

Impact of Impaired Kidney Function on Arrhythmia-Promoting Cardiac Ion Channel Regulation


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
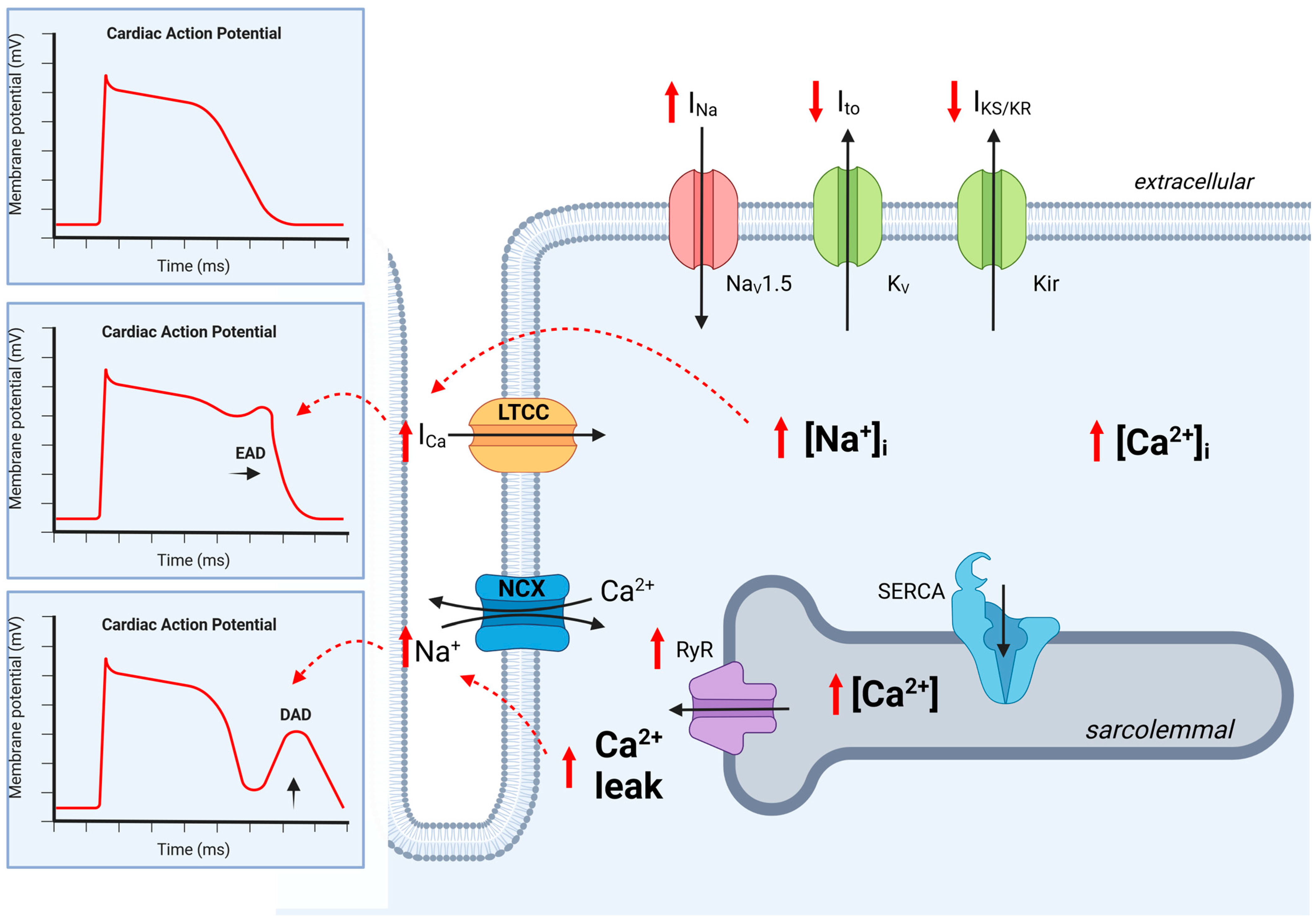
Excitation-Contraction Coupling in Healthy Cardiomyocytes
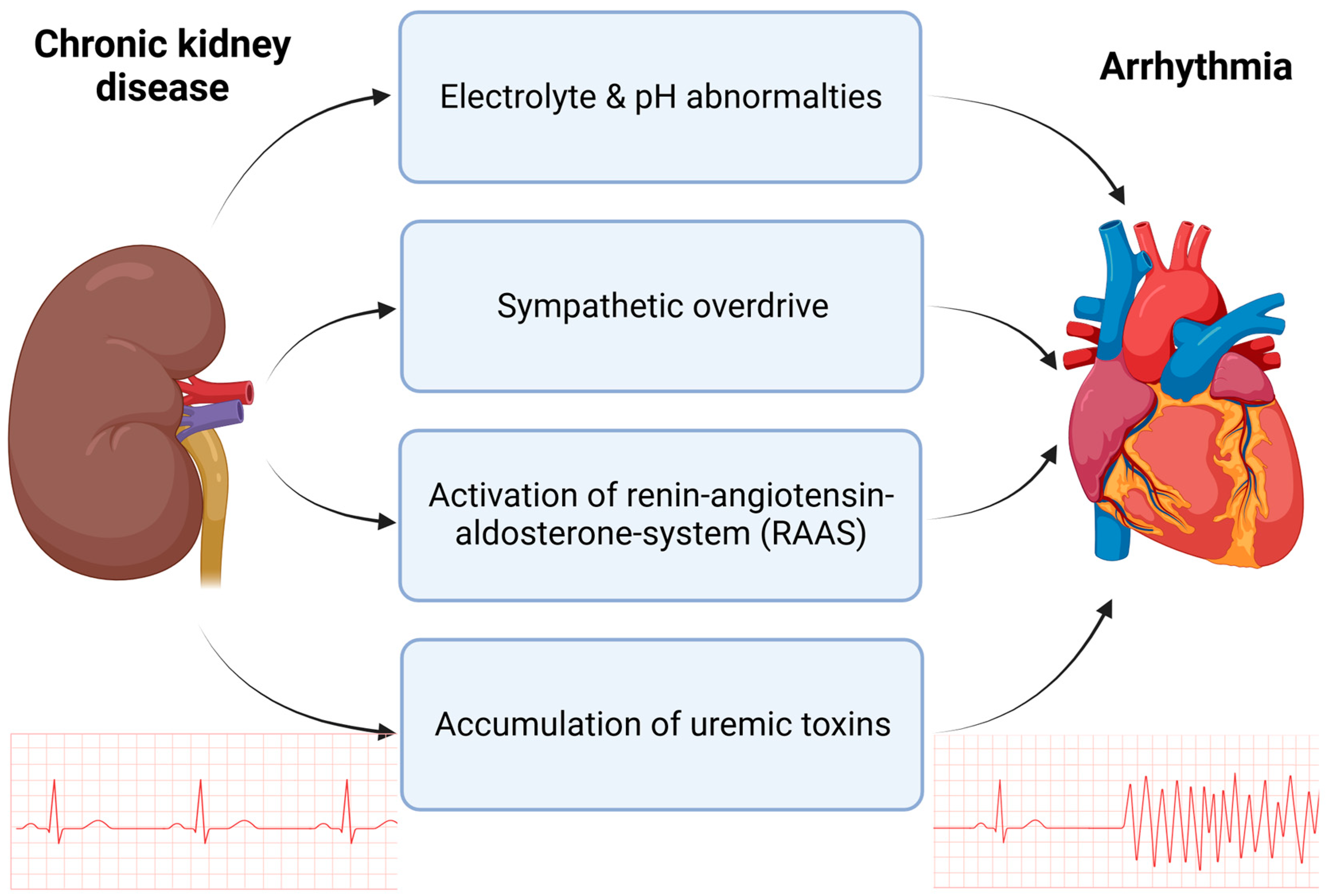
2. Translational Approaches to Arrhythmia in CKD
3. Characteristics of Chronic Kidney Disease
3.1. Electrolyte and pH Abnormalities in CKD
3.2. Sympathetic Nerve System Activation in CKD
3.3. The Renin–Angiotensin–Aldosterone System in CKD
3.4. Uremic Toxins
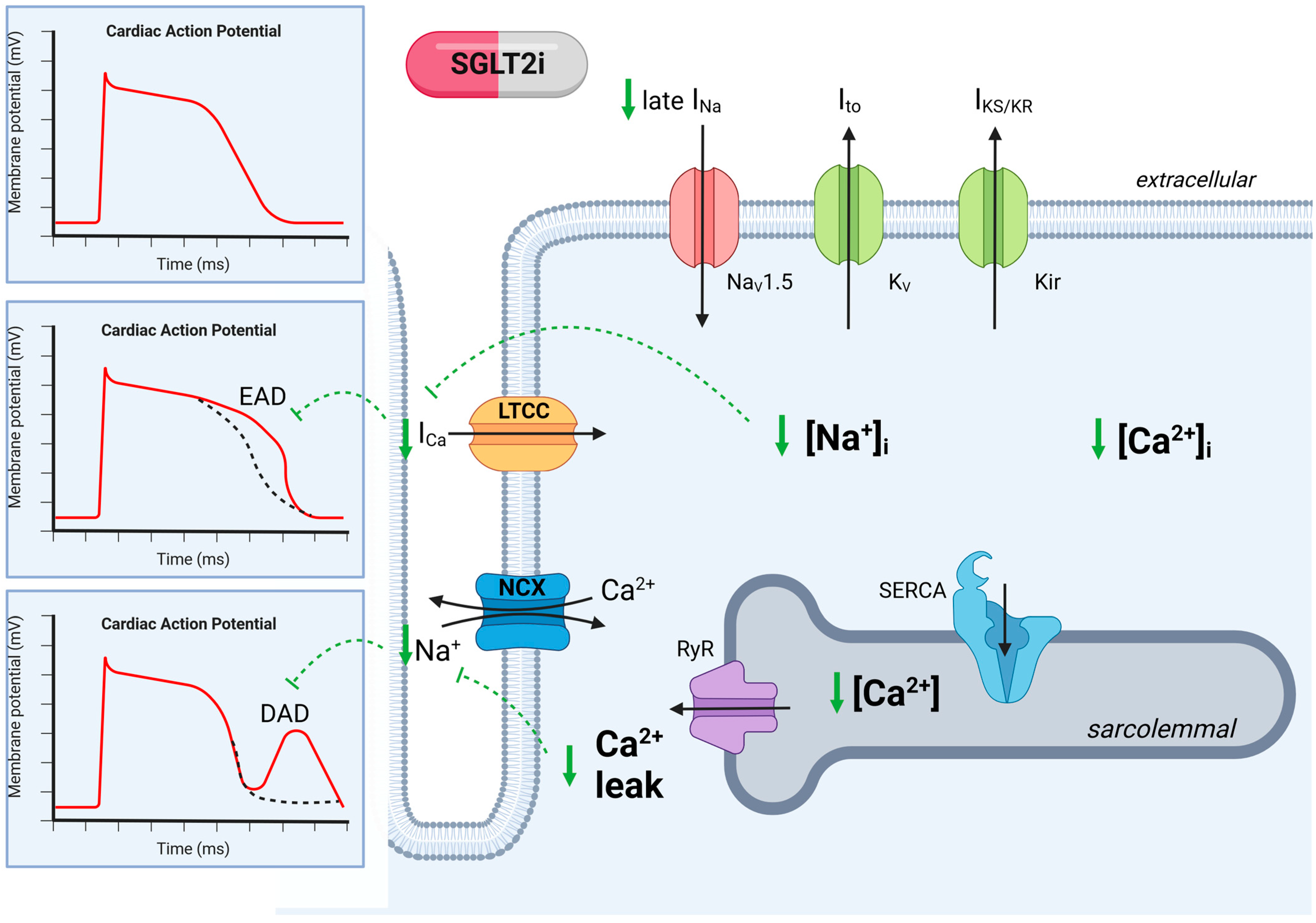
3.5. SGLT2- Inhibitors: Novel Treatment Strategies for Arrhythmia in CKD?
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Levey, A.S.; Eckardt, K.-U.; Tsukamoto, Y.; Levin, A.; Coresh, J.; Rossert, J.; Zeeuw, D.; de Hostetter, T.H.; Lameire, N.; Eknoyan, G. Definition and classification of chronic kidney disease: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2005, 67, 2089–2100. [Google Scholar] [CrossRef] [PubMed]
- Franczyk-Skóra, B.; Gluba-Brzózka, A.; Wranicz, J.K.; Banach, M.; Olszewski, R.; Rysz, J. Sudden cardiac death in CKD patients. Int. Urol. Nephrol. 2015, 47, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.J.; Foley, R.N.; Chavers, B.; Gilbertson, D.; Herzog, C.; Johansen, K.; Kasiske, B.; Kutner, N.; Liu, J.; St Peter, W.; et al. United States Renal Data System 2011 Annual Data Report: Atlas of chronic kidney disease & end-stage renal disease in the United States. Am. J. Kidney Dis. 2012, 59, e1–e420. [Google Scholar]
- Genovesi, S.; Boriani, G.; Covic, A.; Vernooij, R.W.M.; Combe, C.; Burlacu, A.; Davenport, A.; Kanbay, M.; Kirmizis, D.; Schneditz, D.; et al. Sudden cardiac death in dialysis patients: Different causes and management strategies. Nephrol. Dial. Transplant. 2021, 36, 396–405. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Wagner, S.; Maier, L.S.; Bers, D.M. Role of sodium and calcium dysregulation in tachyarrhythmias in sudden cardiac death. Circ. Res. 2015, 116, 1956–1970. [Google Scholar] [CrossRef]
- Andelova, K.; Egan Benova, T.; Szeiffova Bacova, B.; Sykora, M.; Prado, N.J.; Diez, E.R.; Hlivak, P.; Tribulova, N. Cardiac Connexin-43 Hemichannels and Pannexin1 Channels: Provocative Antiarrhythmic Targets. Int. J. Mol. Sci. 2020, 22, 260. [Google Scholar] [CrossRef]
- Danik, S.B.; Liu, F.; Zhang, J.; Suk, H.J.; Morley, G.E.; Fishman, G.I.; Gutstein, D.E. Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ. Res. 2004, 95, 1035–1041. [Google Scholar] [CrossRef]
- Kim, E.D.; Soliman, E.Z.; Coresh, J.; Matsushita, K.; Chen, L.Y. Two-Week Burden of Arrhythmias across CKD Severity in a Large Community-Based Cohort: The ARIC Study. J. Am. Soc. Nephrol. 2021, 32, 629–638. [Google Scholar] [CrossRef]
- Rautavaara, J.; Kerola, T.; Kaartinen, K.; Vilpakka, M.; Aitkoski, A.; Anttonen, O.; Ahvonen, J.; Koistinen, J.; Vääräniemi, K.; Miettinen, M.; et al. Asystole episodes and bradycardia in patients with end-stage renal disease. Nephrol. Dial. Transplant. 2022, 37, 575–583. [Google Scholar] [CrossRef]
- van Ham, W.B.; Cornelissen, C.M.; van Veen, T.A.B. Uremic toxins in chronic kidney disease highlight a fundamental gap in understanding their detrimental effects on cardiac electrophysiology and arrhythmogenesis. Acta Physiol. 2022, 236, e13888. [Google Scholar] [CrossRef]
- Touchberry, C.D.; Green, T.M.; Tchikrizov, V.; Mannix, J.E.; Mao, T.F.; Carney, B.W.; Girgis, M.; Vincent, R.J.; Wetmore, L.A.; Dawn, B.; et al. FGF23 is a novel regulator of intracellular calcium and cardiac contractility in addition to cardiac hypertrophy. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E863–E873. [Google Scholar] [CrossRef]
