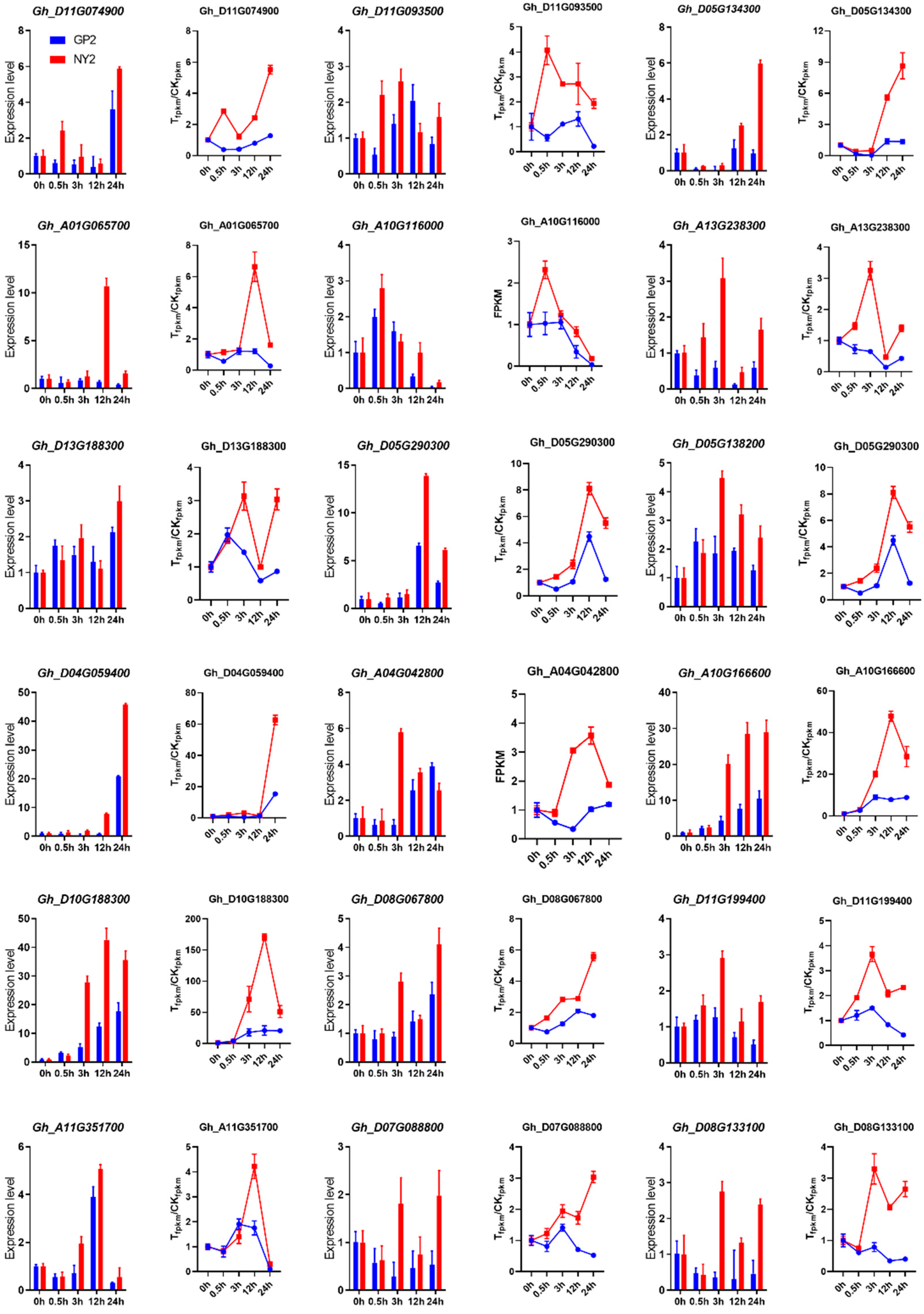
2.10. qRT-PCR Validation of RNA-Seq Data
3. Discussion
3.1. Germplasm of G. purpurascens Collected from South China Sea Islands in China Exhibited Strong Salt Tolerance
3.2. Comparative Analysis of Transcriptome Data and Differentially Expressed Genes Related to Salt Tolerance
3.3. MAPK, SSM, PPI, FB, PHST, and FA Pathways Play Role in Salt Stress Tolerance
3.4. TF, RLK, CAZy, and Reported Salt Tolerance-Related Genes
4. Materials and Methods
4.1. Plant Materials
4.2. Plant Growth Conditions and Salinity Stress
4.3. Physiological and Biochemical Traits
4.4. Transcriptome Profiling
4.5. Library Preparation for Transcriptome Sequencing
4.6. Differential Expression Analysis of Salt-Tolerant Genes
4.7. Construction of Weighted Gene Co-Expression Network (WGCNA)
4.8. Quantitative Real-Time PCR Analysis
4.9. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Annunziata, M.G.; Ciarmiello, L.F.; Woodrow, P.; Maximova, E.; Fuggi, A.; Carillo, P. Durum wheat roots adapt to salinity remodeling the cellular content of nitrogen metabolites and sucrose. Front. Plant Sci. 2017, 7, 2035. [Google Scholar] [CrossRef]
- Mohamed, I.A.; Shalby, N.; Bai, C.; Qin, M.; Agami, R.A.; Jie, K.; Wang, B.; Zhou, G. Stomatal and photosynthetic traits are associated with investigating sodium chloride tolerance of Brassica napus L. cultivars. Plants 2020, 9, 62. [Google Scholar] [CrossRef] [PubMed]
- Rajabi Dehnavi, A.; Zahedi, M.; Ludwiczak, A.; Cardenas Perez, S.; Piernik, A. Effect of salinity on seed germination and seedling development of sorghum (Sorghum bicolor (L.) Moench) genotypes. Agronomy 2020, 10, 859. [Google Scholar] [CrossRef]
- Mohamed, I.A.; Shalby, N.; MA El-Badri, A.; Saleem, M.H.; Khan, M.N.; Nawaz, M.A.; Qin, M.; Agami, R.A.; Kuai, J.; Wang, B. Stomata and xylem vessels traits improved by melatonin application contribute to enhancing salt tolerance and fatty acid composition of Brassica napus L. plants. Agronomy 2020, 10, 1186. [Google Scholar] [CrossRef]
- Jamil, A.; Riaz, S.; Ashraf, M.; Foolad, M.R. Gene Expression Profiling of Plants under Salt Stress. Crit. Rev. Plant Sci. 2011, 30, 435–458. [Google Scholar] [CrossRef]
- Zhang, C.; Luo, W.; Li, Y.; Zhang, X.; Bai, X.; Niu, Z.; Zhang, X.; Li, Z.; Wan, D. Transcriptomic analysis of seed germination under salt stress in two desert sister species (Populus euphratica and P. pruinosa). Front. Genet. 2019, 10, 231. [Google Scholar] [CrossRef]
- Erpen, L.; Devi, H.S.; Grosser, J.W.; Dutt, M. Potential use of the DREB/ERF, MYB, NAC and WRKY transcription factors to improve abiotic and biotic stress in transgenic plants. Plant Cell Tissue Organ Cult. 2018, 132, 1–25. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, L.; Zhu, P.; Cao, Q.; Sun, J.; Li, Z.; Xu, T. Genome-wide identification, characterisation and functional evaluation of WRKY genes in the sweet potato wild ancestor Ipomoea trifida (HBK) G. Don. under abiotic stresses. BMC Genet. 2019, 20, 90. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Sun, J.; Chen, Y.; Zhu, P.; Zhang, L.; Wu, S.; Ma, D.; Cao, Q.; Li, Z.; Xu, T. Genome-wide identification, structural and gene expression analysis of the bZIP transcription factor family in sweet potato wild relative Ipomoea trifida. BMC Genet. 2019, 20, 41. [Google Scholar] [CrossRef]
- Zhu, M.; Meng, X.; Cai, J.; Li, G.; Dong, T.; Li, Z. Basic leucine zipper transcription factor SlbZIP1 mediates salt and drought stress tolerance in tomato. BMC Plant Biol. 2018, 18, 83. [Google Scholar] [CrossRef]
- Su, Y.; Guo, A.; Huang, Y.; Wang, Y.; Hua, J. GhCIPK6a increases salt tolerance in transgenic upland cotton by involving in ROS scavenging and MAPK signaling pathways. BMC Plant Biol. 2020, 20, 421. [Google Scholar] [CrossRef]
