
Gut Microbiota Alterations and Their Functional Differences in Depression According to Enterotypes in Asian Individuals



Abstract
:1. Introduction
2. Results
2.1. Collection of Fecal Bacteria and Enterotypes of the Participants
2.2. Gut Microbiota Composition of Participants with and without Depression in ET-B
2.3. Gut Microbiota Composition of Patients with Depression in ET-L
2.4. Gut Microbiota Composition of Patients with Depression in ET-P
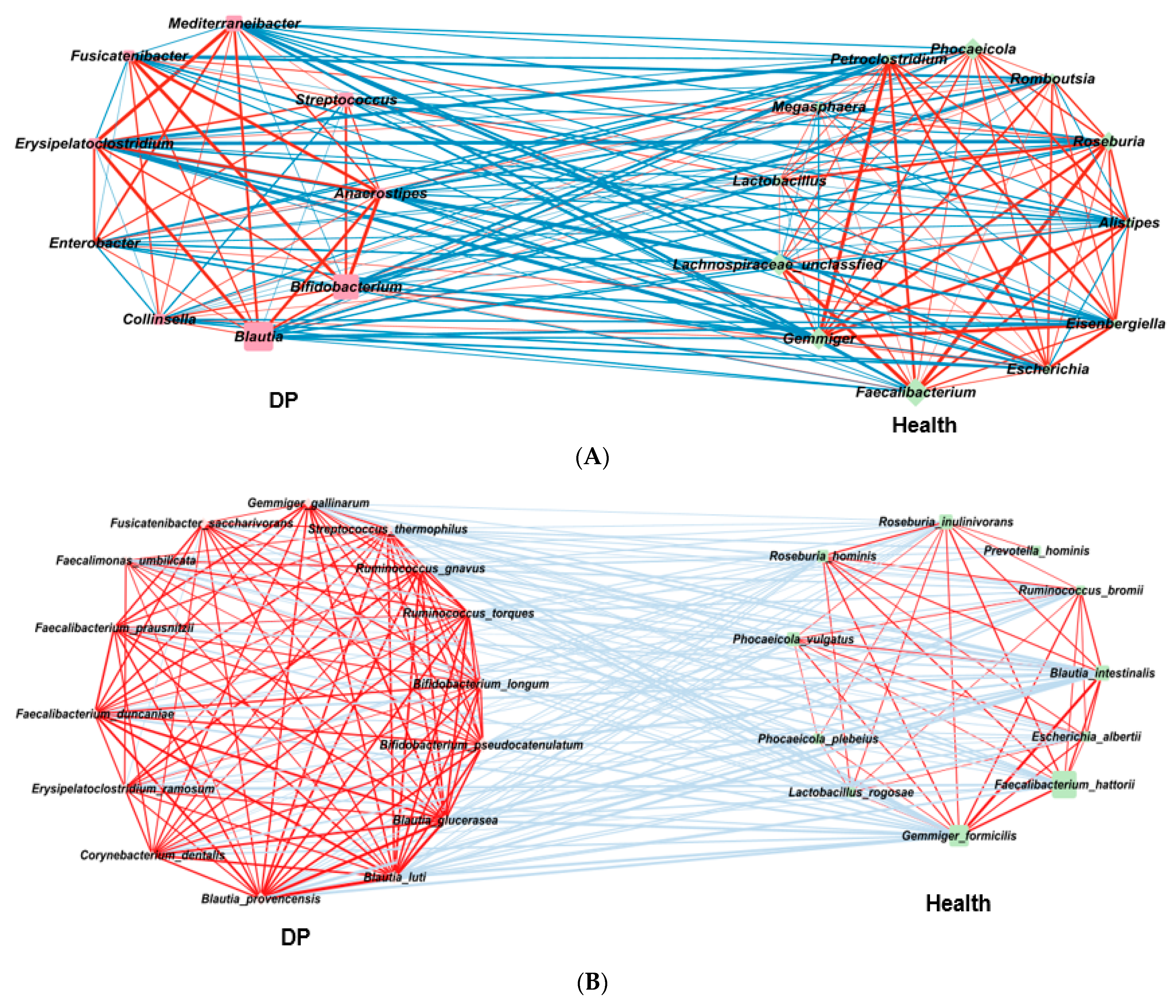
2.5. Gut Microbiota Interaction Network in ET-B
2.6. Gut Microbiota Interaction Network in ET-L
2.7. Metagenome Function in ET-B
2.8. Metagenome Function in ET-L
3. Discussion
4. Methods and Materials
4.1. Collection of FASTA/Q Files of Fecal Bacteria from Depressed and Healthy Adults
4.2. Gut Microbiota Composition and Community Analysis
4.3. Enterotype Classification
4.4. Diversity and LDA Scores of Gut Microbiota
4.5. XGBoost Classifier Training and SHapley Additive exPlanations (SHAP) Interpreter
4.6. Network and Metagenome Function of Gut Microbiota
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Santomauro, D.F.; Mantilla Herrera, A.M.; Shadid, J.; Zheng, P.; Ashbaugh, C.; Pigott, D.M.; Abbafati, C.; Adolph, C.; Amlag, J.O.; Aravkin, A.Y.; et al. Global prevalence and burden of depressive and anxiety disorders in 204 countries and territories in 2020 due to the COVID-19 pandemic. Lancet 2021, 398, 1700–1712. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Dong, W.; Peng, Q.; Bai, Z. Trends in depression incidence in China, 1990–2019. J. Affect Disord. 2022, 296, 291–297. [Google Scholar] [CrossRef]
- Zakaria, F.H.; Samhani, I.; Mustafa, M.Z.; Shafin, N. Pathophysiology of Depression: Stingless Bee Honey Promising as an Antidepressant. Molecules 2022, 27, 5091. [Google Scholar] [CrossRef]
- Łojko, D.; Rybakowski, J.K. Atypical depression: Current perspectives. Neuropsychiatr. Dis. Treat 2017, 13, 2447–2456. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Hu, Z.; Xu, J.; Wang, C. The molecular pathophysiology of depression and the new therapeutics. Med. Comm. 2022, 3, e156. [Google Scholar] [CrossRef] [PubMed]
- Wachsmuth, H.R.; Weninger, S.N.; Duca, F.A. Role of the gut–brain axis in energy and glucose metabolism. Exp. Mol. Med. 2022, 54, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Radjabzadeh, D.; Bosch, J.A.; Uitterlinden, A.G.; Zwinderman, A.H.; Ikram, M.A.; van Meurs, J.B.J.; Luik, A.I.; Nieuwdorp, M.; Lok, A.; van Duijn, C.M.; et al. Gut microbiome-wide association study of depressive symptoms. Nature Comm. 2022, 13, 7128. [Google Scholar] [CrossRef] [PubMed]