- Donohoe, P.; McMahon, A.C.; Walgama, O.V.; Bertaso, F.; Dockrell, M.E.; Cramp, H.A.; Mullen, A.M.; Shattock, M.J.; Hendry, B.M.; James, A.F. L-type calcium current of isolated rat cardiac myocytes in experimental uraemia. Nephrol. Dial. Transplant. 2000, 15, 791–798. [Google Scholar] [CrossRef]
- McMahon, A.C.; Vescovo, G.; Dalla Libera, L.; Wynne, D.G.; Fluck, R.J.; Harding, S.E.; Raine, A.E. Contractile dysfunction of isolated ventricular myocytes in experimental uraemia. Exp. Nephrol. 1996, 4, 144–150. [Google Scholar]
- McMahon, A.C.; Greenwald, S.E.; Dodd, S.M.; Hurst, M.J.; Raine, A.E.G. Prolonged calcium transients and myocardial remodelling in early experimental uraemia. Nephrol. Dial. Transplant. 2002, 17, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.; Omran, E.; Periyasamy, S.M.; Nadoor, J.; Priyadarshi, A.; Willey, J.C.; Malhotra, D.; Xie, Z.; Shapiro, J.I. Effect of chronic renal failure on cardiac contractile function, calcium cycling, and gene expression of proteins important for calcium homeostasis in the rat. J. Am. Soc. Nephrol. 2003, 14, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Einhorn, L.M.; Zhan, M.; van Hsu, D.; Walker, L.D.; Moen, M.F.; Seliger, S.L.; Weir, M.R.; Fink, J.C. The frequency of hyperkalemia and its significance in chronic kidney disease. Arch. Intern. Med. 2009, 169, 1156–1162. [Google Scholar] [CrossRef]
- Weiss, J.N.; Qu, Z.; Shivkumar, K. Electrophysiology of Hypokalemia and Hyperkalemia. Circ. Arrhythm. Electrophysiol. 2017, 10, e004667. [Google Scholar] [CrossRef]
- Parham, W.A.; Mehdirad, A.A.; Biermann, K.M.; Fredman, C.S. Hyperkalemia Revisited. Tex. Heart Inst. J. 2006, 33, 40–47. [Google Scholar] [PubMed]
- Pastore, J.M.; Girouard, S.D.; Laurita, K.R.; Akar, F.G.; Rosenbaum, D.S. Mechanism linking T-wave alternans to the genesis of cardiac fibrillation. Circulation 1999, 99, 1385–1394. [Google Scholar] [CrossRef]
- Hegyi, B.; Chen-Izu, Y.; Izu, L.T.; Bányász, T. Altered K+ current profiles underlie cardiac action potential shortening in hyperkalemia and β-adrenergic stimulation. Can. J. Physiol. Pharmacol. 2019, 97, 773–780. [Google Scholar] [CrossRef]
- Han, B.; Trew, M.L.; Zgierski-Johnston, C.M. Cardiac Conduction Velocity, Remodeling and Arrhythmogenesis. Cells 2021, 10, 2923. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.K.; Peters, C.H.; Allard, C.R.; Claydon, T.W.; Ruben, P.C. Proton sensors in the pore domain of the cardiac voltage-gated sodium channel. J. Biol. Chem. 2013, 288, 4782–4791. [Google Scholar] [CrossRef] [PubMed]
- Murphy, L.; Renodin, D.; Antzelevitch, C.; Di Diego, J.M.; Cordeiro, J.M. Extracellular proton depression of peak and late Na+ current in the canine left ventricle. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H936–H944. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.K.; Peters, C.H.; Tolhurst, S.A.; Claydon, T.W.; Ruben, P.C. Extracellular proton modulation of the cardiac voltage-gated sodium channel, Nav1.5. Biophys. J. 2011, 101, 2147–2156. [Google Scholar] [CrossRef] [PubMed]
- Fabiato, A.; Fabiato, F. Effects of pH on the myofilaments and the sarcoplasmic reticulum of skinned cells from cardiace and skeletal muscles. J. Physiol. 1978, 276, 233–255. [Google Scholar] [CrossRef]
- Orchard, C.H.; Kentish, J.C. Effects of changes of pH on the contractile function of cardiac muscle. Am. J. Physiol. 1990, 258, C967–C981. [Google Scholar] [CrossRef]
- Choi, H.S.; Trafford, A.W.; Orchard, C.H.; Eisner, D.A. The effect of acidosis on systolic Ca2+ and sarcoplasmic reticulum calcium content in isolated rat ventricular myocytes. J. Physiol. 2000, 529 Pt 3, 661–668. [Google Scholar] [CrossRef]
