
Superoxide Anion Chemistry—Its Role at the Core of the Innate Immunity

 ,
,  and
and 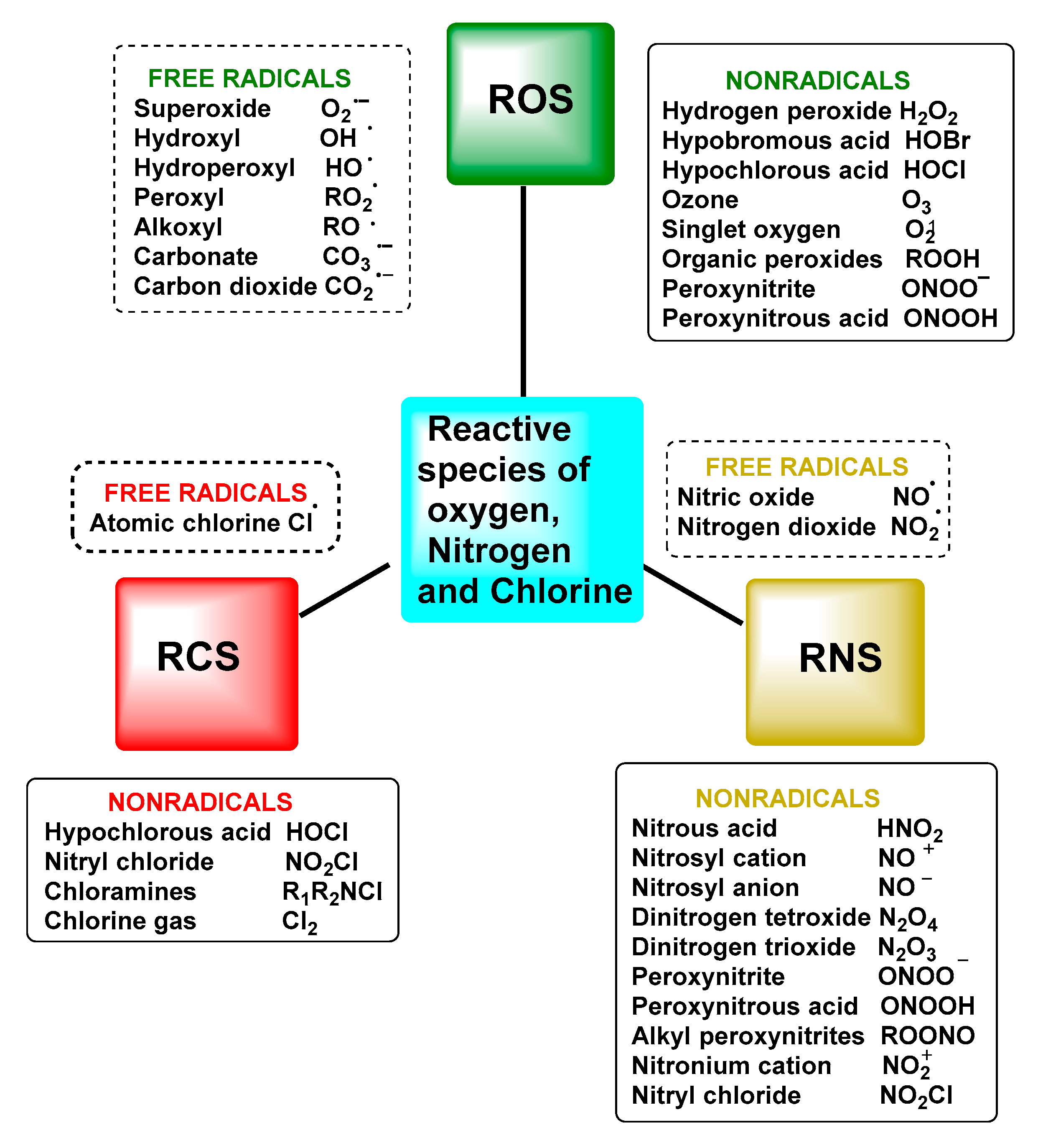{