
In Silico and In Vitro Study towards the Rational Design of 4,4′-Disarylbisthiazoles as a Selective α-Synucleinopathy Biomarker

 ,
, 
 , and
, and 
Abstract
:1. Introduction
2. Results
2.1. Chemical Yields (CYs)
2.2. Competitive Binding Assays and clogP
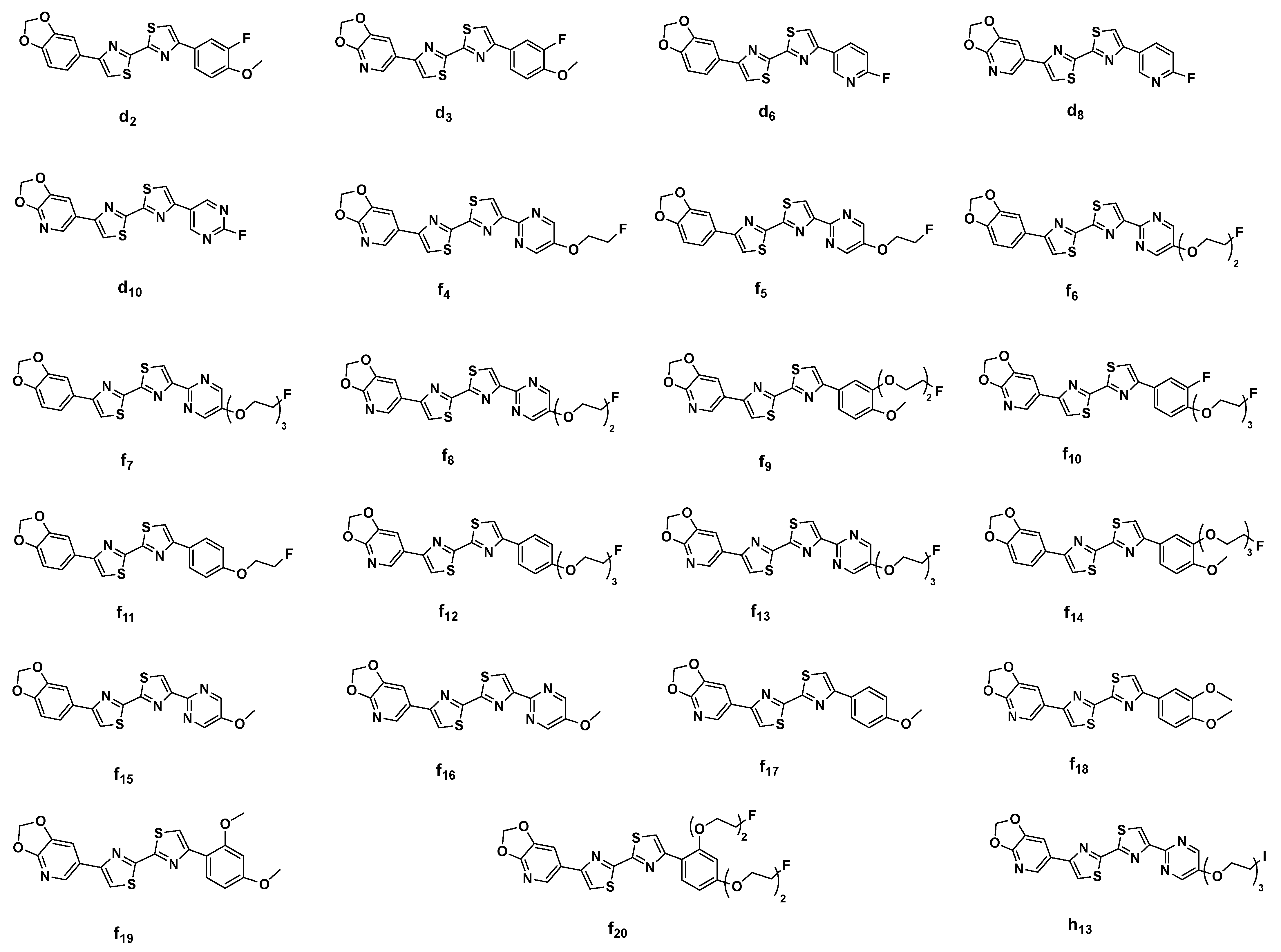
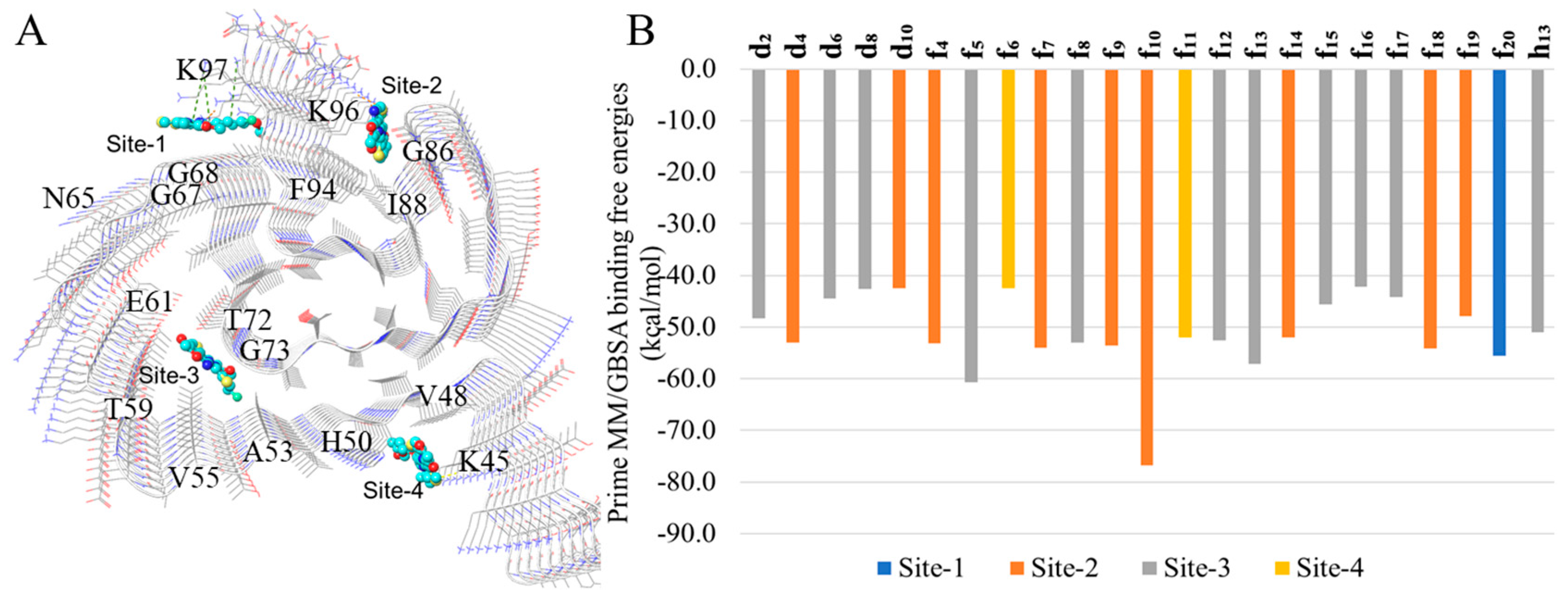
2.3. In Silico Modeling of the Binding of DABTAs to α-syn
3. Discussion
4. Materials and Methods
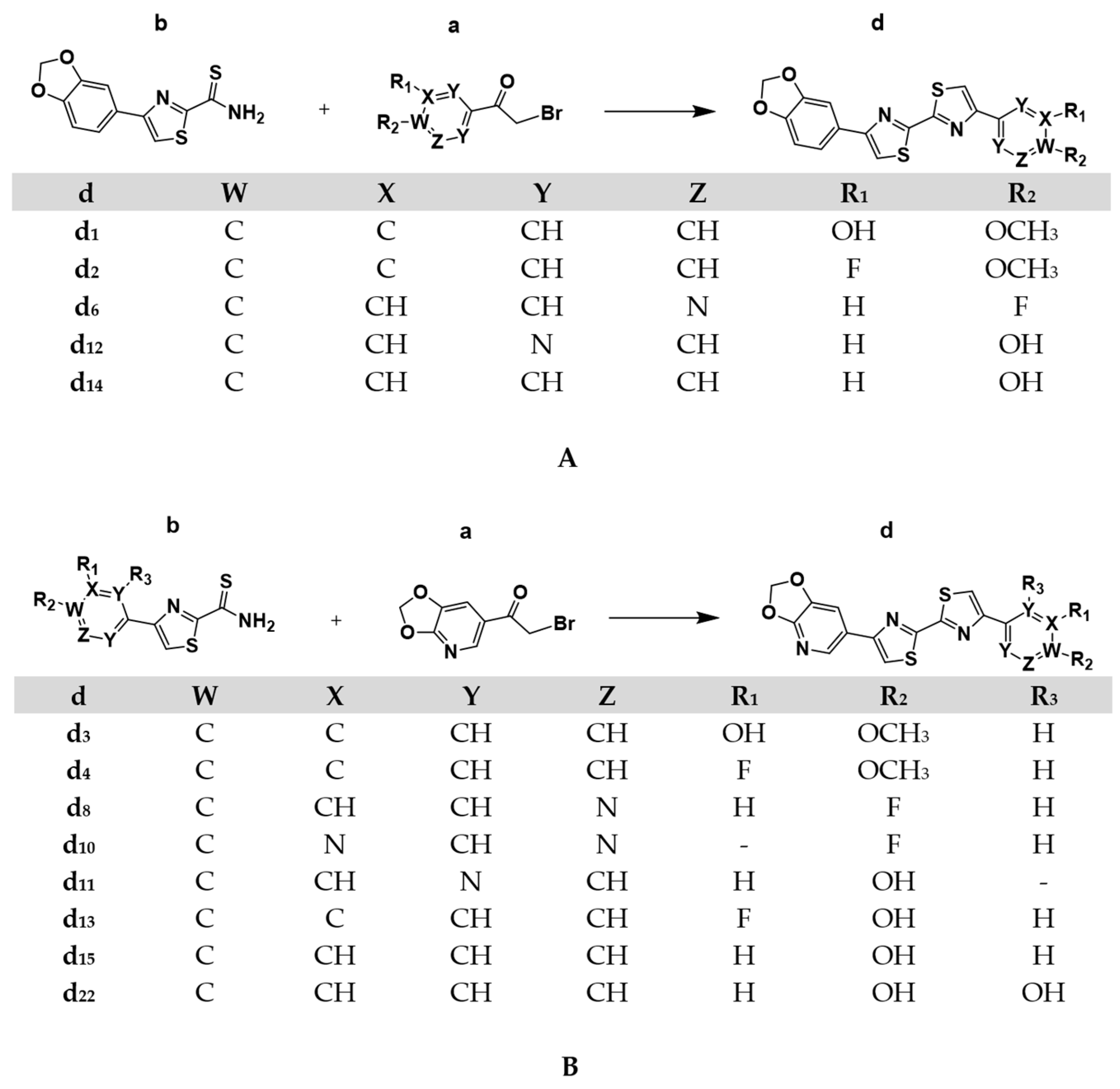
4.1. Chemical Synthesis
4.1.1. General Procedure for the Synthesis of the 4-Aryl/heteroarylthiazole-2-carbothioamide Intermediate, b
4.1.1.1. 4-(Benzo[d][1,3]dioxol-5-yl)thiazole-2-carbothioamide, b1
4.1.1.2. 4-(3-Hydroxy-4-methoxyphenyl)thiazole-2-carbothioamide, b3
4.1.1.3. 4-(3-Fluoro-4-methoxyphenyl)thiazole-2-carbothioamide, b4
4.1.1.4. 4-(6-Fluoropyridin-3-yl)thiazole-2-carbothioamide, b8
4.1.1.5. 4-(2-Fluoropyrimidin-4-yl)thiazole-2-carbothioamide, b10
4.1.1.6. 4-(5-Hydroxypyrimidin-2-yl)thiazole-2-carbothioamide, b11
4.1.1.7. 4-(3-Fluoro-4-hydroxyphenyl)thiazole-2-carbothioamide, b12
4.1.1.8. 4-(4-Hydroxyphenyl)thiazole-2-carbothioamide, b13
4.1.1.9. 4-(2,4-Dihydroxyphenyl)thiazole-2-carbothioamide, b16
4.1.2. General Procedure for the Synthesis of the Asymmetric DABTAs, d
4.1.2.1. 5-(4′-(Benzo[d][1,3]dioxol-5-yl)-[2,2′-bithiazol]-4-yl)-2-methoxyphenol, d1
4.1.2.2. 4-(Benzo[d][1,3]dioxol-5-yl)-4′-(3-fluoro-4-methoxyphenyl)-2,2′-bithiazole, d2
4.1.2.3. 5-(4′-([1,3]Dioxolo[4,5-b]pyridin-6-yl)-[2,2′-bithiazol]-4-yl)-2-methoxyphenol, d3
4.1.2.4. 6-(4′-(3-Fluoro-4-methoxyphenyl)-[2,2′-bithiazol]-4-yl)-[1,3]dioxolo[4,5-b]pyridine, d4
4.1.2.5. 4-(Benzo[d][1,3]dioxol-5-yl)-4′-(6-fluoropyridin-3-yl)-2,2′-bithiazole, d6
4.1.2.6. 6-(4′-(6-Fluoropyridin-3-yl)-[2,2′-bithiazol]-4-yl)-[1,3]dioxolo[4,5-b]pyridine, d8
4.1.2.7. 4-(4′-(2-Fluoropyrimidin-5-yl)-[2,2′-bithiazol]-4-yl)-[1,3]dioxolo[4,5-b]pyridine, d10
4.1.2.8. 2-(4′-([1,3]Dioxolo[4,5-b]pyridin-6-yl)-[2,2′-bithiazol]-4-yl)pyrimidin-5-ol, d11
4.1.2.9. 2-(4′-(Benzo[d][1,3]dioxol-5-yl)-[2,2′-bithiazol]-4-yl)pyrimidin-5-ol, d12