- Li, L.; Watanabe, Y.; Matsuoka, I.; Kimura, J. Acidic preconditioning inhibits Na+/H+ and Na+/Ca2+ exchanger interaction via PKCepsilon in guinea-pig ventricular myocytes. J. Pharmacol. Sci. 2008, 107, 309–316. [Google Scholar] [CrossRef]
- Kreitmeier, K.G.; Tarnowski, D.; Nanadikar, M.S.; Baier, M.J.; Wagner, S.; Katschinski, D.M.; Maier, L.S.; Sag, C.M. CaMKIIδ Met281/282 oxidation is not required for recovery of calcium transients during acidosis. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H1199–H1212. [Google Scholar] [CrossRef]
- Bögeholz, N.; Pauls, P.; Bauer, B.K.; Schulte, J.S.; Dechering, D.G.; Frommeyer, G.; Kirchhefer, U.; Goldhaber, J.I.; Müller, F.U.; Eckardt, L.; et al. Suppression of Early and Late Afterdepolarizations by Heterozygous Knockout of the Na+/Ca2+ Exchanger in a Murine Model. Circ. Arrhythm. Electrophysiol. 2015, 8, 1210–1218. [Google Scholar] [CrossRef] [PubMed]
- Ek-Vitorín, J.F.; Calero, G.; Morley, G.E.; Coombs, W.; Taffet, S.M.; Delmar, M. PH regulation of connexin43: Molecular analysis of the gating particle. Biophys. J. 1996, 71, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Sinovas, A.; Sánchez, J.A.; Valls-Lacalle, L.; Consegal, M.; Ferreira-González, I. Connexins in the Heart: Regulation, Function and Involvement in Cardiac Disease. Int. J. Mol. Sci. 2021, 22, 4413. [Google Scholar] [CrossRef]
- Salameh, A.; Dhein, S. Pharmacology of gap junctions. New pharmacological targets for treatment of arrhythmia, seizure and cancer? Biochim. Biophys. Acta 2005, 1719, 36–58. [Google Scholar] [CrossRef] [PubMed]
- Ewen, S.; Ukena, C.; Linz, D.; Schmieder, R.E.; Böhm, M.; Mahfoud, F. The sympathetic nervous system in chronic kidney disease. Curr. Hypertens. Rep. 2013, 15, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Cohn, J.N. Sympathetic nervous system activity and the heart. Am. J. Hypertens. 1989, 2, 353S–356S. [Google Scholar]
- Myers, R.W.; Pearlman, A.S.; Hyman, R.M.; Goldstein, R.A.; Kent, K.M.; Goldstein, R.E.; Epstein, S.E. Beneficial effects of vagal stimulation and bradycardia during experimental acute myocardial ischemia. Circulation 1974, 49, 943–947. [Google Scholar] [CrossRef]
- Koomans, H.A.; Blankestijn, P.J.; Joles, J.A. Sympathetic hyperactivity in chronic renal failure: A wake-up call. J. Am. Soc. Nephrol. 2004, 15, 524–537. [Google Scholar] [CrossRef]
- Converse, R.L.; Jacobsen, T.N.; Toto, R.D.; Jost, C.M.; Cosentino, F.; Fouad-Tarazi, F.; Victor, R.G. Sympathetic overactivity in patients with chronic renal failure. N. Engl. J. Med. 1992, 327, 1912–1918. [Google Scholar] [CrossRef]
- Baylis, C. Nitric oxide deficiency in chronic kidney disease. Am. J. Physiol. Renal Physiol. 2008, 294, F1–F9. [Google Scholar] [CrossRef]
- Grassi, G.; Seravalle, G.; Ghiadoni, L.; Tripepi, G.; Bruno, R.M.; Mancia, G.; Zoccali, C. Sympathetic nerve traffic and asymmetric dimethylarginine in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 2620–2627. [Google Scholar] [CrossRef] [PubMed]
- Mahfoud, F.; Moon, L.B.; Pipenhagen, C.A.; Jensen, J.A.; Pathak, A.; Papademetriou, V.; Ewen, S.; Linz, D.; Böhm, M. Catheter-based radio-frequency renal nerve denervation lowers blood pressure in obese hypertensive swine model. J. Hypertens. 2016, 34, 1854–1862. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-H.; Lo, L.-W.; Chou, Y.-H.; Lin, W.-L.; Tsai, T.-Y.; Cheng, W.-H.; Yamada, S.; Chen, S.-A. Renal denervation prevents myocardial structural remodeling and arrhythmogenicity in a chronic kidney disease rabbit model. Heart Rhythm. 2021, 18, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Shi, L.; Cui, X.; Yu, Y.; Qi, T.; Chen, C.; Tang, X. Renal denervation decreases susceptibility of the heart to ventricular fibrillation in a canine model of chronic kidney disease. Exp. Physiol. 2017, 102, 1414–1423. [Google Scholar] [CrossRef] [PubMed]