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
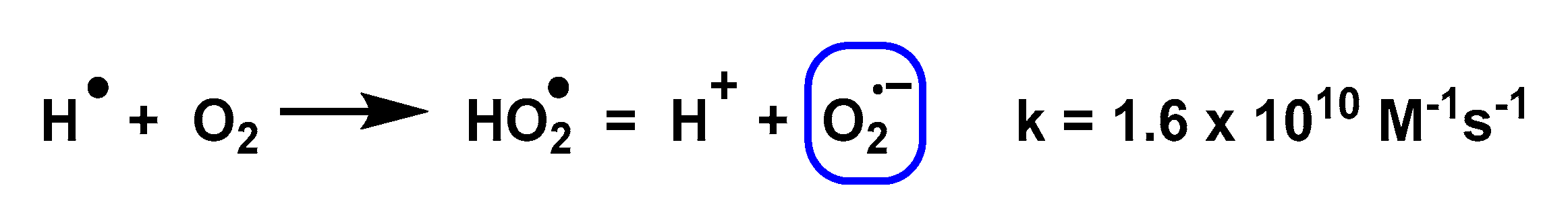{kind=link}
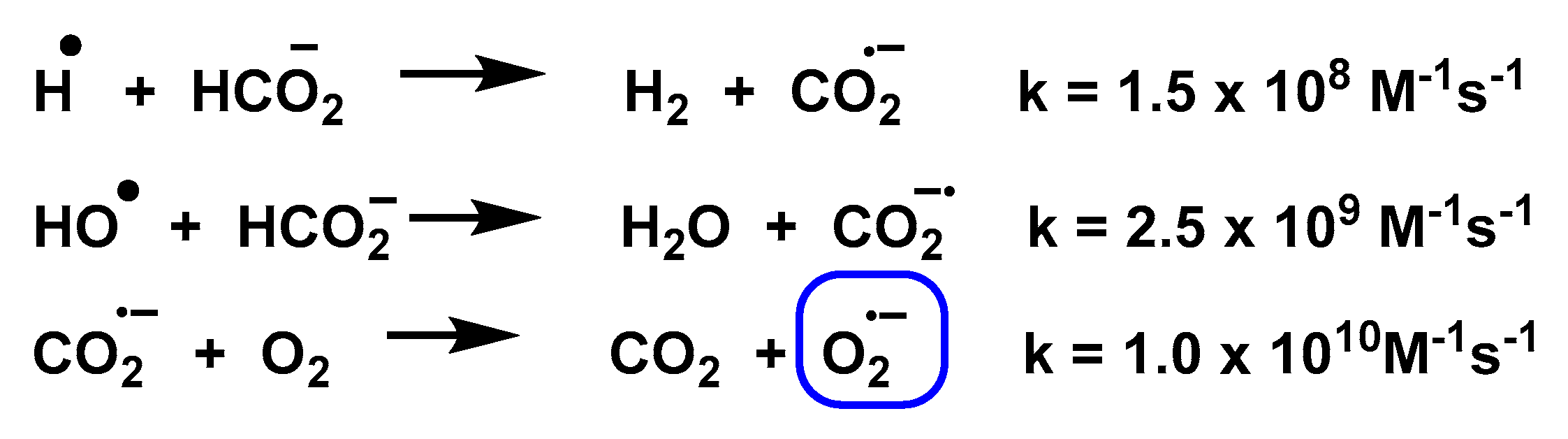{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