- Wang, J.; Li, C.; Yao, X.; Liu, S.; Zhang, P.; Chen, K. The Antarctic moss leucine-rich repeat receptor-like kinase (PnLRR-RLK2) functions in salinity and drought stress adaptation. Polar Biol. 2018, 41, 353–364. [Google Scholar] [CrossRef]
- Lin, F.; Li, S.; Wang, K.; Tian, H.; Gao, J.; Zhao, Q.; Du, C. A leucine-rich repeat receptor-like kinase, OsSTLK, modulates salt tolerance in rice. Plant Sci. 2020, 296, 110465. [Google Scholar] [CrossRef]
- Ibrahim, E.A. Seed priming to alleviate salinity stress in germinating seeds. J. Plant Physiol. 2016, 192, 38–46. [Google Scholar] [CrossRef]
- Dinakar, C.; Bartels, D. Desiccation tolerance in resurrection plants: New insights from transcriptome, proteome and metabolome analysis. Front. Plant Sci. 2013, 4, 482. [Google Scholar] [CrossRef] [PubMed]
- Gapper, N.E.; Giovannoni, J.J.; Watkins, C.B. Understanding development and ripening of fruit crops in an ‘omics’ era. Hortic. Res. 2014, 1, 14034. [Google Scholar] [CrossRef]
- McGettigan, P.A. Transcriptomics in the RNA-seq era. Curr. Opin. Chem. Biol. 2013, 17, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Amirbakhtiar, N.; Ismaili, A.; Ghaffari, M.R.; Nazarian Firouzabadi, F.; Shobbar, Z.-S. Transcriptome response of roots to salt stress in a salinity-tolerant bread wheat cultivar. PLoS ONE 2019, 14, e0213305. [Google Scholar] [CrossRef]
- Nakayama, R.; Safi, M.T.; Ahmadzai, W.; Sato, K.; Kawaura, K. Comparative transcriptome analysis of synthetic and common wheat in response to salt stress. Sci. Rep. 2022, 12, 11534. [Google Scholar] [CrossRef]
- Zhu, Y.; Ren, Y.; Liu, J.A.; Liang, W.; Zhang, Y.; Shen, F.; Ling, J.; Zhang, C. New Genes Identified as Modulating Salt Tolerance in Maize Seedlings Using the Combination of Transcriptome Analysis and BSA. Plants 2023, 12, 1331. [Google Scholar] [CrossRef] [PubMed]
- Chandran, A.K.N.; Kim, J.-W.; Yoo, Y.-H.; Park, H.L.; Kim, Y.-J.; Cho, M.-H.; Jung, K.-H. Transcriptome analysis of rice-seedling roots under soil–salt stress using RNA-Seq method. Plant Biotechnol. Rep. 2019, 13, 567–578. [Google Scholar] [CrossRef]
- Punia, H.; Tokas, J.; Malik, A.; Sangwan, S.; Rani, A.; Yashveer, S.; Alansi, S.; Hashim, M.J.; El-Sheikh, M.A. Genome-wide transcriptome profiling, characterization, and functional identification of NAC transcription factors in sorghum under salt stress. Antioxidants 2021, 10, 1605. [Google Scholar] [CrossRef] [PubMed]
- Zeng, A.; Chen, P.; Korth, K.L.; Ping, J.; Thomas, J.; Wu, C.; Srivastava, S.; Pereira, A.; Hancock, F.; Brye, K. RNA sequencing analysis of salt tolerance in soybean (Glycine max). Genomics 2019, 111, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jiang, L.; Chen, J.; Tao, L.; An, Y.; Cai, H.; Guo, C. Overexpression of the alfalfa WRKY11 gene enhances salt tolerance in soybean. PLoS ONE 2018, 13, e0192382. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, Z.; Ding, Y.; Liu, L.; Han, X.; Zhan, J.; Wei, X.; Diao, Y.; Qin, W.; Wang, P.; et al. Over-expression of an R2R3 MYB Gene, GhMYB73, increases tolerance to salt stress in transgenic Arabidopsis. Plant Sci. 2019, 286, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhang, G.; Zhou, S.; Ren, Y.; Wang, W. The improvement of salt tolerance in transgenic tobacco by overexpression of wheat F-box gene TaFBA1. Plant Sci. 2017, 259, 71–85. [Google Scholar] [CrossRef]
- Feng, L.; Li, Y.; Zhou, Y.-L.; Meng, G.-H.; Ji, Z.-L.; Lin, W.-H.; He, J.-X. Integrative transcriptomic and proteomic analyses reveal a positive role of BES1 in salt tolerance in Arabidopsis. Front. Plant Sci. 2023, 14, 1034393. [Google Scholar] [CrossRef]