4.1.2.10. 4-(4′-([1,3]Dioxolo[4,5-b]pyridin-6-yl)-[2,2′-bithiazol]-4-yl)-2-fluorophenol, d13
4.1.2.11. 4-(4′-(Benzo[d][1,3]dioxol-5-yl)-[2,2′-bithiazol]-4-yl)phenol, d14
4.1.2.12. 4-(4′-([1,3]Dioxolo[4,5-b]pyridin-6-yl)-[2,2′-bithiazol]-4-yl)phenol, d15
4.1.2.13. 4-(4′-([1,3]Dioxolo[4,5-b]pyridin-6-yl)-[2,2′-bithiazol]-4-yl)benzene-1,3-diol, d22
4.1.3. Synthesis of the DABTAs from the Phenol and Pyrimidinol DABTAs
4.1.3.1. 6-(4′-(5-(2-Fluoroethoxy)pyrimidin-2-yl)-[2,2′-bithiazol]-4-yl)-[1,3]dioxolo[4,5-b]pyridine, f4
4.1.3.2. 4-(Benzo[d][1,3]dioxol-5-yl)-4′-(5-(2-fluoroethoxy)pyrimidin-2-yl)-2,2′-bithiazole, f5
4.1.3.3. 4-(Benzo[d][1,3]dioxol-5-yl)-4′-(5-(2-(2-fluoroethoxy)ethoxy)pyrimidin-2-yl)-2,2′-bithiazole, f6
4.1.3.4. 4-(Benzo[d][1,3]dioxol-5-yl)-4′-(5-(2-(2-(2-fluoroethoxy)ethoxy)ethoxy)pyrimidin-2-yl)-2,2′-bithiazole, f7
4.1.3.5. 6-(4′-(5-(2-(2-Fluoroethoxy)ethoxy)pyrimidin-2-yl)-[2,2′-bithiazol]-4-yl)-[1,3]dioxolo[4,5-b]pyridine, f8
4.1.3.6. 6-(4′-(3-(2-(2-Fluoroethoxy)ethoxy)-4-methoxyphenyl)-[2,2′-bithiazol]-4-yl)-[1,3]dioxolo [4,5-b]pyridine, f9
4.1.3.7. 6-(4′-(3-Fluoro-4-(2-(2-(2-fluoroethoxy)ethoxy)ethoxy)phenyl)-[2,2′-bithiazol]-4-yl)-[1,3]dioxolo[4,5-b]pyridine, f10
4.1.3.8. 4-(Benzo[d][1,3]dioxol-5-yl)-4′-(4-(2-fluoroethoxy)phenyl)-2,2′-bithiazole, f11
4.1.3.9. 6-(4′-(4-(2-(2-(2-Fluoroethoxy)ethoxy)ethoxy)phenyl)-[2,2′-bithiazol]-4-yl)-[1,3]dioxolo[4,5-b]pyridine, f12
4.1.3.10. 6-(4′-(5-(2-(2-(2-Fluoroethoxy)ethoxy)ethoxy)pyrimidin-2-yl)-[2,2′-bithiazol]-4-yl)-[1,3]dioxolo[4,5-b]pyridine, f13
4.1.3.11. 4-(Benzo[d][1,3]dioxol-5-yl)-4′-(3-(2-(2-(2-fluoroethoxy)ethoxy)ethoxy)-4-methoxyphenyl)-2,2′-bithiazole, f14
4.1.3.12. 4-(Benzo[d][1,3]dioxol-5-yl)-4′-(5-methoxypyrimidin-2-yl)-2,2′-bithiazole, f15
4.1.3.13. 5-(4′-(5-Methoxypyrimidin-2-yl)-[2,2′-bithiazol]-4-yl)-[1,3]dioxolo[4,5-b]pyridine, f16
4.1.3.14. 6-(4′-(4-Methoxyphenyl)-[2,2′-bithiazol]-4-yl)-[1,3]dioxolo[4,5-b]pyridine, f17
4.1.3.15. 6-(4′-(3,4-Dimethoxyphenyl)-[2,2′-bithiazol]-4-yl)-[1,3]dioxolo[4,5-b]pyridine, f18
4.1.3.16. 6-(4′-(2,4-Dimethoxyphenyl)-[2,2′-bithiazol]-4-yl)-[1,3]dioxolo[4,5-b]pyridine, f19