- Hohl, M.; Selejan, S.-R.; Wintrich, J.; Lehnert, U.; Speer, T.; Schneider, C.; Mauz, M.; Markwirth, P.; Wong, D.W.L.; Boor, P.; et al. Renal Denervation Prevents Atrial Arrhythmogenic Substrate Development in CKD. Circ. Res. 2022, 130, 814–828. [Google Scholar] [CrossRef] [PubMed]
- Zoccali, C.; Mallamaci, F.; Parlongo, S.; Cutrupi, S.; Benedetto, F.A.; Tripepi, G.; Bonanno, G.; Rapisarda, F.; Fatuzzo, P.; Seminara, G.; et al. Plasma norepinephrine predicts survival and incident cardiovascular events in patients with end-stage renal disease. Circulation 2002, 105, 1354–1359. [Google Scholar] [CrossRef]
- Sperelakis, N. Regulation of calcium slow channels of heart by cyclic nucleotides and effects of ischemia. Adv. Pharmacol. 1994, 31, 1–24. [Google Scholar]
- Chu, G.; Lester, J.W.; Young, K.B.; Luo, W.; Zhai, J.; Kranias, E.G. A single site (Ser16) phosphorylation in phospholamban is sufficient in mediating its maximal cardiac responses to beta -agonists. J. Biol. Chem. 2000, 275, 38938–38943. [Google Scholar] [CrossRef]
- Bennett, P.B.; Begenisich, T.B. Catecholamines modulate the delayed rectifying potassium current (IK) in guinea pig ventricular myocytes. Pflugers Arch. 1987, 410, 217–219. [Google Scholar] [CrossRef]
- Taggart, P.; Sutton, P.; Chalabi, Z.; Boyett, M.R.; Simon, R.; Elliott, D.; Gill, J.S. Effect of adrenergic stimulation on action potential duration restitution in humans. Circulation 2003, 107, 285–289. [Google Scholar] [CrossRef]
- Gutierrez, D.A.; Fernandez-Tenorio, M.; Ogrodnik, J.; Niggli, E. NO-dependent CaMKII activation during β-adrenergic stimulation of cardiac muscle. Cardiovasc. Res. 2013, 100, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Myles, R.C.; Wang, L.; Kang, C.; Bers, D.M.; Ripplinger, C.M. Local β-adrenergic stimulation overcomes source-sink mismatch to generate focal arrhythmia. Circ. Res. 2012, 110, 1454–1464. [Google Scholar] [CrossRef] [PubMed]
- Herring, N.; Kalla, M.; Paterson, D.J. The autonomic nervous system and cardiac arrhythmias: Current concepts and emerging therapies. Nat. Rev. Cardiol. 2019, 16, 707–726. [Google Scholar] [CrossRef] [PubMed]
- Shiferaw, Y.; Aistrup, G.L.; Wasserstrom, J.A. Intracellular Ca2+ waves, afterdepolarizations, and triggered arrhythmias. Cardiovasc. Res. 2012, 95, 265–268. [Google Scholar] [CrossRef]
- Szeiffova Bacova, B.; Viczenczova, C.; Andelova, K.; Sykora, M.; Chaudagar, K.; Barancik, M.; Adamcova, M.; Knezl, V.; Egan Benova, T.; Weismann, P.; et al. Antiarrhythmic Effects of Melatonin and Omega-3 Are Linked with Protection of Myocardial Cx43 Topology and Suppression of Fibrosis in Catecholamine Stressed Normotensive and Hypertensive Rats. Antioxidants 2020, 9, 546. [Google Scholar] [CrossRef]
- Peters, N.S. New insights into myocardial arrhythmogenesis: Distribution of gap-junctional coupling in normal, ischaemic and hypertrophied human hearts. Clin. Sci. 1996, 90, 447–452. [Google Scholar] [CrossRef]
- Mircoli, L.; Rivera, R.; Bonforte, G.; Fedele, L.; Genovesi, S.; Surian, M.; Ferrari, A.U. Influence of left ventricular mass, uremia and hypertension on vagal tachycardic reserve. J. Hypertens. 2003, 21, 1547–1553. [Google Scholar] [CrossRef]
- Belardinelli, L.; Isenberg, G. Isolated atrial myocytes: Adenosine and acetylcholine increase potassium conductance. Am. J. Physiol. 1983, 244, H734–H737. [Google Scholar] [CrossRef]