- Wang, R.; Liu, L.; Kong, Z.; Li, S.; Lu, L.; Chen, G.; Zhang, J.; Qanmber, G.; Liu, Z. Identification of GhLOG gene family revealed that GhLOG3 is involved in regulating salinity tolerance in cotton (Gossypium hirsutum L.). Plant Physiol. Biochem. 2021, 166, 328–340. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef]
- Ruan, J.; Dean, A.K.; Zhang, W. A general co-expression network-based approach to gene expression analysis: Comparison and applications. BMC Syst. Biol. 2010, 4, 8. [Google Scholar] [CrossRef]
- Wan, Q.; Tang, J.; Han, Y.; Wang, D. Co-expression modules construction by WGCNA and identify potential prognostic markers of uveal melanoma. Exp. Eye Res. 2018, 166, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Sadhukhan, A.; Tazib, T.; Nakano, Y.; Kusunoki, K.; Kamara, M.; Chaffai, R.; Iuchi, S.; Sahoo, L.; Kobayashi, M. Joint genetic and network analyses identify loci associated with root growth under NaCl stress in Arabidopsis thaliana. Plant Cell Environ. 2016, 39, 918–934. [Google Scholar] [CrossRef]
- Wang, J.; Lv, P.; Yan, D.; Zhang, Z.; Xu, X.; Wang, T.; Wang, Y.; Peng, Z.; Yu, C.; Gao, Y. Exogenous melatonin improves seed germination of wheat (Triticum aestivum L.) under salt stress. Int. J. Mol. Sci. 2022, 23, 8436. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhang, M.; Chen, J.; Qing, C.; He, S.; Zou, C.; Yuan, G.; Yang, C.; Peng, H.; Pan, G. GWAS and WGCNA uncover hub genes controlling salt tolerance in maize (Zea mays L.) seedlings. Theor. Appl. Genet. 2021, 134, 3305–3318. [Google Scholar] [CrossRef]
- Zhu, M.; Xie, H.; Wei, X.; Dossa, K.; Yu, Y.; Hui, S.; Tang, G.; Zeng, X.; Yu, Y.; Hu, P. WGCNA analysis of salt-responsive core transcriptome identifies novel hub genes in rice. Genes 2019, 10, 719. [Google Scholar] [CrossRef]
- Xu, Y.; Magwanga, R.O.; Yang, X.; Jin, D.; Cai, X.; Hou, Y.; Wei, Y.; Zhou, Z.; Wang, K.; Liu, F. Genetic regulatory networks for salt-alkali stress in Gossypium hirsutum with differing morphological characteristics. BMC Genom. 2020, 21, 15. [Google Scholar] [CrossRef]
- Rojo-Gutiérrez, E.; Buenrostro-Figueroa, J.; López-Martínez, L.; Sepúlveda, D.; Baeza-Jiménez, R. Biotechnological potential of cottonseed, a by-product of cotton production. In Valorisation of Agro-Industrial Residues–Volume II: Non-Biological Approaches; Springer: Berlin/Heidelberg, Germany, 2020; pp. 63–82. [Google Scholar]
- Abdelraheem, A.; Esmaeili, N.; O’Connell, M.; Zhang, J. Progress and perspective on drought and salt stress tolerance in cotton. Ind. Crops Prod. 2019, 130, 118–129. [Google Scholar] [CrossRef]
- Wang, N.; Wang, X.; Shi, J.; Liu, X.; Xu, Q.; Zhou, H.; Song, M.; Yan, G. Mepiquat chloride-priming induced salt tolerance during seed germination of cotton (Gossypium hirsutum L.) through regulating water transport and K+/Na+ homeostasis. Environ. Exp. Bot. 2019, 159, 168–178. [Google Scholar] [CrossRef]
- Watt, G. The Wild and Cultivated Cotton Plants of the World: A Revision of the Genus Gossypium, Framed Primarily with the Object of Aiding Planters and Investigators Who May Contemplate the Systematic Improvement of the Cotton Staple; Longmans, Green, and Company: New York, NY, USA, 1907. [Google Scholar]
- Watt, G. Gossypium. Bull. Misc. Inf. R. Bot. Gard. Kew 1927, 1927, 321–356. [Google Scholar] [CrossRef]
- Harland, S. New polyploids in cotton by the use of colchicine. Trop. Agric. 1940, 17, 53–54. [Google Scholar]
- Hutchinson, J.; Stephens, S. Note on the “French” or “small-seeded” cotton grown in the west indies in the 18th century. Trop. Agric. 1944, 21, 123–125. [Google Scholar]