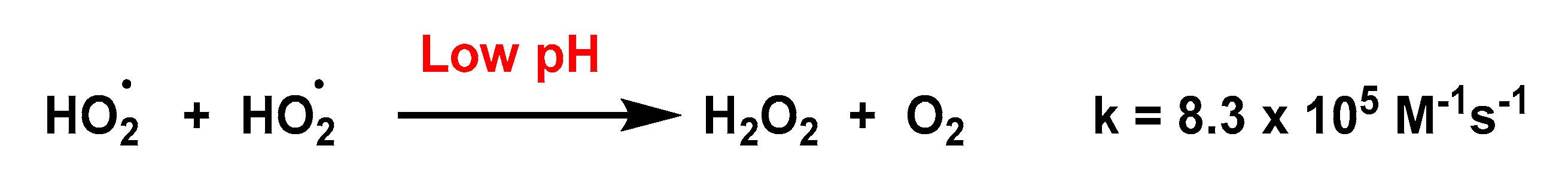{kind=link}
{kind=link}
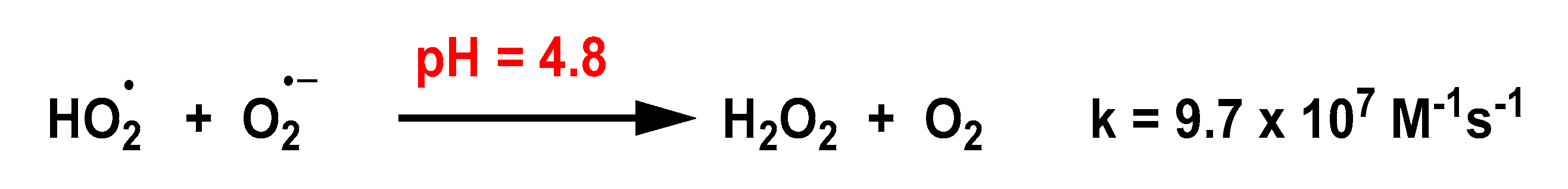{