- Radford-Smith, D.E.; Anthony, D.C. Prebiotic and Probiotic Modulation of the Microbiota-Gut-Brain Axis in Depression. Nutrients 2023, 15, 1880. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Unno, T.; Kang, S.; Park, S. A Korean-Style Balanced Diet Has a Potential Connection with Ruminococcaceae Enterotype and Reduction of Metabolic Syndrome Incidence in Korean Adults. Nutrients 2021, 13, 495. [Google Scholar] [CrossRef]
- Park, S.; Wu, X. Modulation of the Gut Microbiota in Memory Impairment and Alzheimer’s Disease via the Inhibition of the Parasympathetic Nervous System. Int. J. Mol. Sci. 2022, 23, 13574. [Google Scholar] [CrossRef]
- Costea, P.I.; Hildebrand, F.; Arumugam, M.; Bäckhed, F.; Blaser, M.J.; Bushman, F.D.; de Vos, W.M.; Ehrlich, S.D.; Fraser, C.M.; Hattori, M.; et al. Enterotypes in the landscape of gut microbial community composition. Nat. Microbiol. 2018, 3, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Song, E.J.; Han, K.; Lim, T.J.; Lim, S.; Chung, M.J.; Nam, M.H.; Kim, H.; Nam, Y.D. Effect of probiotics on obesity-related markers per enterotype: A double-blind, placebo-controlled, randomized clinical trial. EPMA J. 2020, 11, 31–51. [Google Scholar] [CrossRef]
- Wu, X.; Park, S. Fecal Bacterial Community and Metagenome Function in Asians with Type 2 Diabetes, According to Enterotypes. Biomedicines 2022, 10, 2998. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, Y.; Wu, J.; Zhang, H.; Huang, Y.; Tan, X.; Wen, L.; Zhou, X.; Xie, P.; Olasunkanmi, O.I.; et al. Changes of gut microbiota reflect the severity of major depressive disorder: A cross-sectional study. Transl. Psychiatry 2023, 13, 137. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Huh, J.R.; Shah, K. Microbiota and the gut-brain-axis: Implications for new therapeutic design in the CNS. EBioMedicine 2022, 77, 103908. [Google Scholar] [CrossRef] [PubMed]
- Irum, N.; Afzal, T.; Faraz, M.H.; Aslam, Z.; Rasheed, F. The role of gut microbiota in depression: An analysis of the gut-brain axis. Front Behav. Neurosci. 2023, 17, 1185522. [Google Scholar] [CrossRef] [PubMed]
- Suganya, K.; Koo, B.-S. Gut–Brain Axis: Role of Gut Microbiota on Neurological Disorders and How Probiotics/Prebiotics Beneficially Modulate Microbial and Immune Pathways to Improve Brain Functions. Int. J. Mol. Sci. 2020, 21, 7551. [Google Scholar] [CrossRef]