- Hutchinson, J. Intra-specific differentiation in Gossypium hirsutum. Heredity 1951, 5, 161–193. [Google Scholar] [CrossRef]
- Yu, Q. A brief description of the investigation and research on cotton seed in china. Acta Agric. 1941, 6, 715. [Google Scholar]
- Cheng, Y.; Huang, C.; Hu, Y.; Jin, S.; Zhang, X.; Si, Z.; Zhao, T.; Chen, J.; Fang, L.; Dai, F.; et al. Gossypium purpurascens genome provides insight into the origin and domestication of upland cotton. J. Adv. Res. 2023. [Google Scholar] [CrossRef]
- Gharaghanipor, N.; Arzani, A.; Rahimmalek, M.; Ravash, R. Physiological and transcriptome indicators of salt tolerance in wild and cultivated barley. Front. Plant Sci. 2022, 13, 819282. [Google Scholar] [CrossRef] [PubMed]
- Nazir, M.F.; He, S.; Ahmed, H.; Sarfraz, Z.; Jia, Y.; Li, H.; Sun, G.; Iqbal, M.S.; Pan, Z.; Du, X. Genomic insight into the divergence and adaptive potential of a forgotten landrace G. hirsutum L. purpurascens. J. Genet. Genom. 2021, 48, 473–484. [Google Scholar] [CrossRef]
- Huang, M.; Zhang, Z.; Sheng, Z.; Zhu, C.; Zhai, Y.; Lu, P.; Brinkman, D. Soil salinity and maize growth under cycle irrigation in coastal soils. Agron. J. 2019, 111, 2276–2286. [Google Scholar] [CrossRef]
- Walia, H.; Wilson, C.; Wahid, A.; Condamine, P.; Cui, X.; Close, T.J. Expression analysis of barley (Hordeum vulgare L.) during salinity stress. Funct. Integr. Genom. 2006, 6, 143–156. [Google Scholar] [CrossRef]
- Darko, E.; Gierczik, K.; Hudak, O.; Forgó, P.; Pál, M.; Türkösi, E.; Kovács, V.; Dulai, S.; Majlath, I.; Molnar, I. Differing metabolic responses to salt stress in wheat-barley addition lines containing different 7H chromosomal fragments. PLoS ONE 2017, 12, e0174170. [Google Scholar] [CrossRef]
- Khan, M.S.; Akther, T.; Hemalatha, S. Impact of panchagavya on Oryza sativa L. grown under saline stress. J. Plant Growth Regul. 2017, 36, 702–713. [Google Scholar] [CrossRef]
- Xiao, C.; Ji, Q.; Zhang, F.; Li, Y.; Fan, J.; Hou, X.; Yan, F.; Liu, X.; Gong, K. Effects of various soil water potential thresholds for drip irrigation on soil salinity, seed cotton yield and water productivity of cotton in northwest China. Agric. Water Manag. 2023, 279, 108172. [Google Scholar] [CrossRef]
- Singh, J.; Singh, V.; Sharma, P. Elucidating the role of osmotic, ionic and major salt responsive transcript components towards salinity tolerance in contrasting chickpea (Cicer arietinum L.) genotypes. Physiol. Mol. Biol. Plants 2018, 24, 441–453. [Google Scholar] [CrossRef]
- Arif, Y.; Singh, P.; Siddiqui, H.; Bajguz, A.; Hayat, S. Salinity induced physiological and biochemical changes in plants: An omic approach towards salt stress tolerance. Plant Physiol. Biochem. 2020, 156, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Shabala, S.; Cuin, T.A. Potassium transport and plant salt tolerance. Physiol. Plant. 2008, 133, 651–669. [Google Scholar] [CrossRef] [PubMed]
- Cotsaftis, O.; Plett, D.; Johnson, A.A.; Walia, H.; Wilson, C.; Ismail, A.M.; Close, T.J.; Tester, M.; Baumann, U. Root-specific transcript profiling of contrasting rice genotypes in response to salinity stress. Mol. Plant 2011, 4, 25–41. [Google Scholar] [CrossRef]
- Walia, H.; Wilson, C.; Zeng, L.; Ismail, A.M.; Condamine, P.; Close, T.J. Genome-wide transcriptional analysis of salinity stressed japonica and indica rice genotypes during panicle initiation stage. Plant Mol. Biol. 2007, 63, 609–623. [Google Scholar] [CrossRef]
- Kumar, N.; Soren, K.R.; Bharadwaj, C.; P.R., S.P.; Shrivastava, A.K.; Pal, M.; Roorkiwal, M.; Kumar, K.; Patil, B.S.; Soni, A.; et al. Genome-wide transcriptome analysis and physiological variation modulates gene regulatory networks acclimating salinity tolerance in chickpea. Environ. Exp. Bot. 2021, 187, 104478. [Google Scholar] [CrossRef]