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
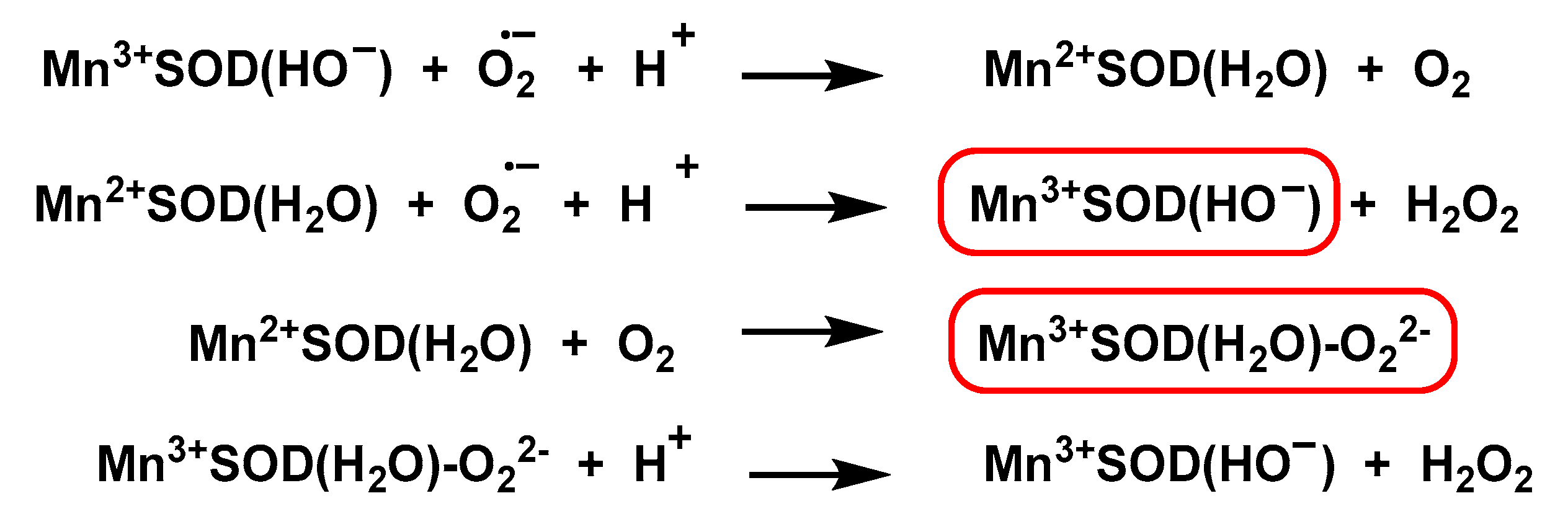{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
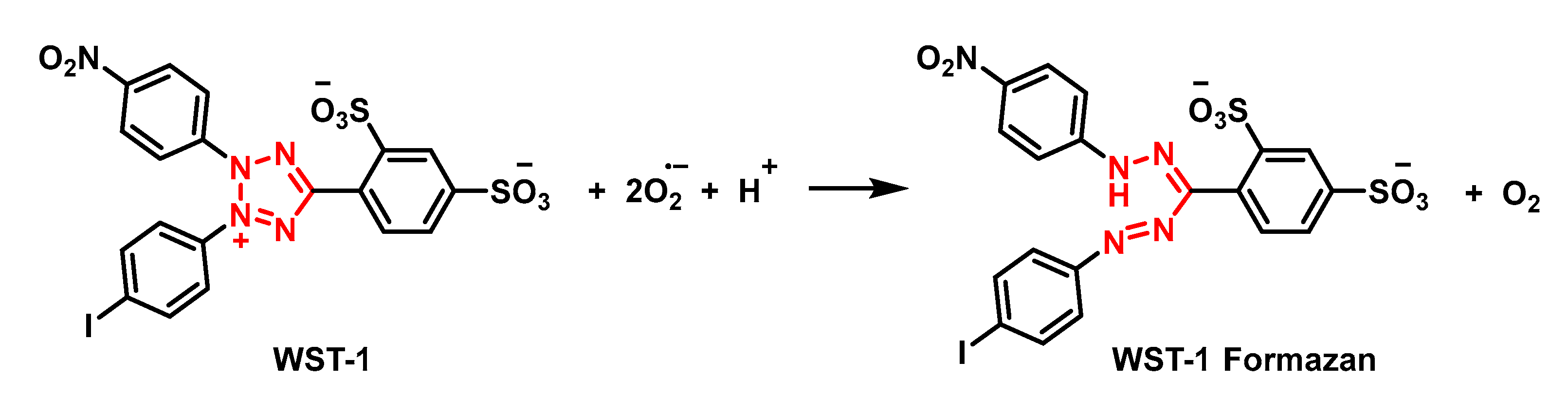