- Liang, B.; Nissen, J.D.; Laursen, M.; Wang, X.; Skibsbye, L.; Hearing, M.C.; Andersen, M.N.; Rasmussen, H.B.; Wickman, K.; Grunnet, M.; et al. G-protein-coupled inward rectifier potassium current contributes to ventricular repolarization. Cardiovasc. Res. 2014, 101, 175–184. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, Y.; Liang, B.; Liu, J.; Li, J.; Grunnet, M.; Olesen, S.-P.; Rasmussen, H.B.; Ellinor, P.T.; Gao, L.; et al. Identification of a Kir3.4 mutation in congenital long QT syndrome. Am. J. Hum. Genet. 2010, 86, 872–880. [Google Scholar] [CrossRef]
- Schweda, F.; Kurtz, A. Regulation of renin release by local and systemic factors. Rev. Physiol. Biochem. Pharmacol. 2011, 161, 1–44. [Google Scholar] [PubMed]
- Schweda, F.; Friis, U.; Wagner, C.; Skott, O.; Kurtz, A. Renin release. Physiology 2007, 22, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, P.D.; Purohit, A.; Hund, T.J.; Anderson, M.E. Calmodulin-dependent protein kinase II: Linking heart failure and arrhythmias. Circ. Res. 2012, 110, 1661–1677. [Google Scholar] [CrossRef] [PubMed]
- Palomeque, J.; Rueda, O.V.; Sapia, L.; Valverde, C.A.; Salas, M.; Petroff, M.V.; Mattiazzi, A. Angiotensin II-induced oxidative stress resets the Ca2+ dependence of Ca2+-calmodulin protein kinase II and promotes a death pathway conserved across different species. Circ. Res. 2009, 105, 1204–1212. [Google Scholar] [CrossRef]
- Wagner, S.; Dantz, C.; Flebbe, H.; Azizian, A.; Sag, C.M.; Engels, S.; Möllencamp, J.; Dybkova, N.; Islam, T.; Shah, A.M.; et al. NADPH oxidase 2 mediates angiotensin II-dependent cellular arrhythmias via PKA and CaMKII. J. Mol. Cell Cardiol. 2014, 75, 206–215. [Google Scholar] [CrossRef]
- Zhao, Z.; Fefelova, N.; Shanmugam, M.; Bishara, P.; Babu, G.J.; Xie, L.-H. Angiotensin II induces afterdepolarizations via reactive oxygen species and calmodulin kinase II signaling. J. Mol. Cell Cardiol. 2011, 50, 128–136. [Google Scholar] [CrossRef]
- Caballero, R.; Gómez, R.; Moreno, I.; Nuñez, L.; González, T.; Arias, C.; Guizy, M.; Valenzuela, C.; Tamargo, J.; Delpón, E. Interaction of angiotensin II with the angiotensin type 2 receptor inhibits the cardiac transient outward potassium current. Cardiovasc. Res. 2004, 62, 86–95. [Google Scholar] [CrossRef]
- Matsuda, H.; Kurata, Y.; Imanishi, S.; Sato, R.; Shibamoto, T. Effects of angiotensin II on sustained outward currents in rat ventricular myocytes. Pflugers Arch. 2004, 448, 54–62. [Google Scholar] [CrossRef]
- Bénitah, J.P.; Vassort, G. Aldosterone upregulates Ca(2+) current in adult rat cardiomyocytes. Circ. Res. 1999, 85, 1139–1145. [Google Scholar] [CrossRef]
- Kashihara, T.; Nakada, T.; Kojima, K.; Takeshita, T.; Yamada, M. Angiotensin II activates CaV 1.2 Ca2+ channels through β-arrestin2 and casein kinase 2 in mouse immature cardiomyocytes. J. Physiol. 2017, 595, 4207–4225. [Google Scholar] [CrossRef]
- Mello WC, de. Intracellular angiotensin II regulates the inward calcium current in cardiac myocytes. Hypertension 1998, 32, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Kaibara, M.; Mitarai, S.; Yano, K.; Kameyama, M. Involvement of Na(+)-H+ antiporter in regulation of L-type Ca2+ channel current by angiotensin II in rabbit ventricular myocytes. Circ. Res. 1994, 75, 1121–1125. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M.; Morotti, S. Ca(2+) current facilitation is CaMKII-dependent and has arrhythmogenic consequences. Front. Pharmacol. 2014, 5, 144. [Google Scholar] [CrossRef] [PubMed]
- Mattiazzi, A.; Kranias, E.G. The role of CaMKII regulation of phospholamban activity in heart disease. Front. Pharmacol. 2014, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Mustroph, J.; Neef, S.; Maier, L.S. CaMKII as a target for arrhythmia suppression. Pharmacol. Ther. 2017, 176, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Smet, R.; de Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; Deyn PP de Deppisch, R.; Descamps-Latscha, B.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef]