- Flowers, T.J.; Gaur, P.M.; Gowda, C.L.; Krishnamurthy, L.; Samineni, S.; Siddique, K.H.; Turner, N.C.; Vadez, V.; Varshney, R.K.; Colmer, T.D. Salt sensitivity in chickpea. Plant Cell Environ. 2010, 33, 490–509. [Google Scholar] [CrossRef]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef]
- Ebrahim, F.; Arzani, A.; Rahimmalek, M.; Sun, D.; Peng, J. Salinity tolerance of wild barley Hordeum vulgare ssp. spontaneum. Plant Breed. 2020, 139, 304–316. [Google Scholar] [CrossRef]
- Arefian, M.; Malekzadeh Shafaroudi, S. Physiological and gene expression analysis of extreme chickpea (Cicer arietinum L.) genotypes in response to salinity stress. Acta Physiol. Plant. 2015, 37, 193. [Google Scholar] [CrossRef]
- Negrão, S.; Schmöckel, S.; Tester, M. Evaluating physiological responses of plants to salinity stress. Ann. Bot. 2017, 119, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Yi, H.; Hu, L.; Fu, J. Stomatal and metabolic limitations to photosynthesis resulting from NaCl stress in perennial ryegrass genotypes differing in salt tolerance. J. Am. Soc. Hortic. Sci. 2013, 138, 350–357. [Google Scholar] [CrossRef]
- Bahieldin, A.; Atef, A.; Sabir, J.S.; Gadalla, N.O.; Edris, S.; Alzohairy, A.M.; Radhwan, N.A.; Baeshen, M.N.; Ramadan, A.M.; Eissa, H.F. RNA-Seq analysis of the wild barley (H. spontaneum) leaf transcriptome under salt stress. Comptes Rendus Biol. 2015, 338, 285–297. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, P.; Cui, F.; Zhang, F.; Luo, X.; Xie, J. Transcriptome analysis of salt stress responsiveness in the seedlings of Dongxiang wild rice (Oryza rufipogon Griff.). PLoS ONE 2016, 11, e0146242. [Google Scholar] [CrossRef]
- Li, J.; Gao, Z.; Zhou, L.; Li, L.; Zhang, J.; Liu, Y.; Chen, H. Comparative transcriptome analysis reveals K+ transporter gene contributing to salt tolerance in eggplant. BMC Plant Biol. 2019, 19, 67. [Google Scholar] [CrossRef]
- Luo, Y.; Reid, R.; Freese, D.; Li, C.; Watkins, J.; Shi, H.; Zhang, H.; Loraine, A.; Song, B.-H. Salt tolerance response revealed by RNA-Seq in a diploid halophytic wild relative of sweet potato. Sci. Rep. 2017, 7, 9624. [Google Scholar] [CrossRef]
- Shinde, H.; Tanaka, K.; Dudhate, A.; Tsugama, D.; Mine, Y.; Kamiya, T.; Gupta, S.K.; Liu, S.; Takano, T. Comparative de novo transcriptomic profiling of the salinity stress responsiveness in contrasting pearl millet lines. Environ. Exp. Bot. 2018, 155, 619–627. [Google Scholar] [CrossRef]
- Han, M.; Lu, X.; Yu, J.; Chen, X.; Wang, X.; Malik, W.A.; Wang, J.; Wang, D.; Wang, S.; Guo, L. Transcriptome analysis reveals cotton (Gossypium hirsutum) genes that are differentially expressed in cadmium stress tolerance. Int. J. Mol. Sci. 2019, 20, 1479. [Google Scholar] [CrossRef]
- Guo, J.; Li, C.; Zhang, X.; Li, Y.; Zhang, D.; Shi, Y.; Song, Y.; Li, Y.; Yang, D.; Wang, T. Transcriptome and GWAS analyses reveal candidate gene for seminal root length of maize seedlings under drought stress. Plant Sci. 2020, 292, 110380. [Google Scholar] [CrossRef]
- Walia, H.; Wilson, C.; Condamine, P.; Liu, X.; Ismail, A.M.; Zeng, L.; Wanamaker, S.I.; Mandal, J.; Xu, J.; Cui, X. Comparative transcriptional profiling of two contrasting rice genotypes under salinity stress during the vegetative growth stage. Plant Physiol. 2005, 139, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Verma, N.; Pandey, C.; Verma, D.; Bhagat, P.K.; Noryang, S.; Singh, K.; Tayyeba, S.; Banerjee, G.; Sinha, A.K. MAP kinase as regulators for stress responses in plants: An overview. In Protein Kinases and Stress Signaling in Plants: Functional Genomic Perspective; Wiley: Hoboken, NJ, USA, 2020; pp. 369–392. [Google Scholar]