{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
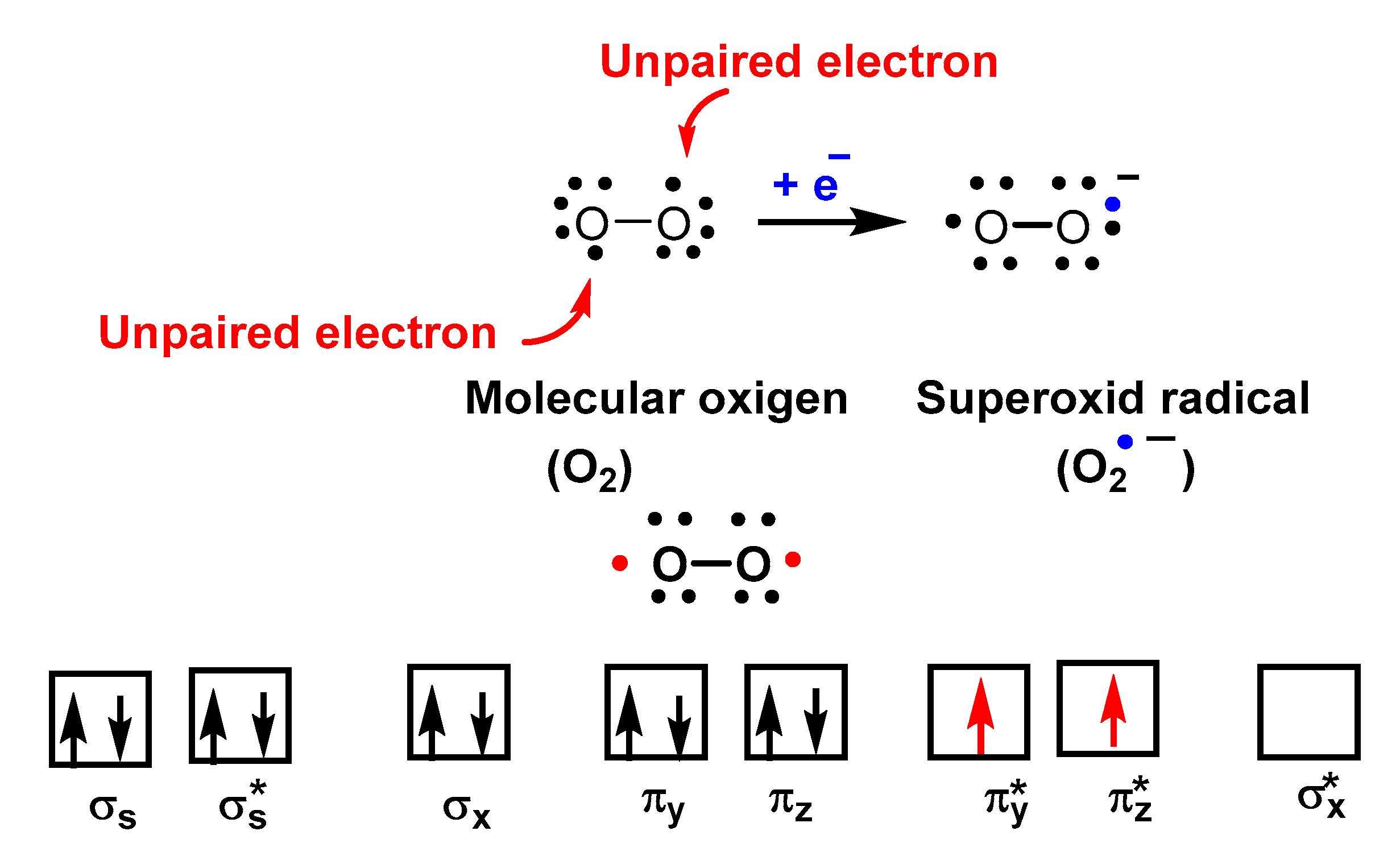
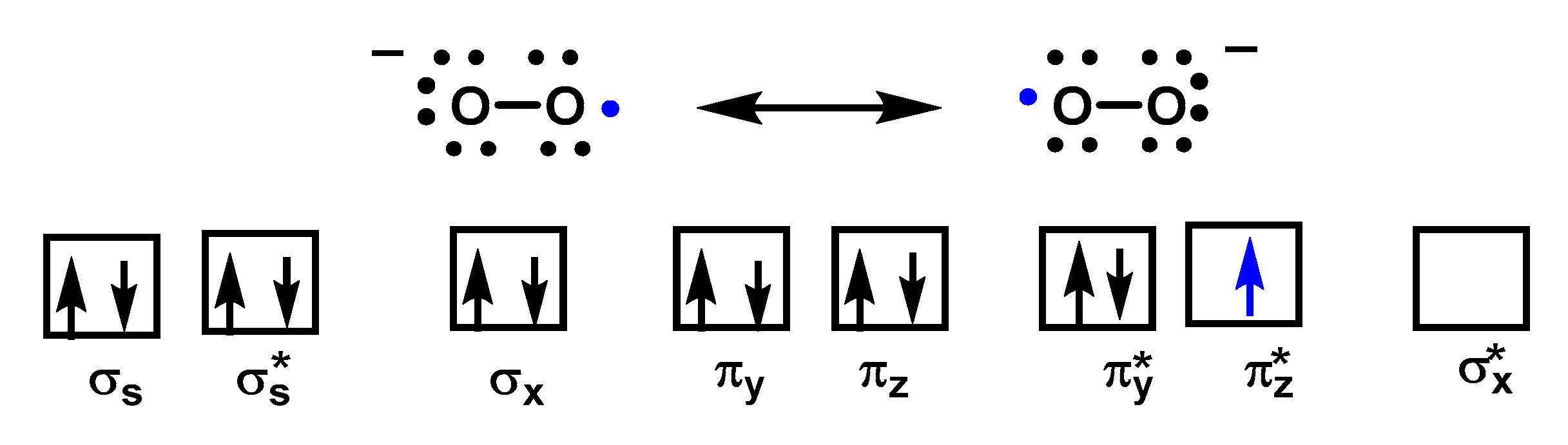
