
Exposure of Cultured Hippocampal Neurons to the Mitochondrial Uncoupler Carbonyl Cyanide Chlorophenylhydrazone Induces a Rapid Growth of Dendritic Processes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
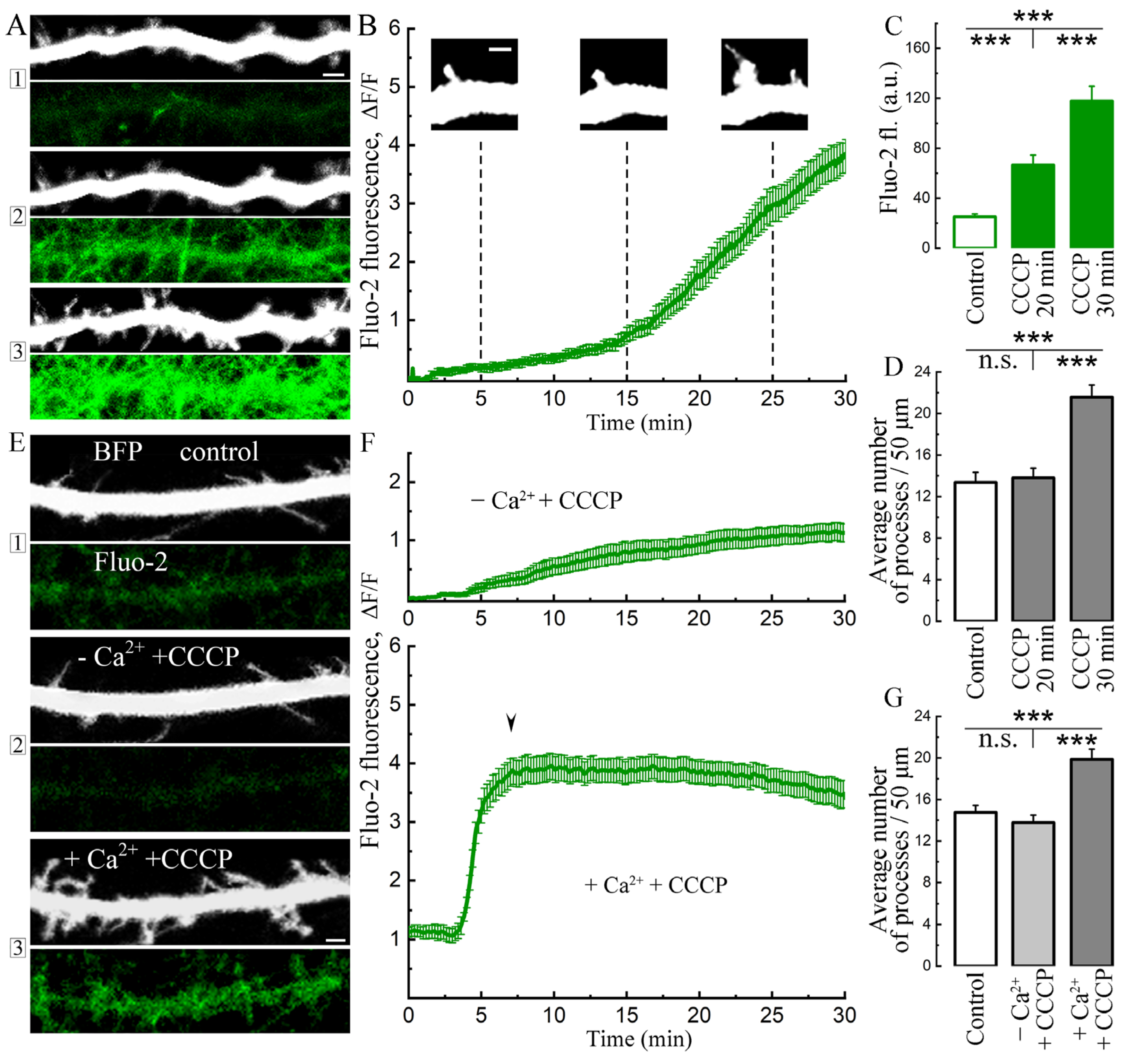
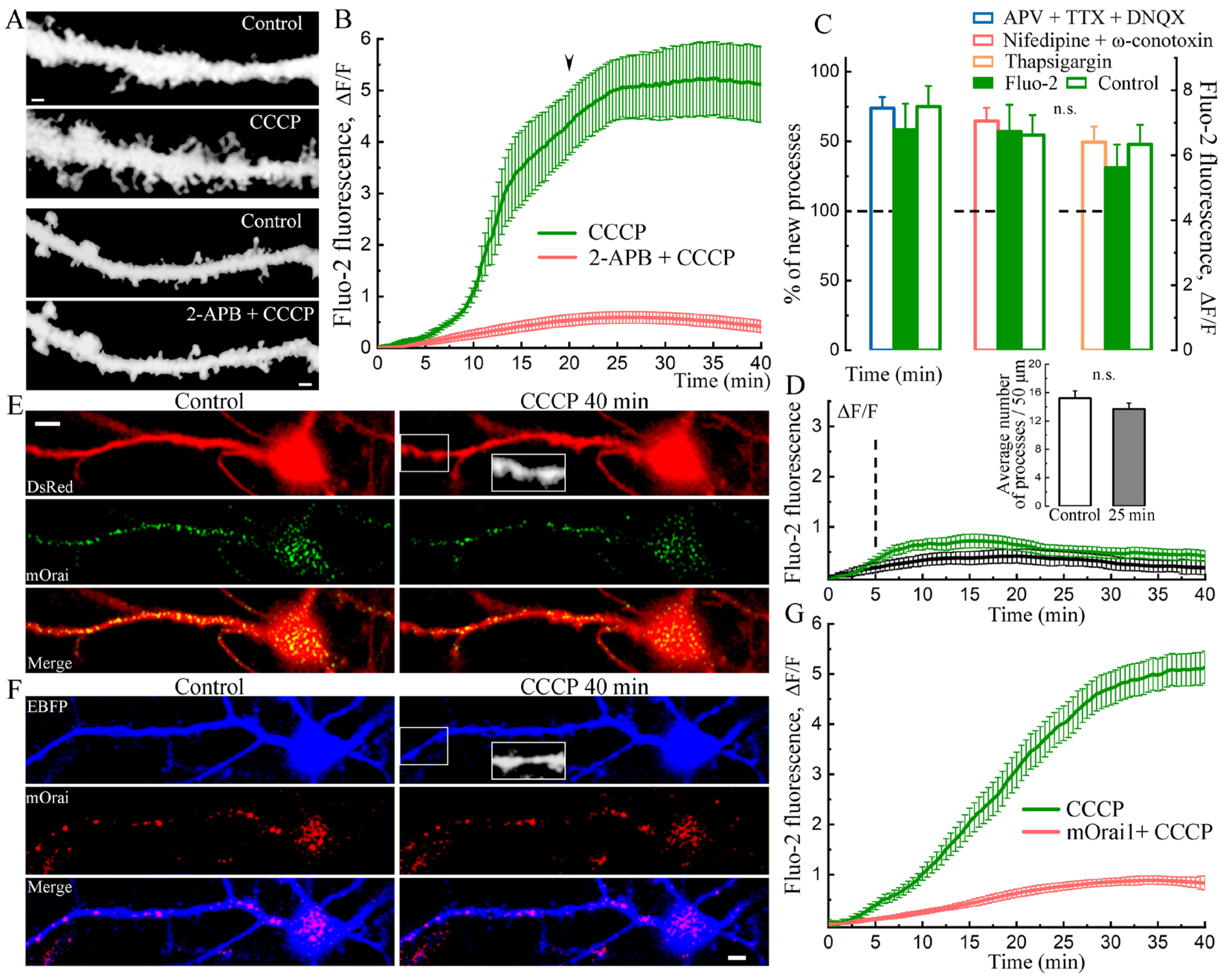
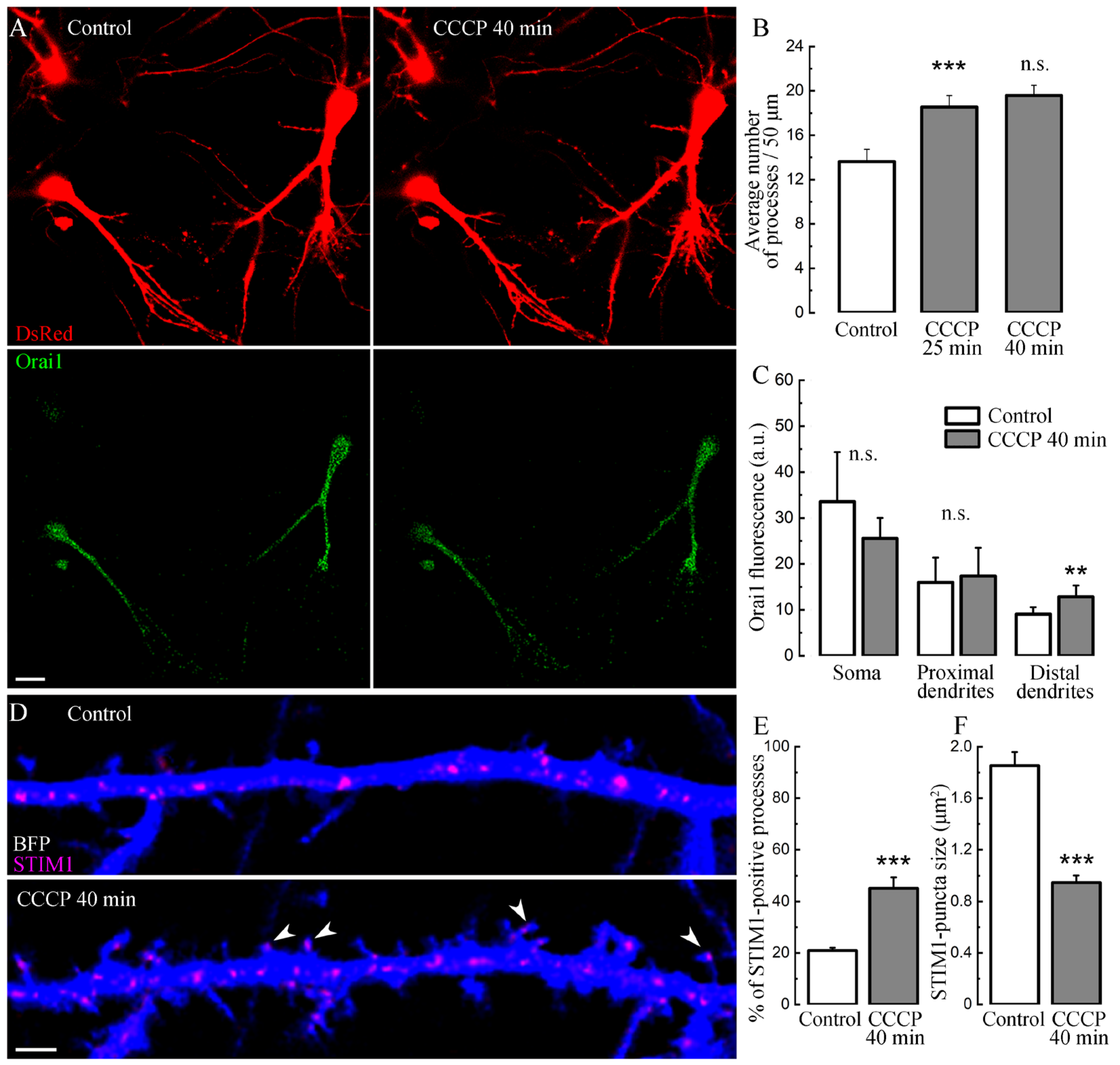
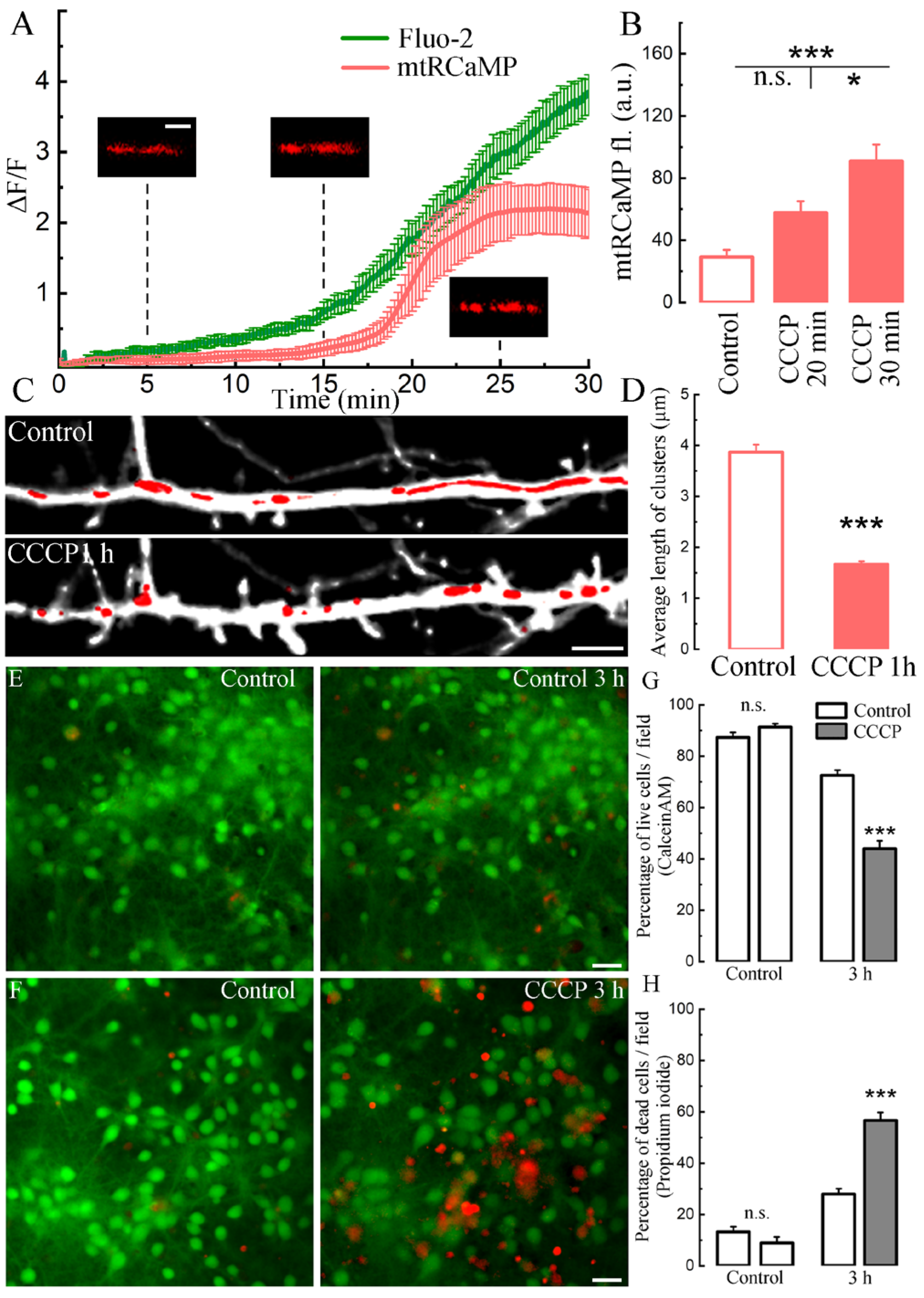
2. Results
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kushnireva, L.; Korkotian, E.; Segal, M. Calcium Sensors STIM1 and STIM2 Regulate Different Calcium Functions in Cultured Hippocampal Neurons. Front. Synaptic Neurosci. 2021, 12, 573714. [Google Scholar] [CrossRef] [PubMed]
- Bogeski, I.; Kilch, T.; Niemeyer, B.A. ROS and SOCE: Recent advances and controversies in the regulation of STIM and Orai. J. Physiol. 2012, 590, 4193–4200. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.-H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Skibinska-Kijek, A.; Wisniewska, M.B.; Gruszczynska-Biegala, J.; Methner, A.; Kuznicki, J. Immunolocalization of STIM1 in the mouse brain. Acta Neurobiol. Exp. 2009, 69, 413–428. [Google Scholar]
- Klejman, M.E.; Gruszczynska-Biegala, J.; Skibinska-Kijek, A.; Wisniewska, M.B.; Misztal, K.; Blazejczyk, M.; Bojarski, L.; Kuznicki, J. Expression of STIM1 in brain and puncta-like co-localization of STIM1 and ORAI1 upon depletion of Ca2+ store in neurons. Neurochem. Int. 2009, 54, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, C.B.; Gasperini, R.J.; Small, D.H.; Foa, L. STIM1 is necessary for store-operated calcium entry in turning growth cones. J. Neurochem. 2012, 122, 1155–1166. [Google Scholar] [CrossRef]