- Chang, L.; Wei, Y.; Hashimoto, K. Brain–gut–microbiota axis in depression: A historical overview and future directions. Brain Res. Bull 2022, 182, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.A.; Álvarez-Mon, M.A.; García-Montero, C.; Fraile-Martínez, Ó.; Monserrat, J.; Martinez-Rozas, L.; Rodríguez-Jiménez, R.; Álvarez-Mon, M.; Lahera, G. Microbiota–gut–brain axis mechanisms in the complex network of bipolar disorders: Potential clinical implications and translational opportunities. Mol. Psychiatry 2023. [Google Scholar] [CrossRef]
- Lowe, V.M.; Chaplin, M.; Sgambato, D. Major depressive disorder and the gut microbiome: What is the link? Gen. Psychiatr. 2023, 36, e100973. [Google Scholar] [CrossRef]
- Cheung, S.G.; Goldenthal, A.R.; Uhlemann, A.-C.; Mann, J.J.; Miller, J.M.; Sublette, M.E. Systematic Review of Gut Microbiota and Major Depression. Front. Psychiatry 2019, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Pramanik, J.; Goyal, N.; Chauhan, D.; Sivamaruthi, B.S.; Prajapati, B.G.; Chaiyasut, C. Gut Microbiota in Anxiety and Depression: Unveiling the Relationships and Management Options. Pharmaceuticals 2023, 16, 565. [Google Scholar] [CrossRef] [PubMed]
- Gebrayel, P.; Nicco, C.; Al Khodor, S.; Bilinski, J.; Caselli, E.; Comelli, E.M.; Egert, M.; Giaroni, C.; Karpinski, T.M.; Loniewski, I.; et al. Microbiota medicine: Towards clinical revolution. J. Transl. Med. 2022, 20, 111. [Google Scholar] [CrossRef]
- Lee, S.-H.; Yoon, S.-H.; Jung, Y.; Kim, N.; Min, U.; Chun, J.; Choi, I. Emotional well-being and gut microbiome profiles by enterotype. Sci. Rep. 2020, 10, 20736. [Google Scholar] [CrossRef] [PubMed]
- Barberio, B.; Zamani, M.; Black, C.J.; Savarino, E.V.; Ford, A.C. Prevalence of symptoms of anxiety and depression in patients with inflammatory bowel disease: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2021, 6, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Pan, C.; Cai, Q.; Zhao, Y.; He, D.; Wei, W.; Zhang, N.; Shi, S.; Chu, X.; Zhang, F. Assessing the effect of interaction between gut microbiome and inflammatory bowel disease on the risks of depression. Brain Behav. Immun. Health 2022, 26, 100557. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Chen, Y.; Chen, Y.; Wang, C. Depression and Anxiety Disorders in Patients With Inflammatory Bowel Disease. Front. Psychiatry 2021, 12, 714057. [Google Scholar] [CrossRef]