4.1.3.17. 6-(4′-(2,4-Bis(2-(2-fluoroethoxy)ethoxy)phenyl)-[2,2′-bithiazol]-4-yl)-[1,3]dioxolo[4,5-b]pyridine, f20
4.1.3.18. 6-(4′-(5-(2-(2-(2-Iodoethoxy)ethoxy)ethoxy)pyrimidin-2-yl)-[2,2′-bithiazol]-4-yl)-[1,3]dioxolo[4,5-b]pyridine, h13
4.2. In Vitro Binding Assays
4.2.1. Preparation of the Amyloid Protein Aggregates
4.2.1.1. Preparation of Recombinant α-syn Fibrils
4.2.1.2. Preparation of Recombinant β-Amyloid Fibrils
4.2.1.3. Preparation of Recombinant Tau Fibrils
4.2.2. Preparation of α-Synuclein, β-Amyloid1−42, and Tau Fibrils for Competition Binding Assays
4.2.3. Competition Binding Assays
4.2.3.1. Competition Binding Assays with the α-syn Fibrils
4.2.3.2. Competition Binding Assays for Aβ1−42 and Tau Fibrils
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uzuegbunam, B.C.; Li, J.; Paslawski, W.; Weber, W.; Svenningsson, P.; Ågren, H.; Yousefi, B.H. Toward Novel 18FFluorine-Labeled Radiotracers for the Imaging of α-Synuclein Fibrils. Front. Aging Neurosci. 2022, 14, 830704. [Google Scholar] [CrossRef]
- Verdurand, M.; Levigoureux, E.; Lancelot, S.; Zeinyeh, W.; Billard, T.; Quadrio, I.; Perret-Liaudet, A.; Zimmer, L.; Chauveau, F. Amyloid-Beta Radiotracer 18FBF-227 Does Not Bind to Cytoplasmic Glial Inclusions of Postmortem Multiple System Atrophy Brain Tissue. Contrast Media Mol. Imaging 2018, 2018, 9165458. [Google Scholar] [CrossRef] [PubMed]
- Verdurand, M.; Levigoureux, E.; Zeinyeh, W.; Berthier, L.; Mendjel-Herda, M.; Cadarossanesaib, F.; Bouillot, C.; Iecker, T.; Terreux, R.; Lancelot, S.; et al. In Silico, in Vitro, and in Vivo Evaluation of New Candidates for α-Synuclein PET Imaging. Mol. Pharm. 2018, 15, 3153–3166. [Google Scholar] [CrossRef] [PubMed]
- Parkinson Disease: Practice Essentials, Background, Anatomy. Available online: https://emedicine.medscape.com/article/1831191-overview?icd=login_success_email_match_norm#a4 (accessed on 8 June 2023).
- Forno, L.S. Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef]