- Pavez, M.; Thompson, A.C.; Arnott, H.J.; Mitchell, C.B.; D’Atri, I.; Don, E.K.; Chilton, J.K.; Scott, E.K.; Lin, J.Y.; Young, K.M.; et al. STIM1 Is Required for Remodeling of the Endoplasmic Reticulum and Microtubule Cytoskeleton in Steering Growth Cones. J. Neurosci. 2019, 39, 5095–5114. [Google Scholar] [CrossRef]
- Park, C.Y.; Shcheglovitov, A.; Dolmetsch, R. The CRAC Channel Activator STIM1 Binds and Inhibits L-Type Voltage-Gated Calcium Channels. Science 2010, 330, 101–105. [Google Scholar] [CrossRef]
- Steinbeck, J.A.; Henke, N.; Opatz, J.; Gruszczynska-Biegala, J.; Schneider, L.; Theiss, S.; Hamacher, N.; Steinfarz, B.; Golz, S.; Brüstle, O.; et al. Store-operated calcium entry modulates neuronal network activity in a model of chronic epilepsy. Exp. Neurol. 2011, 232, 185–194. [Google Scholar] [CrossRef]
- Zhang, M.; Song, J.-N.; Wu, Y.; Zhao, Y.-L.; Pang, H.-G.; Fu, Z.-F.; Zhang, B.-F.; Ma, X.-D. Suppression of STIM1 in the early stage after global ischemia attenuates the injury of delayed neuronal death by inhibiting store-operated calcium entry-induced apoptosis in rats. Neuroreport 2014, 25, 507–513. [Google Scholar] [CrossRef]
- Henke, N.; Albrecht, P.; Bouchachia, I.; Ryazantseva, M.; Knoll, K.; Lewerenz, J.; Kaznacheyeva, E.; Maher, P.; Methner, A. The plasma membrane channel ORAI1 mediates detrimental calcium influx caused by endogenous oxidative stress. Cell Death Dis. 2013, 24, e470. [Google Scholar] [CrossRef] [PubMed]