- He, H.; Sun, Y.; Zhang, S.; Zhang, H.; Su, W.; Guo, Z.; Zhang, Y.; Wen, J.; Li, X.; Hu, J.; et al. Arabinogalactan, Bifidobacterium longum, and Faecalibacterium prausnitzii improve insulin resistance in high-fat diet-induced C57BL/6J mice. eFood 2022, 3, e1. [Google Scholar] [CrossRef]
- Guo, Y.; Xie, J.P.; Deng, K.; Li, X.; Yuan, Y.; Xuan, Q.; Xie, J.; He, X.M.; Wang, Q.; Li, J.J.; et al. Prophylactic Effects of Bifidobacterium adolescentis on Anxiety and Depression-Like Phenotypes After Chronic Stress: A Role of the Gut Microbiota-Inflammation Axis. Front. Behav. Neurosci. 2019, 13, 126. [Google Scholar] [CrossRef] [PubMed]
- Dugas, L.R.; Bernabé, B.P.; Priyadarshini, M.; Fei, N.; Park, S.J.; Brown, L.; Plange-Rhule, J.; Nelson, D.; Toh, E.C.; Gao, X.; et al. Decreased microbial co-occurrence network stability and SCFA receptor level correlates with obesity in African-origin women. Sci. Rep. 2018, 8, 17135. [Google Scholar] [CrossRef] [PubMed]
- Ohlsson, L.; Gustafsson, A.; Lavant, E.; Suneson, K.; Brundin, L.; Westrin, Å.; Ljunggren, L.; Lindqvist, D. Leaky gut biomarkers in depression and suicidal behavior. Acta Psychiatr. Scand. 2019, 139, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Chakaroun, R.M.; Massier, L.; Kovacs, P. Gut Microbiome, Intestinal Permeability, and Tissue Bacteria in Metabolic Disease: Perpetrators or Bystanders? Nutrients 2020, 12, 1082. [Google Scholar] [CrossRef] [PubMed]
- Appleton, J. The Gut-Brain Axis: Influence of Microbiota on Mood and Mental Health. Integr. Med. 2018, 17, 28–32. [Google Scholar]
- Qiu, L.; Yang, Q.; Zhao, W.; Xing, Y.; Li, P.; Zhou, X.; Ning, H.; Shi, R.; Gou, S.; Chen, Y.; et al. Dysfunction of the energy sensor NFE2L1 triggers uncontrollable AMPK signaling and glucose metabolism reprogramming. Cell Death Dis. 2022, 13, 501. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, J.; Chen, Y. Regulation of Neurotransmitters by the Gut Microbiota and Effects on Cognition in Neurological Disorders. Nutrients 2021, 13, 2099. [Google Scholar] [CrossRef]
- Liu, M.; Nieuwdorp, M.; de Vos, W.M.; Rampanelli, E. Microbial Tryptophan Metabolism Tunes Host Immunity, Metabolism, and Extraintestinal Disorders. Metabolites 2022, 12, 834. [Google Scholar] [CrossRef] [PubMed]
- Koning, E.; Vorstman, J.; McIntyre, R.S.; Brietzke, E. Characterizing eating behavioral phenotypes in mood disorders: A narrative review. Psychol. Med. 2022, 52, 2885–2898. [Google Scholar] [CrossRef] [PubMed]