- Fraire-Velázquez, S.; Rodríguez-Guerra, R.; Sánchez-Calderón, L. Abiotic and biotic stress response crosstalk in plants. In Abiotic Stress Response in Plants—Physiological, Biochemical and Genetic Perspectives; IntechOpen: London, UK, 2011; pp. 3–26. [Google Scholar]
- Zhu, J.-K. Abiotic stress signaling and responses in plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Meng, Y.; Bai, Y.; Yu, H.; Qian, Y.; Zhang, D.; Zhou, Y. Starch and Sucrose Metabolism and Plant Hormone Signaling Pathways Play Crucial Roles in Aquilegia Salt Stress Adaption. Int. J. Mol. Sci. 2023, 24, 3948. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.G.; Kobayashi, Y.; Sanuki, A.; Kondo, S.; Fukuda, N.; Ezura, H.; Sugaya, S.; Matsukura, C. Salinity induces carbohydrate accumulation and sugar-regulated starch biosynthetic genes in tomato (Solanum lycopersicum L. cv. ‘Micro-Tom’) fruits in an ABA- and osmotic stress-independent manner. J. Exp. Bot. 2010, 61, 563–574. [Google Scholar] [CrossRef]
- Tocher, D.R.; Castell, J.D.; Dick, J.R.; Sargent, J.R. Effects of salinity on the fatty acid compositions of total lipid and individual glycerophospholipid classes of Atlantic salmon (Salmo salar) and turbot (Scophthalmus maximus) cells in culture. Fish Physiol. Biochem. 1995, 14, 125–137. [Google Scholar] [CrossRef]
- Goyal, E.; Amit, S.K.; Singh, R.S.; Mahato, A.K.; Chand, S.; Kanika, K. Transcriptome profiling of the salt-stress response in Triticum aestivum cv. Kharchia Local. Sci. Rep. 2016, 6, 27752. [Google Scholar] [CrossRef]
- Zhu, J.; Fan, Y.; Li, C.; Shabala, S.; Zhao, C.; Hong, Y.; Lv, C.; Guo, B.; Xu, R.; Zhou, M. Candidate genes for salinity tolerance in barley revealed by RNA-seq analysis of near-isogenic lines. Plant Growth Regul. 2020, 92, 571–582. [Google Scholar] [CrossRef]
- Heydarian, Z.; Yu, M.; Gruber, M.; Coutu, C.; Robinson, S.J.; Hegedus, D.D. Changes in gene expression in Camelina sativa roots and vegetative tissues in response to salinity stress. Sci. Rep. 2018, 8, 9804. [Google Scholar] [CrossRef]
- Hu, L.; Li, H.; Chen, L.; Lou, Y.; Amombo, E.; Fu, J. RNA-seq for gene identification and transcript profiling in relation to root growth of bermudagrass (Cynodon dactylon) under salinity stress. BMC Genom. 2015, 16, 575. [Google Scholar] [CrossRef]
- Tsukagoshi, H.; Suzuki, T.; Nishikawa, K.; Agarie, S.; Ishiguro, S.; Higashiyama, T. RNA-seq analysis of the response of the halophyte, Mesembryanthemum crystallinum (ice plant) to high salinity. PLoS ONE 2015, 10, e0118339. [Google Scholar] [CrossRef]
- Wang, S.; Cao, M.; Ma, X.; Chen, W.; Zhao, J.; Sun, C.; Tan, L.; Liu, F. Integrated RNA sequencing and QTL mapping to identify candidate genes from Oryza rufipogon associated with salt tolerance at the seedling stage. Front. Plant Sci. 2017, 8, 1427. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, J.; Zuo, D.; Wang, Q.; Malik, W.; Zhang, Y.; Abid, M.A.; Cheng, H.; Yang, Q.; Song, G. Recent insights into cotton functional genomics: Progress and future perspectives. Plant Biotechnol. J. 2018, 16, 699–713. [Google Scholar] [CrossRef]
- Peng, Z.; He, S.; Gong, W.; Sun, J.; Pan, Z.; Xu, F.; Lu, Y.; Du, X. Comprehensive analysis of differentially expressed genes and transcriptional regulation induced by salt stress in two contrasting cotton genotypes. BMC Genom. 2014, 15, 760. [Google Scholar] [CrossRef]
- Zhou, B.; Zhang, L.; Ullah, A.; Jin, X.; Yang, X.; Zhang, X. Identification of multiple stress responsive genes by sequencing a normalized cDNA library from sea-land cotton (Gossypium barbadense L.). PLoS ONE 2016, 11, e0152927. [Google Scholar] [CrossRef] [PubMed]