- Keil, J.M.; Shen, Z.; Briggs, S.P.; Patrick, G.N. Regulation of STIM1 and SOCE by the Ubiquitin-Proteasome System (UPS). PLoS ONE 2010, 5, e13465. [Google Scholar] [CrossRef] [PubMed]
- Korkotian, E.; Oni-Biton, E.; Segal, M. The role of the store-operated calcium entry channel Orai1 in cultured rat hippocampal synapse formation and plasticity. J. Physiol. 2017, 595, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Kushnireva, L.; Basnayake, K.; Holcman, D.; Segal, M.; Korkotian, E. Dynamic Regulation of Mitochondrial [Ca2+] in Hippocampal Neurons. Int. J. Mol. Sci. 2022, 23, 12321. [Google Scholar] [CrossRef] [PubMed]
- Poncer, J.-C.; McKinney, R.A.; Gähwiler, B.H.; Thompson, S.M. Either N- or P-type Calcium Channels Mediate GABA Release at Distinct Hippocampal Inhibitory Synapses. Neuron 1997, 18, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Peckys, D.B.; Gaa, D.; Alansary, D.; Niemeyer, B.A.; de Jonge, N. Supra-Molecular Assemblies of ORAI1 at Rest Precede Local Accumulation into Puncta after Activation. Int. J. Mol. Sci. 2021, 22, 799. [Google Scholar] [CrossRef] [PubMed]
- Korkotian, E.; Segal, M. Roles of Calcium Stores and Store-Operated Channels in Plasticity of Dendritic Spines. Neuroscientist 2016, 22, 477–485. [Google Scholar] [CrossRef]
- Slepian, M.J.; Massia, S.P.; Whitesell, L. Pre-Conditioning of Smooth Muscle Cells via Induction of the Heat Shock Response Limits Proliferation Following Mechanical Injury. Biochem. Biophys. Res. Commun. 1996, 225, 600–607. [Google Scholar] [CrossRef]