- Berg, A.H.; Kumar, S.; Karumanchi, S.A. Indoxyl sulfate in uremia: An old idea with updated concepts. J. Clin. Invest. 2022, 132, e155860. [Google Scholar] [CrossRef]
- Aoki, K.; Teshima, Y.; Kondo, H.; Saito, S.; Fukui, A.; Fukunaga, N.; Nawata, T.; Shimada, T.; Takahashi, N.; Shibata, H. Role of Indoxyl Sulfate as a Predisposing Factor for Atrial Fibrillation in Renal Dysfunction. J. Am. Heart Assoc. 2015, 4, e002023. [Google Scholar] [CrossRef]
- Tang, W.-H.; Wang, C.-P.; Chung, F.-M.; Huang, L.L.H.; Yu, T.-H.; Hung, W.-C.; Lu, L.-F.; Chen, P.-Y.; Luo, C.-H.; Lee, K.-T.; et al. Uremic retention solute indoxyl sulfate level is associated with prolonged QTc interval in early CKD patients. PLoS ONE 2015, 10, e0119545. [Google Scholar] [CrossRef]
- van Ham, W.B.; Cornelissen, C.M.; Polyakova, E.; van der Voorn, S.M.; Ligtermoet, M.L.; Monshouwer-Kloots, J.; Vos, M.A.; Bossu, A.; van Rooij, E.; van der Heyden, M.A.G.; et al. Pro-Arrhythmic Potential of Accumulated Uremic Toxins Is Mediated via Vulnerability of Action Potential Repolarization. Int. J. Mol. Sci. 2023, 24, 5373. [Google Scholar] [CrossRef]
- Tsai, I.-T.; Hsu, C.-C.; Hung, W.-C.; Wang, C.-P.; Yu, T.-H.; Houng, J.-Y.; Lee, K.-T.; Tang, W.-H. The Arrhythmogenic Effect of Protein-Bound Uremic Toxin p-Cresylsulfate: An In Vitro Study. Acta Cardiol. Sin. 2019, 35, 641–648. [Google Scholar] [PubMed]
- Changchien, C.-Y.; Sung, M.-H.; Chang, H.-H.; Tsai, W.-C.; Peng, Y.-S.; Chen, Y. Uremic toxin indoxyl sulfate suppresses myocardial Cx43 assembly and expression via JNK activation. Chem. Biol. Interact. 2020, 319, 108979. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-T.; Chen, Y.-C.; Hsieh, M.-H.; Huang, S.-Y.; Kao, Y.-H.; Chen, Y.-A.; Lin, Y.-K.; Chen, S.-A.; Chen, Y.-J. The uremic toxin indoxyl sulfate increases pulmonary vein and atrial arrhythmogenesis. J. Cardiovasc. Electrophysiol. 2015, 26, 203–210. [Google Scholar] [CrossRef]
- Mehta, R.; Cai, X.; Lee, J.; Scialla, J.J.; Bansal, N.; Sondheimer, J.H.; Chen, J.; Hamm, L.L.; Ricardo, A.C.; Navaneethan, S.D.; et al. Association of Fibroblast Growth Factor 23 with Atrial Fibrillation in Chronic Kidney Disease, From the Chronic Renal Insufficiency Cohort Study. JAMA Cardiol. 2016, 1, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.B.; Ning, H.; Boer IH de Kestenbaum, B.; Lima, J.A.C.; Mehta, R.; Allen, N.B.; Shah, S.J.; Lloyd-Jones, D.M. Fibroblast Growth Factor 23 and Long-Term Cardiac Function: The Multi-Ethnic Study of Atherosclerosis. Circ. Cardiovasc. Imaging 2020, 13, e011925. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.M.; Vallejo, J.A.; Hamill, C.S.; Wang, D.; Ahuja, R.; Patel, S.; Faul, C.; Wacker, M.J. Fibroblast growth factor 23 (FGF23) induces ventricular arrhythmias and prolongs QTc interval in mice in an FGF receptor 4-dependent manner. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H2283–H2294. [Google Scholar] [CrossRef]
- Navarro-García, J.A.; Rueda, A.; Romero-García, T.; Aceves-Ripoll, J.; Rodríguez-Sánchez, E.; González-Lafuente, L.; Zaragoza, C.; Fernández-Velasco, M.; Kuro, O.M.; Ruilope, L.M.; et al. Enhanced Klotho availability protects against cardiac dysfunction induced by uraemic cardiomyopathy by regulating Ca2+ handling. Br. J. Pharmacol. 2020, 177, 4701–4719. [Google Scholar] [CrossRef]