- Munns, R.; Gilliham, M. Salinity tolerance of crops–what is the cost? New Phytol. 2015, 208, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-K. Plant salt tolerance. Trends Plant Sci. 2001, 6, 66–71. [Google Scholar] [CrossRef]
- Zhang, F.; Zhu, G.; Du, L.; Shang, X.; Cheng, C.; Yang, B.; Hu, Y.; Cai, C.; Guo, W. Genetic regulation of salt stress tolerance revealed by RNA-Seq in cotton diploid wild species, Gossypium davidsonii. Sci. Rep. 2016, 6, 20582. [Google Scholar] [CrossRef]
- Harb, A.; Simpson, C.; Guo, W.; Govindan, G.; Kakani, V.G.; Sunkar, R. The Effect of Drought on Transcriptome and Hormonal Profiles in Barley Genotypes with Contrasting Drought Tolerance. Front. Plant Sci. 2020, 11, 618491. [Google Scholar] [CrossRef]
- Zhao, J.; Gao, Y.; Zhang, Z.; Chen, T.; Guo, W.; Zhang, T. A receptor-like kinase gene (GbRLK) from Gossypium barbadense enhances salinity and drought-stress tolerance in Arabidopsis. BMC Plant Biol. 2013, 13, 110. [Google Scholar] [CrossRef]
- Noh, G.; Kim, J.-H.; Cho, S.W.; Kim, Y.-H.; Jung, J.-Y.; Hong, W.-J.; Jung, K.-H.; Park, G.; Son, H.-J.; Jo, I.H.; et al. Overexpression of the ginseng GH18 gene confers salinity tolerance in Arabidopsis. Plant Biotechnol. Rep. 2022, 16, 683–696. [Google Scholar] [CrossRef]
- Nishiyama, R.; Le, D.T.; Watanabe, Y.; Matsui, A.; Tanaka, M.; Seki, M.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.-S.P. Transcriptome analyses of a salt-tolerant cytokinin-deficient mutant reveal differential regulation of salt stress response by cytokinin deficiency. PLoS ONE 2012, 7, e32124. [Google Scholar] [CrossRef] [PubMed]
- Hong-Bo, S.; Zong-Suo, L.; Ming-An, S. LEA proteins in higher plants: Structure, function, gene expression and regulation. Colloids Surf. B. Biointerfaces 2005, 45, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Leon, D.; Vermeuel, M.; Gupta, P.; Aschoff, R.; Bunagan, M.R. The Effect of Salt and Temperature on the Conformational Changes of P1LEA-22, a Repeat Unit of Plantlate Embryogenesis Abundant Proteins. Biophys. J. 2020, 118, 354a. [Google Scholar] [CrossRef]
- Chourey, K.; Ramani, S.; Apte, S.K. Accumulation of LEA proteins in salt (NaCl) stressed young seedlings of rice (Oryza sativa L.) cultivar Bura Rata and their degradation during recovery from salinity stress. J. Plant Physiol. 2003, 160, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Jyothsnakumari, G.; Thippeswamy, M.; Veeranagamallaiah, G.; Sudhakar, C. Differential expression of LEA proteins in two genotypes of mulberry under salinity. Biol. Plant. 2009, 53, 145–150. [Google Scholar] [CrossRef]
- Liang, J.; Zhou, M.; Zhou, X.; Jin, Y.; Xu, M.; Lin, J. JcLEA, a Novel LEA-Like Protein from Jatropha curcas, Confers a High Level of Tolerance to Dehydration and Salinity in Arabidopsis thaliana. PLoS ONE 2014, 8, e83056. [Google Scholar] [CrossRef]
- Naot, D.; Ben-Hayyim, G.; Eshdat, Y.; Holland, D. Drought, heat and salt stress induce the expression of a citrus homologue of an atypical late-embryogenesis Lea5 gene. Plant Mol. Biol. 1995, 27, 619–622. [Google Scholar] [CrossRef]
- Aduse Poku, S.; Nkachukwu Chukwurah, P.; Aung, H.H.; Nakamura, I. Over-expression of a melon Y3SK2-type LEA gene confers drought and salt tolerance in transgenic tobacco plants. Plants 2020, 9, 1749. [Google Scholar] [CrossRef]
- Mahajan, S.; Pandey, G.K.; Tuteja, N. Calcium-and salt-stress signaling in plants: Shedding light on SOS pathway. Arch. Biochem. Biophys. 2008, 471, 146–158. [Google Scholar] [CrossRef]