2. Superoxide Radical Anion O2•−
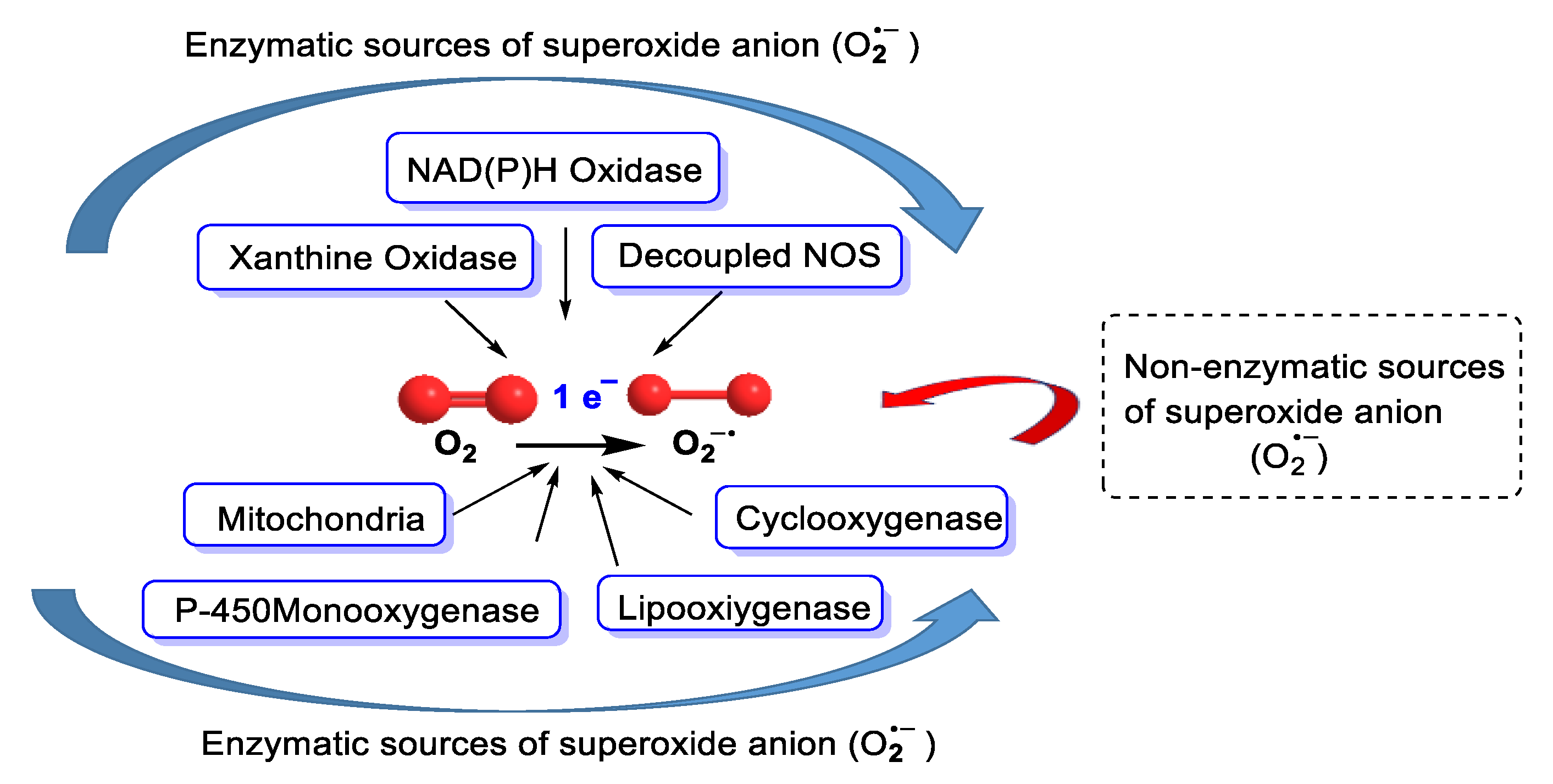
3. Sources of Superoxide Anion
3.1. Biological Sources
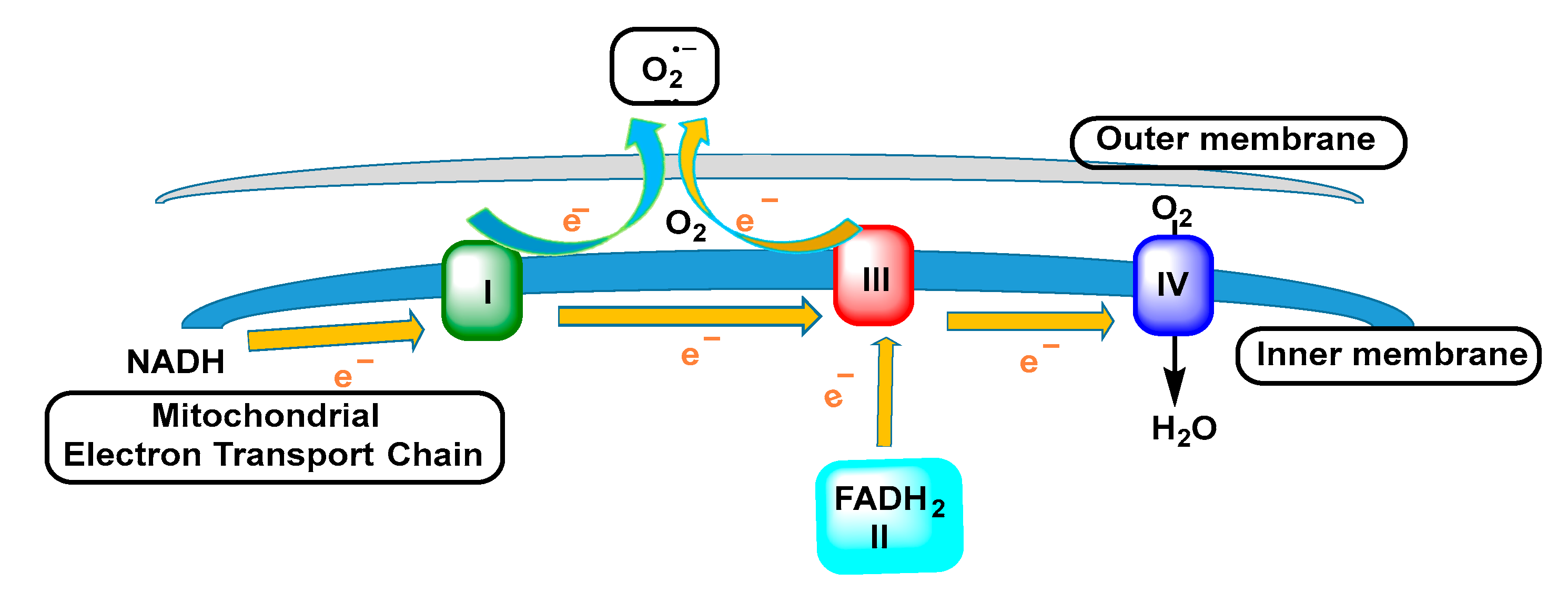