- Lindner, M.; Mehel, H.; David, A.; Leroy, C.; Burtin, M.; Friedlander, G.; Terzi, F.; Mika, D.; Fischmeister, R.; Prié, D. Fibroblast growth factor 23 decreases PDE4 expression in heart increasing the risk of cardiac arrhythmia; Klotho opposes these effects. Basic. Res. Cardiol. 2020, 115, 51. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.J.V.; Claggett, B.; Boer RA de DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; Shah, S.J.; et al. Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N. Engl. J. Med. 2022, 387, 1089–1098. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.-F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner-La Rocca, H.-P.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Aklilu, A.M.; Kumar, S.; Yamamoto, Y.; Moledina, D.G.; Sinha, F.; Testani, J.M.; Wilson, F.P. Outcomes Associated with Sodium-Glucose Cotransporter-2 Inhibitor Use in Acute Heart Failure Hospitalizations Complicated by Acute Kidney Injury. Kidney360 2023. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-L.; Lip, G.Y.H.; Feng, Q.; Fei, Y.; Tse, Y.-K.; Wu, M.-Z.; Ren, Q.-W.; Tse, H.-F.; Cheung, B.-M.Y.; Yiu, K.-H. Sodium-glucose cotransporter 2 inhibitors (SGLT2i) and cardiac arrhythmias: A systematic review and meta-analysis. Cardiovasc. Diabetol. 2021, 20, 100. [Google Scholar] [CrossRef]
- Fernandes, G.C.; Fernandes, A.; Cardoso, R.; Penalver, J.; Knijnik, L.; Mitrani, R.D.; Myerburg, R.J.; Goldberger, J.J. Association of SGLT2 inhibitors with arrhythmias and sudden cardiac death in patients with type 2 diabetes or heart failure: A meta-analysis of 34 randomized controlled trials. Heart Rhythm. 2021, 18, 1098–1105. [Google Scholar] [CrossRef]
- Sfairopoulos, D.; Zhang, N.; Wang, Y.; Chen, Z.; Letsas, K.P.; Tse, G.; Li, G.; Lip, G.Y.H.; Liu, T.; Korantzopoulos, P. Association between sodium-glucose cotransporter-2 inhibitors and risk of sudden cardiac death or ventricular arrhythmias: A meta-analysis of randomized controlled trials. Europace 2022, 24, 20–30. [Google Scholar] [CrossRef]
- Oates, C.P.; Santos-Gallego, C.G.; Smith, A.; Basyal, B.; Moss, N.; Kawamura, I.; Musikantow, D.R.; Turagam, M.K.; Miller, M.A.; Whang, W.; et al. SGLT2 inhibitors reduce sudden cardiac death risk in heart failure: Meta-analysis of randomized clinical trials. J. Cardiovasc. Electrophysiol. 2023, 34, 1277–1285. [Google Scholar] [CrossRef]
- Neuen, B.L.; Oshima, M.; Agarwal, R.; Arnott, C.; Cherney, D.Z.; Edwards, R.; Langkilde, A.M.; Mahaffey, K.W.; McGuire, D.K.; Neal, B.; et al. Sodium-Glucose Cotransporter 2 Inhibitors and Risk of Hyperkalemia in People with Type 2 Diabetes: A Meta-Analysis of Individual Participant Data From Randomized, Controlled Trials. Circulation 2022, 145, 1460–1470. [Google Scholar] [CrossRef]
- Sinha, F.; Federlein, A.; Biesold, A.; Schwarzfischer, M.; Krieger, K.; Schweda, F.; Tauber, P. Empagliflozin increases kidney weight due to increased cell size in the proximal tubule S3 segment and the collecting duct. Front. Pharmacol. 2023, 14, 1118358. [Google Scholar] [CrossRef]
- Herat, L.Y.; Magno, A.L.; Rudnicka, C.; Hricova, J.; Carnagarin, R.; Ward, N.C.; Arcambal, A.; Kiuchi, M.G.; Head, G.A.; Schlaich, M.P.; et al. SGLT2 Inhibitor-Induced Sympathoinhibition: A Novel Mechanism for Cardiorenal Protection. JACC Basic. Transl. Sci. 2020, 5, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Kravtsova, O.; Bohovyk, R.; Levchenko, V.; Palygin, O.; Klemens, C.A.; Rieg, T.; Staruschenko, A. SGLT2 inhibition effect on salt-induced hypertension, RAAS, and Na+ transport in Dahl SS rats. Am. J. Physiol. Renal Physiol. 2022, 322, F692–F707. [Google Scholar] [CrossRef] [PubMed]
- Tauber, P.; Sinha, F.; Berger, R.S.; Gronwald, W.; Dettmer, K.; Kuhn, M.; Trum, M.; Maier, L.S.; Wagner, S.; Schweda, F. Empagliflozin Reduces Renal Hyperfiltration in Response to Uninephrectomy, but Is Not Nephroprotective in UNx/DOCA/Salt Mouse Models. Front. Pharmacol. 2021, 12, 761855. [Google Scholar] [CrossRef] [PubMed]