- Hasanuzzaman, M.; Bhuyan, M.; Mahmud, J.; Nahar, K.; Mohsin, S.; Parvin, K.; Fujita, M. Interaction of sulfur with phytohormones and signaling molecules in conferring abiotic stress tolerance to plants. Plant Signal. Behav. 2018, 13, e1477905. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, X.; Lu, X.; Shu, N.; Wang, X.; Yang, X.; Wang, S.; Wang, J.; Guo, L.; Wang, D. Transcriptome analysis of Gossypium hirsutum L. reveals different mechanisms among NaCl, NaOH and Na2CO3 stress tolerance. Sci. Rep. 2018, 8, 13527. [Google Scholar] [CrossRef]
- Han, M.; Cui, R.; Wang, D.; Huang, H.; Rui, C.; Malik, W.A.; Wang, J.; Zhang, H.; Xu, N.; Liu, X. Combined transcriptomic and metabolomic analyses elucidate key salt-responsive biomarkers to regulate salt tolerance in cotton. BMC Plant Biol. 2023, 23, 245. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Feng, L.; Zhou, C.; Wan, H.; Zhou, B. Transcriptome Revealed GhPP2C43-A Negatively Regulates Salinity Tolerance in an Introgression Line from a Semi-wild Upland Cotton. Plant Cell Physiol. 2023, 64, 786–802. [Google Scholar] [CrossRef] [PubMed]
- Ju, F.; Sun, L.; Xiong, C.; Wang, Z.; Yu, H.; Pang, J.; Bai, H.; Zhao, W.; Zhou, Z.; Chen, B. Weighted gene co-expression network analysis revealed the key pathways and hub genes of potassium regulating cotton root adaptation to salt stress. Front. Plant Sci. 2023, 14, 1132877. [Google Scholar] [CrossRef] [PubMed]
- Gavlak, R.G.; Horneck, D.A.; Miller, R.O. Soil, Plant and Water Reference Methods for the Western Region; WCC-103 Publication: Fort Collins, CO, USA, 2003. [Google Scholar]
- Jones, J.B., Jr.; Case, V.W. Sampling, handling, and analyzing plant tissue samples. Soil Test. Plant Anal. 1990, 3, 389–427. [Google Scholar]
- Huang, W.; Wang, R.; Zhou, Y.; Chen, Y.; Yang, R. Improved principal component analysis for 3D seismic data simultaneous reconstruction and denoising. In SEG Technical Program Expanded Abstracts 2016; Society of Exploration Geophysicists: Houston, TX, USA, 2016; pp. 4102–4106. [Google Scholar]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Yang, Z.; Ge, X.; Yang, Z.; Qin, W.; Sun, G.; Wang, Z.; Li, Z.; Liu, J.; Wu, J.; Wang, Y.; et al. Extensive intraspecific gene order and gene structural variations in upland cotton cultivars. Nat. Commun. 2019, 10, 2989. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. In Proceedings of the International AAAI Conference on Web and Social Media, San Jose, CA, USA, 17–20 May 2009; pp. 361–362. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, Z.; Rehman, A.; Li, X.; Jiang, X.; Tian, C.; Wang, X.; Li, H.; Wang, Z.; He, S.; Du, X. Comprehensive Evaluation and Transcriptome Analysis Reveal the Salt Tolerance Mechanism in Semi-Wild Cotton (Gossypium purpurascens). Int. J. Mol. Sci. 2023, 24, 12853. https://doi.org/10.3390/ijms241612853
Peng Z, Rehman A, Li X, Jiang X, Tian C, Wang X, Li H, Wang Z, He S, Du X. Comprehensive Evaluation and Transcriptome Analysis Reveal the Salt Tolerance Mechanism in Semi-Wild Cotton (Gossypium purpurascens). International Journal of Molecular Sciences. 2023; 24(16):12853. https://doi.org/10.3390/ijms241612853
Chicago/Turabian StylePeng, Zhen, Abdul Rehman, Xiawen Li, Xuran Jiang, Chunyan Tian, Xiaoyang Wang, Hongge Li, Zhenzhen Wang, Shoupu He, and Xiongming Du. 2023. "Comprehensive Evaluation and Transcriptome Analysis Reveal the Salt Tolerance Mechanism in Semi-Wild Cotton (Gossypium purpurascens)" International Journal of Molecular Sciences 24, no. 16: 12853. https://doi.org/10.3390/ijms241612853
APA StylePeng, Z., Rehman, A., Li, X., Jiang, X., Tian, C., Wang, X., Li, H., Wang, Z., He, S., & Du, X. (2023). Comprehensive Evaluation and Transcriptome Analysis Reveal the Salt Tolerance Mechanism in Semi-Wild Cotton (Gossypium purpurascens). International Journal of Molecular Sciences, 24(16), 12853. https://doi.org/10.3390/ijms241612853