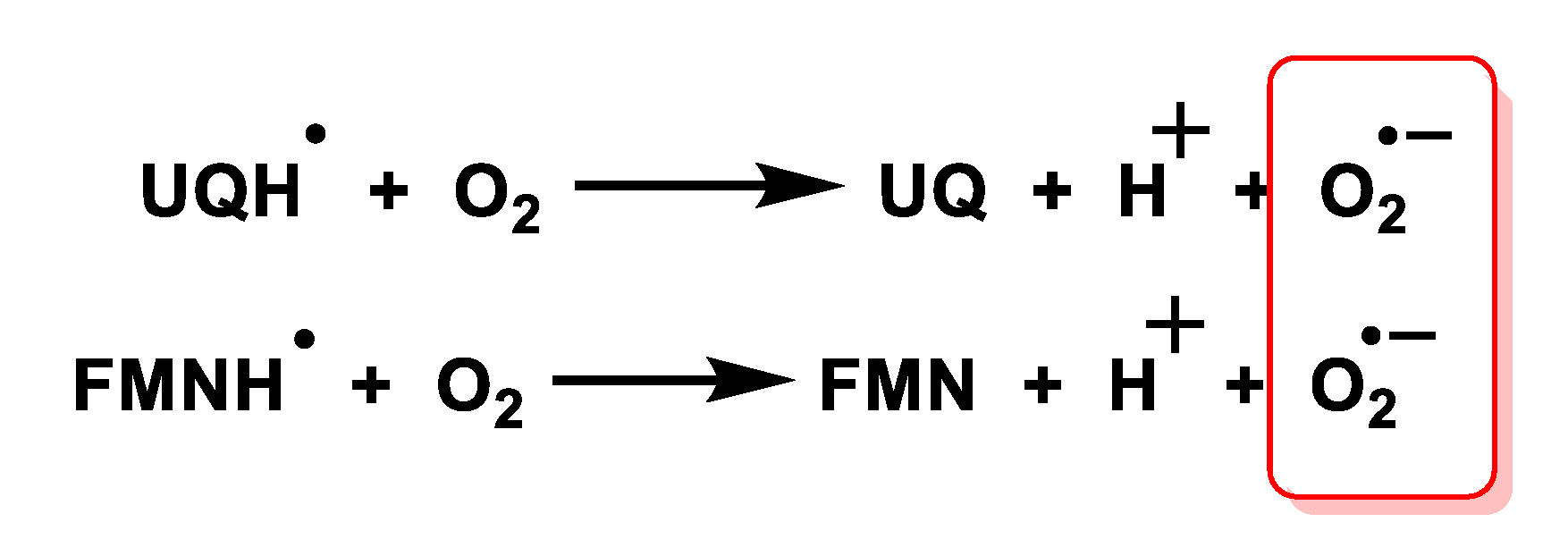
3.2. Mitochondrial Respiratory Chain
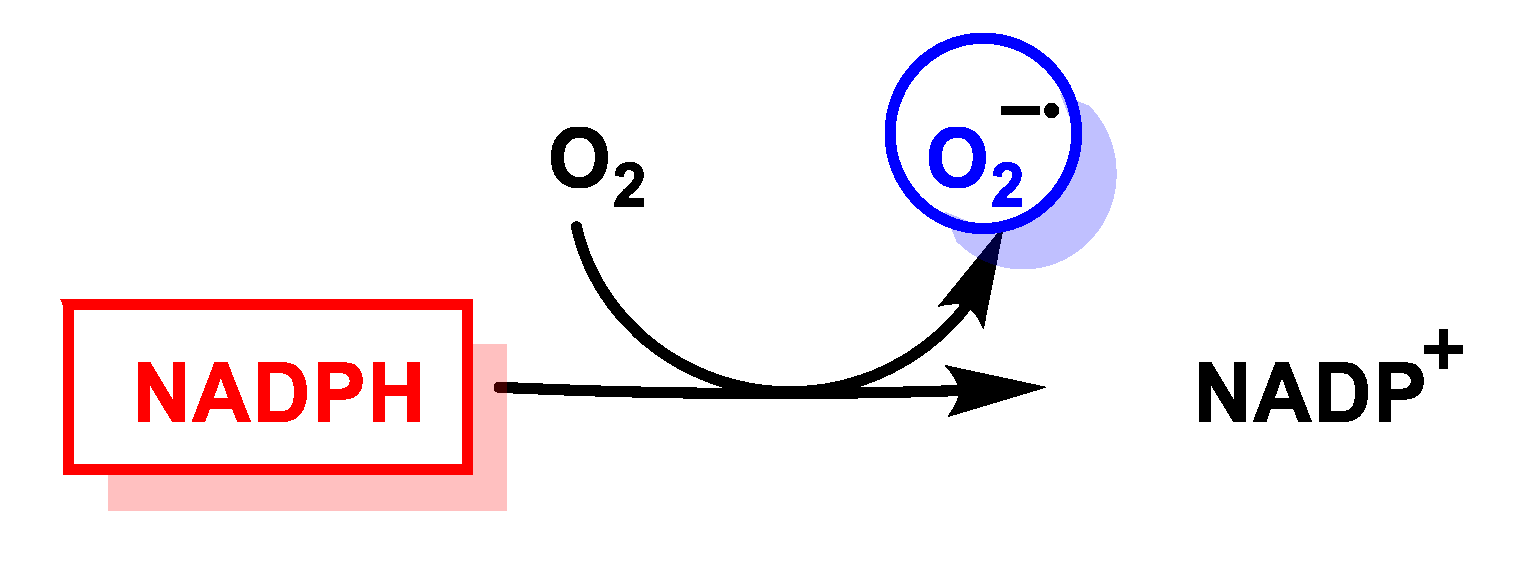