- Sadeh, N.; Oni-Biton, E.; Segal, M. Acute Live/Dead Assay for the Analysis of Toxic Effects of Drugs on Cultured Neurons. Bio-Protocol 2016, 6, e1889. [Google Scholar] [CrossRef]
- Shishkin, V.; Potapenko, E.; Kostyuk, E.; Girnyk, O.; Voitenko, N.; Kostyuk, P. Role of mitochondria in intracellular calcium signaling in primary and secondary sensory neurones of rats. Cell Calcium 2002, 32, 121–130. [Google Scholar] [CrossRef]
- De Oliveira, R.B.; Gravina, F.S.; Lim, R.; Brichta, A.M.; Callister, R.J.; van Helden, D.F. Heterogeneous Responses to Antioxidants in Noradrenergic Neurons of the Locus Coeruleus Indicate Differing Susceptibility to Free Radical Content. Oxidative Med. Cell. Longev. 2012, 2012, 820285. [Google Scholar] [CrossRef] [PubMed]
- Komori, Y.; Tanaka, M.; Kuba, M.; Ishii, M.; Abe, M.; Kitamura, N.; Verkhratsky, A.; Shibuya, I.; Dayanithi, G. Ca2+ homeostasis; Ca2+ signalling and somatodendritic vasopressin release in adult rat supraoptic nucleus neurones. Cell Calcium 2010, 48, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Babcock, D.F.; Herrington, J.; Goodwin, P.C.; Park, Y.B.; Hille, B.; Zöllner, O.; Lenter, M.C.; Blanks, J.E.; Borges, E.; Steegmaier, M.; et al. Mitochondrial Participation in the Intracellular Ca2+ Network. J. Cell Biol. 1997, 136, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.P.; Spiegel, C.; Keren, Y.; Danieli, T.; Melamed-Book, N.; Pal, R.R.; Zlotkin-Rivkin, E.; Rosenshine, I.; Aroeti, B. Mitochondrial Targeting of the Enteropathogenic Escherichia coli Map Triggers Calcium Mobilization, ADAM10-MAP Kinase Signaling, and Host Cell Apoptosis. mBio 2020, 11, e01397-20. [Google Scholar] [CrossRef] [PubMed]