- Mathis, C.A.; Lopresti, B.J.; Ikonomovic, M.D.; Klunk, W.E. Small-molecule PET Tracers for Imaging Proteinopathies. Semin. Nucl. Med. 2017, 47, 553–575. [Google Scholar] [CrossRef] [PubMed]
- Eberling, J.L.; Dave, K.D.; Frasier, M.A. α-synuclein imaging: A critical need for Parkinson’s disease research. J. Parkinsons. Dis. 2013, 3, 565–567. [Google Scholar] [CrossRef] [PubMed]
- Taïeb, D.; Hicks, R.J.; Hindié, E.; Guillet, B.A.; Avram, A.; Ghedini, P.; Timmers, H.J.; Scott, A.T.; Elojeimy, S.; Rubello, D.; et al. European Association of Nuclear Medicine Practice Guideline/Society of Nuclear Medicine and Molecular Imaging Procedure Standard 2019 for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2112–2137. [Google Scholar] [CrossRef]
- Irwin, D.J.; Grossman, M.; Weintraub, D.; Hurtig, H.I.; Duda, J.E.; Xie, S.X.; Lee, E.B.; van Deerlin, V.M.; Lopez, O.L.; Kofler, J.K.; et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: A retrospective analysis. Lancet Neurol. 2017, 16, 55–65. [Google Scholar] [CrossRef]
- Bagchi, D.P.; Yu, L.; Perlmutter, J.S.; Xu, J.; Mach, R.H.; Tu, Z.; Kotzbauer, P.T. Binding of the radioligand SIL23 to α-synuclein fibrils in Parkinson disease brain tissue establishes feasibility and screening approaches for developing a Parkinson disease imaging agent. PLoS ONE 2013, 8, e55031. [Google Scholar] [CrossRef]
- Korat, Š.; Bidesi, N.S.R.; Bonanno, F.; Di Nanni, A.; Hoàng, A.N.N.; Herfert, K.; Maurer, A.; Battisti, U.M.; Bowden, G.D.; Thonon, D.; et al. Alpha-Synuclein PET Tracer Development-An Overview about Current Efforts. Pharmaceuticals 2021, 14, 847. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.; Capotosti, F.; Schain, M.; Ohlsson, T.; Touilloux, T.; Hliva, V.; Vokali, E.; Luthi-Carter, R.; Molette, J.; Dimitrakopoulos, I.K.; et al. Initial clinical scans using [18 F]ACI-12589, a novel α-synuclein PET-tracer. Alzheimer’s Dement. 2022, 18, e065394. [Google Scholar] [CrossRef]