- Xiong, R.-G.; Li, J.; Cheng, J.; Zhou, D.-D.; Wu, S.-X.; Huang, S.-Y.; Saimaiti, A.; Yang, Z.-J.; Gan, R.-Y.; Li, H.-B. The Role of Gut Microbiota in Anxiety, Depression, and Other Mental Disorders as Well as the Protective Effects of Dietary Components. Nutrients 2023, 15, 3258. [Google Scholar]
- Fernandes, B.S.; Salagre, E.; Enduru, N.; Grande, I.; Vieta, E.; Zhao, Z. Insulin resistance in depression: A large meta-analysis of metabolic parameters and variation. Neurosci. Biobehav. Rev. 2022, 139, 104758. [Google Scholar] [CrossRef]
- Jurewicz, I. Mental health in young adults and adolescents-supporting general physicians to provide holistic care. Clin. Med. 2015, 15, 151–154. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
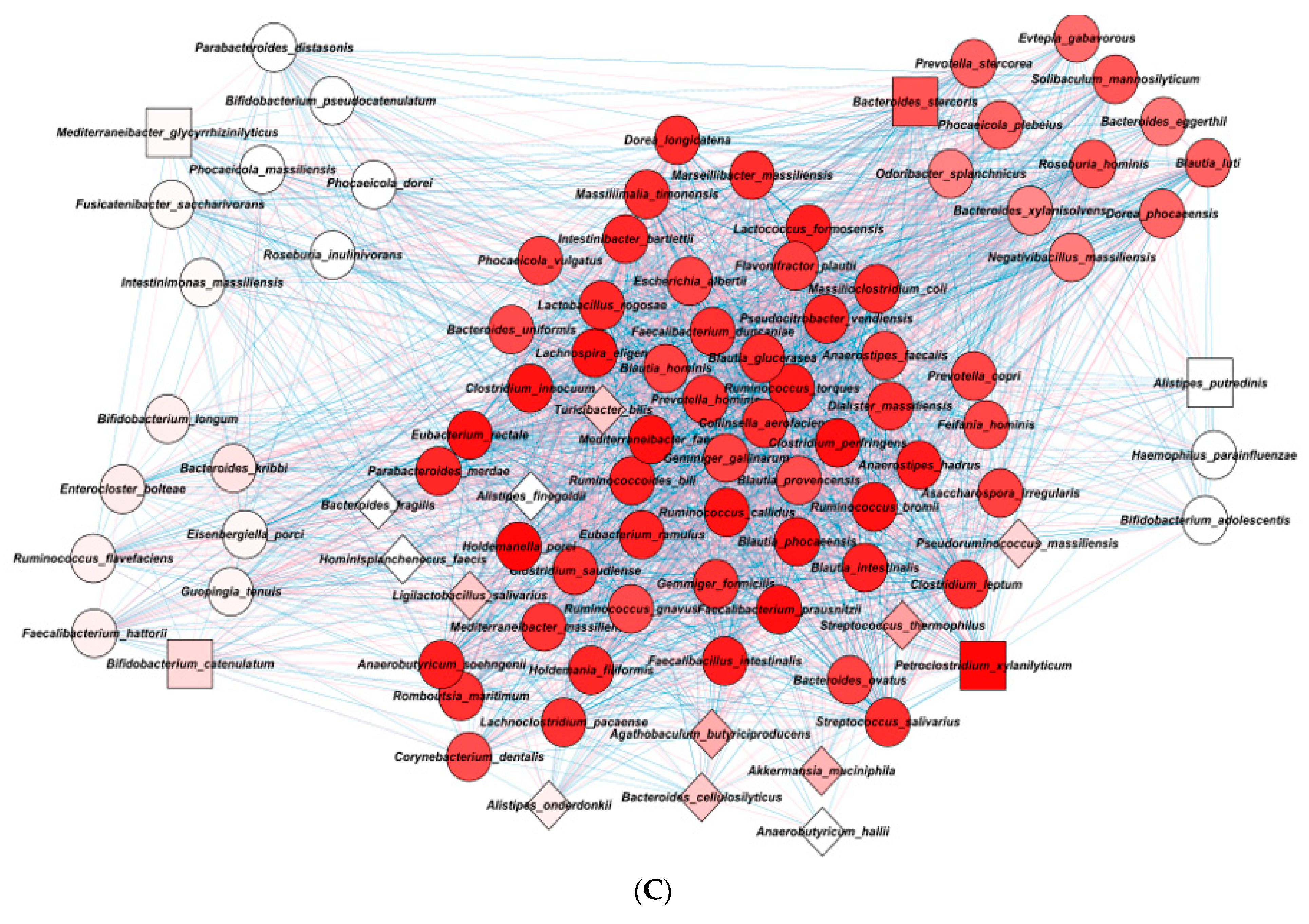
| Mean | Group | MCODE | Degree Layout | p Value | ||
|---|---|---|---|---|---|---|
| Cluster 1 | ||||||
| Corynebacterium dentalis | 0.457 | DP | 25.22 | 46 | 8.36 × 10−5 | |
| Collinsella aerofaciens | 1.302 | DP | 27.69 | 73 | 3.57 × 10−7 | |
| Blautia provencensis | 2.847 | DP | 25.28 | 58 | 1.41 × 10−11 | |
| Gemmiger gallinarum | 0.952 | DP | 26.18 | 45 | 1.77 × 10−5 | |
| Faecalibacterium prausnitzii | 0.636 | DP | 30.12 | 70 | 1.82 × 10−5 | |
| Faecalibacterium duncaniae | 1.403 | DP | 27.51 | 60 | 1.39 × 10−7 | |
| Anaerostipes hadrus | 0.981 | Health | 29.17 | 48 | 2.68 × 10−8 | |
| Blautia glucerasea | 1.283 | DP | 27.81 | 59 | 1.78 × 10−10 | |
| Ruminococcus gnavus | 1.867 | DP | 25.56 | 54 | 7.43 × 10−7 | |
| Petroclostridium xylanilyticum | 0.112 | Health | 30.94 | 60 | 0.00112 | Seed |
| Cluster 2 | ||||||
| Evtepia gabavorous | 0.184 | DP | 22.72 | 41 | 4.29 × 10−5 | |
| Blautia luti | 2.829 | DP | 23.44 | 54 | 2.21 × 10−10 | |
| Solibaculum mannosilyticum | 0.225 | DP | 24.62 | 51 | 9 × 10−5 | |
| Bacteroides stercoris | 0.963 | DP | 24.42 | 53 | 0.503 | Seed |
| Cluster 3 | ||||||
| Enterocloster bolteae | 0.396 | DP | 11.71 | 33 | 3.02 × 10−9 | |
| Bifidobacterium catenulatum | 1.072 | Health | 13.38 | 28 | 0.475 | Seed |
| Cluster 4 | ||||||
| Fusicatenibacter saccharivorans | 1.54 | DP | 10.45 | 33 | 3.01 × 10−7 | |
| Bifidobacterium pseudocatenulatum | 3.8 | DP | 10 | 19 | 2.35 × 10−5 | |
| Mediterraneibacter glycyrrhizinilyticus | 0.209 | DP | 10.5 | 32 | 0.058 | Seed |
| ET-L | ET-B | |||
|---|---|---|---|---|
| Health | Depression | Health | Depression | |