- Lytvyn, Y.; Kimura, K.; Peter, N.; Lai, V.; Tse, J.; Cham, L.; Perkins, B.A.; Soleymanlou, N.; Cherney, D.Z.I. Renal and Vascular Effects of Combined SGLT2 and Angiotensin-Converting Enzyme Inhibition. Circulation 2022, 146, 450–462. [Google Scholar] [CrossRef]
- Xue, G.; Yang, X.; Zhan, G.; Wang, X.; Gao, J.; Zhao, Y.; Wang, X.; Li, J.; Pan, Z.; Xia, Y. Sodium-Glucose cotransporter 2 inhibitor empagliflozin decreases ventricular arrhythmia susceptibility by alleviating electrophysiological remodeling post-myocardial-infarction in mice. Front. Pharmacol. 2022, 13, 988408. [Google Scholar] [CrossRef]
- Jhuo, S.-J.; Liu, I.-H.; Tasi, W.-C.; Chou, T.-W.; Lin, Y.-H.; Wu, B.-N.; Lee, K.-T.; Lai, W.-T. Characteristics of Ventricular Electrophysiological Substrates in Metabolic Mice Treated with Empagliflozin. Int. J. Mol. Sci. 2021, 22, 6105. [Google Scholar] [CrossRef]
- Silva Dos Santos, D.; Turaça, L.T.; Da Coutinho, K.C.S.; Barbosa, R.A.Q.; Polidoro, J.Z.; Kasai-Brunswick, T.H.; Campos de Carvalho, A.C.; Girardi, A.C.C. Empagliflozin reduces arrhythmogenic effects in rat neonatal and human iPSC-derived cardiomyocytes and improves cytosolic calcium handling at least partially independent of NHE1. Sci. Rep. 2023, 13, 8689. [Google Scholar] [CrossRef]
- Maier, L.S.; Sossalla, S. The late Na current as a therapeutic target: Where are we? J. Mol. Cell Cardiol. 2013, 61, 44–50. [Google Scholar] [CrossRef]
- Mustroph, J.; Wagemann, O.; Lücht, C.M.; Trum, M.; Hammer, K.P.; Sag, C.M.; Lebek, S.; Tarnowski, D.; Reinders, J.; Perbellini, F.; et al. Empagliflozin reduces Ca/calmodulin-dependent kinase II activity in isolated ventricular cardiomyocytes. ESC Heart Fail. 2018, 5, 642–648. [Google Scholar] [CrossRef]
- Mustroph, J.; Baier, M.J.; Pabel, S.; Stehle, T.; Trum, M.; Provaznik, Z.; Mohler, P.J.; Musa, H.; Hund, T.J.; Sossalla, S.; et al. Empagliflozin Inhibits Cardiac Late Sodium Current by Ca/Calmodulin-Dependent Kinase II. Circulation 2022, 146, 1259–1261. [Google Scholar] [CrossRef]
- Mustroph, J.; Wagemann, O.; Trum, M.; Lebek, S.; Tarnowski, D.; Reinders, J.; Schmid, C.; Schopka, S.; Hilker, M.; Graf, B.; et al. 3145Empagliflozin potently reduces sarcoplasmic Ca leak and increases Ca transient amplitude of human failing ventricular cardiomyocytes. Eur. Heart J. 2018, 39, ehy563.3145. [Google Scholar] [CrossRef]
- Baris, V.; Gedikli, E.; Dincsoy, A.B.; Erdem, A. Empagliflozin significantly prevents QTc prolongation due to amitriptyline intoxication via intracellular calcium regulation. Eur. Heart J. 2021, 42, 1–5. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinha, F.; Schweda, F.; Maier, L.S.; Wagner, S. Impact of Impaired Kidney Function on Arrhythmia-Promoting Cardiac Ion Channel Regulation. Int. J. Mol. Sci. 2023, 24, 14198. https://doi.org/10.3390/ijms241814198
Sinha F, Schweda F, Maier LS, Wagner S. Impact of Impaired Kidney Function on Arrhythmia-Promoting Cardiac Ion Channel Regulation. International Journal of Molecular Sciences. 2023; 24(18):14198. https://doi.org/10.3390/ijms241814198
Chicago/Turabian StyleSinha, Frederick, Frank Schweda, Lars S. Maier, and Stefan Wagner. 2023. "Impact of Impaired Kidney Function on Arrhythmia-Promoting Cardiac Ion Channel Regulation" International Journal of Molecular Sciences 24, no. 18: 14198. https://doi.org/10.3390/ijms241814198
APA StyleSinha, F., Schweda, F., Maier, L. S., & Wagner, S. (2023). Impact of Impaired Kidney Function on Arrhythmia-Promoting Cardiac Ion Channel Regulation. International Journal of Molecular Sciences, 24(18), 14198. https://doi.org/10.3390/ijms241814198