- Kuang, G.; Murugan, N.A.; Ågren, H. Mechanistic Insight into the Binding Profile of DCVJ and α-Synuclein Fibril Revealed by Multiscale Simulations. ACS Chem. Neurosci. 2019, 10, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Hohenstein, E.G.; Sherrill, C.D. Effects of heteroatoms on aromatic pi-pi interactions: Benzene-pyridine and pyridine dimer. J. Phys. Chem. A 2009, 113, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Martinez, C.R.; Iverson, B.L. Rethinking the term “pi-stacking”. Chem. Sci. 2012, 3, 2191. [Google Scholar] [CrossRef]
- Hunter, C.A. Meldola Lecture. The role of aromatic interactions in molecular recognition. Chem. Soc. Rev. 1994, 23, 101. [Google Scholar] [CrossRef]
- Khavasi, H.R.; Mir Mohammad Sadegh, B. Influence of N-heteroaromatic π-π stacking on supramolecular assembly and coordination geometry; effect of a single-atom change in the ligand. Dalton Trans. 2015, 44, 5488–5502. [Google Scholar] [CrossRef]
- Huber, R.G.; Margreiter, M.A.; Fuchs, J.E.; von Grafenstein, S.; Tautermann, C.S.; Liedl, K.R.; Fox, T. Heteroaromatic π-stacking energy landscapes. J. Chem. Inf. Model. 2014, 54, 1371–1379. [Google Scholar] [CrossRef]
- Antonschmidt, L.; Matthes, D.; Dervişoğlu, R.; Frieg, B.; Dienemann, C.; Leonov, A.; Nimerovsky, E.; Sant, V.; Ryazanov, S.; Giese, A.; et al. The clinical drug candidate anle138b binds in a cavity of lipidic α-synuclein fibrils. Nat. Commun. 2022, 13, 5385. [Google Scholar] [CrossRef]
- Li, J.; Kumar, A.; Långström, B.; Nordberg, A.; Ågren, H. Insight into the Binding of First- and Second-Generation PET Tracers to 4R and 3R/4R Tau Protofibrils. ACS Chem. Neurosci. 2023, 14, 3528–3539. [Google Scholar] [CrossRef]
- Paslawski, W.; Lorenzen, N.; Otzen, D.E. Formation and Characterization of α-Synuclein Oligomers. Methods Mol. Biol. 2016, 1345, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Klunk, W.E.; Wang, Y.; Huang, G.-F.; Debnath, M.L.; Holt, D.P.; Shao, L.; Hamilton, R.L.; Ikonomovic, M.D.; DeKosky, S.T.; Mathis, C.A. The Binding of 2-(4′-Methylaminophenyl)Benzothiazole to Postmortem Brain Homogenates Is Dominated by the Amyloid Component. J. Neurosci. 2003, 23, 2086–2092. [Google Scholar] [CrossRef] [PubMed]