| No. of node | 196 | 285 | 166 | 163 |
| No. of edge | 3599 | 8454 | 2000 | 1273 |
| Average degree | 36.7 | 55.1 | 24.1 | 15.6 |
| Average path length | 0.777 | 1.13 | 1.08 | 1.16 |
| Graphic density | 0.188 | 0.209 | 0.146 | 0.0964 |
| Clustering coefficient | 0.306 | 0.355 | 0.252 | 0.183 |
| Negative edge ratio | 0.452 | 0.463 | 0.456 | 0.352 |
| Mean | Group | MCODE | Degree Layout | p Value | ||
|---|---|---|---|---|---|---|
| Cluster 1 | ||||||
| Butyricicoccus_faecihominis | 0.14407 | DP | 11.93939 | 0.004319 | ||
| Eggerthella_lenta | 0.380625 | DP | 12 | 0.007643 | ||
| Clostridium_colinum | 0.538333 | Health | 12.90196 | 0.017457 | ||
| Alistipes_putredinis | 0.800764 | Health | 16 | 0.39137 | seed | |
| Cluster 2 | ||||||
| Bacteroides_stercoris | 0.962871 | DP | 10.59692 | 0.030355 | seed | |
| Cluster 3 | ||||||
| Bacteroides_fragilis | 0.466068 | DP | 6.019048 | 0.012808 | ||
| Haemophilus_parainfluenzae | 0.290275 | Health | 5.666667 | 0.230277 | Seed | |
| Cluster 4 | ||||||
| Escherichia_albertii | 1.071582 | Health | 3.333333 | 0.028736 | ||
| Akkermansia_muciniphila | 0.264366 | DP | 3.928571 | 0.889247 | seed | |
| Cluster 5 | ||||||
| Ligilactobacillus_salivarius | 0.393043 | Health | 8.423529 | 0.087931 | seed | |
| Collinsella_aerofaciens | 1.302479 | DP | 7.73975 | 0.078893 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.; Li, C.; Wu, X.; Zhang, T. Gut Microbiota Alterations and Their Functional Differences in Depression According to Enterotypes in Asian Individuals. Int. J. Mol. Sci. 2023, 24, 13329. https://doi.org/10.3390/ijms241713329
Park S, Li C, Wu X, Zhang T. Gut Microbiota Alterations and Their Functional Differences in Depression According to Enterotypes in Asian Individuals. International Journal of Molecular Sciences. 2023; 24(17):13329. https://doi.org/10.3390/ijms241713329
Chicago/Turabian StylePark, Sunmin, Chen Li, Xuangao Wu, and Tianshun Zhang. 2023. "Gut Microbiota Alterations and Their Functional Differences in Depression According to Enterotypes in Asian Individuals" International Journal of Molecular Sciences 24, no. 17: 13329. https://doi.org/10.3390/ijms241713329
APA StylePark, S., Li, C., Wu, X., & Zhang, T. (2023). Gut Microbiota Alterations and Their Functional Differences in Depression According to Enterotypes in Asian Individuals. International Journal of Molecular Sciences, 24(17), 13329. https://doi.org/10.3390/ijms241713329