- Herrington, J.; Park, Y.B.; Babcock, D.F.; Hille, B. Dominant Role of Mitochondria in Clearance of Large Ca2+ Loads from Rat Adrenal Chromaffin Cells. Neuron 1996, 16, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.B.; Tisi, R.; Fietto, L.G.; Cardoso, A.S.; França, M.M.; Carvalho, F.M.; Trópia, M.J.M.; Martegani, E.; Castro, I.M.; Brandão, R.L. Carbonyl cyanide m-chlorophenylhydrazone induced calcium signaling and activation of plasma membrane H+-ATPase in the yeast Saccharomyces cerevisiae. FEMS Yeast Res. 2008, 8, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Plumbly, W.; Brandon, N.; Deeb, T.Z.; Hall, J.; Harwood, A.J. L-type voltage-gated calcium channel regulation of in vitro human cortical neuronal networks. Sci. Rep. 2019, 9, 13810. [Google Scholar] [CrossRef]
- Hannon, H.E.; Atchison, W.D. Omega-Conotoxins as Experimental Tools and Therapeutics in Pain Management. Mar. Drugs 2013, 11, 680–699. [Google Scholar] [CrossRef]
- Putney, J.W. Pharmacology of Store-operated Calcium Channels. Mol. Interv. 2010, 10, 209–218. [Google Scholar] [CrossRef]
- Abe, K.; Chisaka, O.; Van Roy, F.; Takeichi, M. Stability of dendritic spines and synaptic contacts is controlled by αN-catenin. Nat. Neurosci. 2004, 7, 357–363. [Google Scholar] [CrossRef]