- Uzuegbunam, B.C.; Librizzi, D.; Hooshyar Yousefi, B. PET Radiopharmaceuticals for Alzheimer’s Disease and Parkinson’s Disease Diagnosis, the Current and Future Landscape. Molecules 2020, 25, 977. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Velasco, A.; Fraser, G.; Beach, T.G.; Sue, L.; Osredkar, T.; Libri, V.; Spillantini, M.G.; Goedert, M.; Lockhart, A. In vitro high affinity alpha-synuclein binding sites for the amyloid imaging agent PIB are not matched by binding to Lewy bodies in postmortem human brain. J. Neurochem. 2008, 105, 1428–1437. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | DABTA | Step I/II CY [%] | Step III CY [%] | Overall CY [%] |
|---|---|---|---|---|
| 1. | d1 | 57.4 ± 0.91, (n = 2) | - | 57.4 ± 0.91, (n = 2) |
| 2. | d2 | 47.7 (n = 1) | - | 47.7 (n = 1) |
| 3. | d3 | 47.7 ± 3.23, (n = 3) | - | 47.7 ± 3.23, (n = 3) |
| 4. | d4 | 43.4, (n = 1) | - | 43.4, (n = 1) |
| 5. | d6 | 57.0, (n = 1) | - | 57.0, (n = 1) |
| 6. | d8 | 70.9 ± 1.63, (n = 3) | - | 70.9 ± 1.63, (n = 3) |
| 7. | d10 | 15.6, (n = 1) | - | 15.6, (n = 1) |
| 8. | d11 | 47.3 ± 3.11, (n = 2) | - | 47.3 ± 2.28, (n = 2) |
| 9. | d12 | 59.5 ± 2.28, (n = 8) | - | 59.5 ± 2.28, (n = 8) |
| 10. | d13 | 55.5, (n = 1) | - | 55.5, (n = 1) |
| 11. | d14 | 44.0, (n = 1) | - | 44.0, (n = 1) |
| 12. | d15 | 30.5, (n = 1) | - | 30.5, (n = 1) |
| 13. | d22 | 63.5, (n = 1) | - | 63.5, (n = 1) |
| 14. | f1 | 57.4 ± 0.91, (n = 2) | 58.0, (n = 1) | 33.3, (n = 1) |
| 15. | f3 | 47.7 ± 3.23, (n = 3) | 58.0, (n = 1) | 27.7, (n = 1) |
| 16. | f4 | 47.3 ± 2.28, (n = 2) | 85.9 ± 15.56, (n = 2) | 40.6 ± 7.37, (n = 2) |
| 17. | f5 | 59.5 ± 2.28, (n = 8) | 89.3, (n = 1) | 53.1, (n = 1) |
| 18. | f6 | 59.5 ± 2.28, (n = 8) | 77.5 ± 24.81, (n = 2) | 46.7 ± 14.98, (n = 2) |
| 17. | f7 | 59.5 ± 2.28, (n = 8) | 84.9 ± 0.71, (n = 2) | 51.2 ± 0.43, (n = 2) |
| 18. | f8 | 47.3 ± 2.28, (n = 2) | 82.0, (n = 1) | 38.8, (n = 1) |
| 19. | f9 | 47.7 ± 3.23, (n = 3) | 54.3 ± 1.03, (n = 2) | 25.9 ± 0.49, (n = 2) |
| 20. | f10 | 55.2, (n = 1) | 66.3 ± 4.94, (n = 2) | 36.6 ± 2.72, (n = 2) |
| 21. | f11 | 42.0, (n = 1) | 91.1, (n = 1) | 38.3, (n = 1) |
| 22. | f12 | 30.5, (n = 1) | 80.0, (n = 1) | 24.5, (n = 1) |
| 23. | f13 | 47.3 ± 2.28, (n = 2) | 57.0, (n = 1) | 27.0, (n = 1) |
| 24. | f14 | 57.4 ± 0.91, (n = 2) | 91.2, (n = 1) | 52.3, (n = 1) |
| 25. | f15 | 59.5 ± 2.28, (n = 8) | 85.5, (n = 1) | 50.9, (n = 1) |
| 26. | f16 | 57.4 ± 0.91, (n = 2) | 77.2, (n = 1) | 44.3, (n = 1) |
| 27. | f17 | 30.5, (n = 1) | 96.4, (n = 1) | 29.4, (n = 1) |
| 28. | f18 | 47.7 ± 3.23, (n = 3) | 87.0, (n = 1) | 41.4, (n = 1) |
| 29. | f19 | 63.5, (n = 1) | 98.3, (n = 1) | 62.4, (n = 1) |
| 30. | f20 | 63.5, (n = 1) | 95.2, (n = 1) | 60.1, (n = 1) |
| 31. | h13 | 47.3 ± 2.28, (n = 2) | 80.7, (n = 1) | 38.2, (n = 1) |
| No. | DABTA | Ki (nM) | Selectivity | clogP | tPSA | |||
|---|---|---|---|---|---|---|---|---|
| α-syn | Aβ | tau | Aβ/α-syn | tau/α-syn | ||||
| 1 | d2 | 1.22 * | 241.7 * | >1000 * | 198.1 | >819.7 | 5.47 | 52.4 |
| 2.32 | 147.1 | 680.7 | 63.4 | 293.4 | ||||
| 2 | d4 | 0.66 * | 275.3 * | >1000 * | 417.1 | >1515.2 | 4.78 | 64.8 |
| 2.56 | 87.4 | 364 | 34.1 | 142.2 | ||||
| 3 | d6 | 1.21 * | 237.2 * | 966 * | 196.0 | 798.3 | 4.19 | 55.5 |
| 2.51 | 150.9 | 798.7 | 60.1 | 318.2 | ||||
| 4 | d8 | 0.10 * | 386.3 * | >1000 * | 3863.0 | >10,000.0 | 3.50 | 67.9 |
| 1.01 | 127.1 | >1000 | 125.8 | >990.1 | ||||
| 5 | d10 | 1.36 * | 406.8 * | >1000 * | 299.1 | >735.3 | 2.50 | 80.4 |
| 4.75 | 214.3 | 434.4 | 45.1 | 91.5 | ||||
| 6 | f4 | 0.99 * | 250.6 * | 464.9 * | 253.1 | 469.6 | 3.35 | 89.5 |
| 1.51 | 185.2 | 766.4 | 122.6 | 507.5 | ||||
| 7 | f5 | 2.27 * | 133.7 * | 515.2 * | 58.9 | 227.0 | 4.04 | 77.1 |
| 2.88 | 154.9 | 516.2 | 53.8 | 179.2 | ||||
| 8 | f6 | 0.50 * | 197.8 * | 437.2 * | 395.6 | 874.4 | 3.79 | 86.4 |
| 4.89 | 96.7 | 362.5 | 19.8 | 74.1 | ||||
| 9 | f7 | 1.51 * | ND | ND | - | - | 3.61 | 95.6 |
| 1.17 | 76.4 | 242.1 | 65.3 | 206.9 | ||||
| 10 | f8 | 2.30 | ND | ND | - | - | 3.10 | 98.7 |
| 11 | f9 | 0.84 * | ND | ND | - | - | 4.48 | 83.2 |
| 0.96 | 47.6 | 1130.0 | 49.6 | 1177.1 | ||||
| 12 | f10 | 1.00 * | ND | ND | - | - | 4.61 | 83.2 |
| 0.93 | 37.3 | 615 | 40.1 | 134.4 | ||||