- Brustovetsky, T.; Li, V.; Brustovetsky, N. Stimulation of glutamate receptors in cultured hippocampal neurons causes Ca2+- dependent mitochondrial contraction. Cell Calcium 2009, 46, 18–29. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, A.O.; van den Heuvel, L.P.; Dijkman, H.B.; De Abreu, R.A.; Birkenkamp, K.U.; de Witte, T.; van der Reijden, B.A.; Smeitink, J.A.; Jansen, J.H. Bcl-2 prevents loss of mitochondria in CCCP-induced apoptosis. Exp. Cell Res. 2004, 299, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.S.; Paris, A.; Codron, P.; Cassereau, J.; Procaccio, V.; Lenaers, G.; Reynier, P.; Chevrollier, A. Current mechanistic insights into the CCCP-induced cell survival response. Biochem. Pharmacol. 2018, 148, 100–110. [Google Scholar] [CrossRef]
- Henke, N.; Albrecht, P.; Pfeiffer, A.; Toutzaris, D.; Zanger, K.; Methner, A. Stromal Interaction Molecule 1 (STIM1) Is Involved in the Regulation of Mitochondrial Shape and Bioenergetics and Plays a Role in Oxidative Stress. J. Biol. Chem. 2012, 287, 42042–42052. [Google Scholar] [CrossRef] [PubMed]
- Vlachos, A.; Korkotian, E.; Schonfeld, E.; Copanaki, E.; Deller, T.; Segal, M. Synaptopodin Regulates Plasticity of Dendritic Spines in Hippocampal Neurons. J. Neurosci. 2009, 29, 1017–1033. [Google Scholar] [CrossRef] [PubMed]
- Korkotian, E.; Meshcheriakova, A.; Segal, M. Presenilin 1 Regulates [Ca2+]i and Mitochondria/ER Interaction in Cultured Rat Hippocampal Neurons. Oxidative Med. Cell. Longev. 2019, 2019, 7284967. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; Yeromin, A.V.; Hu, J.; Amcheslavsky, A.; Zheng, H.; Cahalan, M.D. Mutations in Orai1 transmembrane segment 1 cause STIM1-independent activation of Orai1 channels at glycine 98 and channel closure at arginine 91. Proc. Natl. Acad. Sci. USA 2011, 108, 17838–17843. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kushnireva, L.; Korkotian, E.; Segal, M. Exposure of Cultured Hippocampal Neurons to the Mitochondrial Uncoupler Carbonyl Cyanide Chlorophenylhydrazone Induces a Rapid Growth of Dendritic Processes. Int. J. Mol. Sci. 2023, 24, 12940. https://doi.org/10.3390/ijms241612940
Kushnireva L, Korkotian E, Segal M. Exposure of Cultured Hippocampal Neurons to the Mitochondrial Uncoupler Carbonyl Cyanide Chlorophenylhydrazone Induces a Rapid Growth of Dendritic Processes. International Journal of Molecular Sciences. 2023; 24(16):12940. https://doi.org/10.3390/ijms241612940
Chicago/Turabian StyleKushnireva, Liliia, Eduard Korkotian, and Menahem Segal. 2023. "Exposure of Cultured Hippocampal Neurons to the Mitochondrial Uncoupler Carbonyl Cyanide Chlorophenylhydrazone Induces a Rapid Growth of Dendritic Processes" International Journal of Molecular Sciences 24, no. 16: 12940. https://doi.org/10.3390/ijms241612940
APA StyleKushnireva, L., Korkotian, E., & Segal, M. (2023). Exposure of Cultured Hippocampal Neurons to the Mitochondrial Uncoupler Carbonyl Cyanide Chlorophenylhydrazone Induces a Rapid Growth of Dendritic Processes. International Journal of Molecular Sciences, 24(16), 12940. https://doi.org/10.3390/ijms241612940