| 13 | f11 | 3.71 | 99.6 | 498.7 | 26.9 | 174.0 | 5.70 | 52.4 |
| 14 | f12 | 0.92 | 38.7 | 983.0 | 42.1 | 1068.5 | 4.59 | 83.2 |
| 15 | f13 | 0.38 | 46.6 | 120.0 | 122.6 | 315.8 | 2.93 | 108.0 |
| 16 | f14 | 1.74 | 210.2 | 662.2 | 120.8 | 380.6 | 4.99 | 88.1 |
| 17 | f15 | 2.45 | 59.3 | 707.0 | 24.2 | 288.6 | 3.78 | 77.1 |
| 18 | f16 | ND | ND | ND | ND | ND | 3.10 | 89.5 |
| 19 | f17 | 1.78 | 167.5 | 512.7 | 94.1 | 288.0 | 4.76 | 64.8 |
| 20 | f18 | 4.99 | ND | ND | - | - | 4.48 | 74.0 |
| 21 | f19 | 1.35 | 148.7 | 671.3 | 110.1 | 497.3 | 4.27 | 74.0 |
| 22 | f20 | 1.31 | 221.2 | 814.6 | 168.9 | 621.8 | 4.27 | 92.5 |
| 23 | h13 | 1.17 | ND | ND | - | - | 3.80 | 108.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uzuegbunam, B.C.; Li, J.; Paslawski, W.; Weber, W.; Svenningsson, P.; Ågren, H.; Hooshyar Yousefi, B. In Silico and In Vitro Study towards the Rational Design of 4,4′-Disarylbisthiazoles as a Selective α-Synucleinopathy Biomarker. Int. J. Mol. Sci. 2023, 24, 16445. https://doi.org/10.3390/ijms242216445
Uzuegbunam BC, Li J, Paslawski W, Weber W, Svenningsson P, Ågren H, Hooshyar Yousefi B. In Silico and In Vitro Study towards the Rational Design of 4,4′-Disarylbisthiazoles as a Selective α-Synucleinopathy Biomarker. International Journal of Molecular Sciences. 2023; 24(22):16445. https://doi.org/10.3390/ijms242216445
Chicago/Turabian StyleUzuegbunam, Bright C., Junhao Li, Wojciech Paslawski, Wolfgang Weber, Per Svenningsson, Hans Ågren, and Behrooz Hooshyar Yousefi. 2023. "In Silico and In Vitro Study towards the Rational Design of 4,4′-Disarylbisthiazoles as a Selective α-Synucleinopathy Biomarker" International Journal of Molecular Sciences 24, no. 22: 16445. https://doi.org/10.3390/ijms242216445
APA StyleUzuegbunam, B. C., Li, J., Paslawski, W., Weber, W., Svenningsson, P., Ågren, H., & Hooshyar Yousefi, B. (2023). In Silico and In Vitro Study towards the Rational Design of 4,4′-Disarylbisthiazoles as a Selective α-Synucleinopathy Biomarker. International Journal of Molecular Sciences, 24(22), 16445. https://doi.org/10.3390/ijms242216445