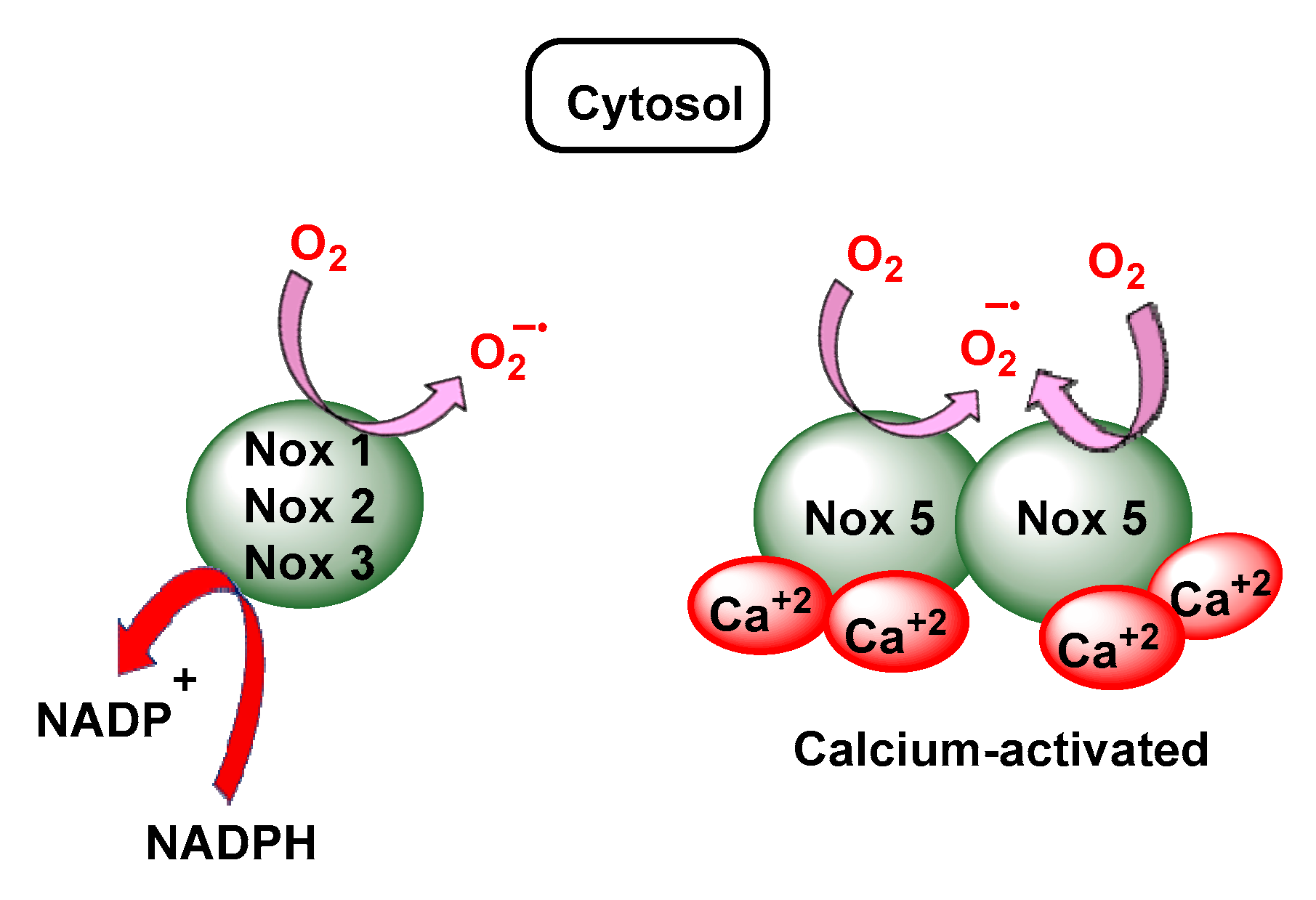
3.3. NADPH Oxidases
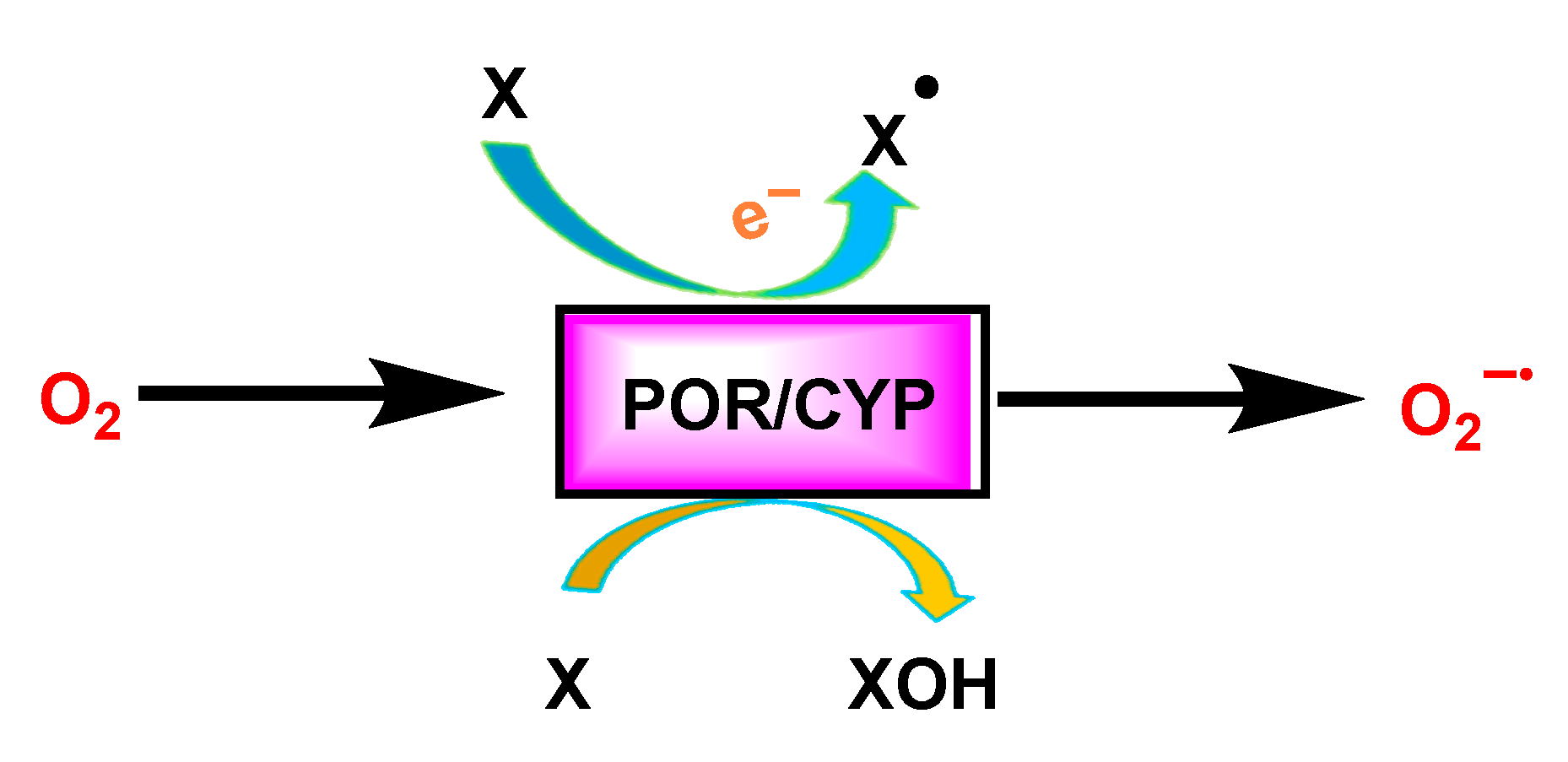
3.4. Cytochrome P450 CYP/Cytochrome P450 Reductase POR System
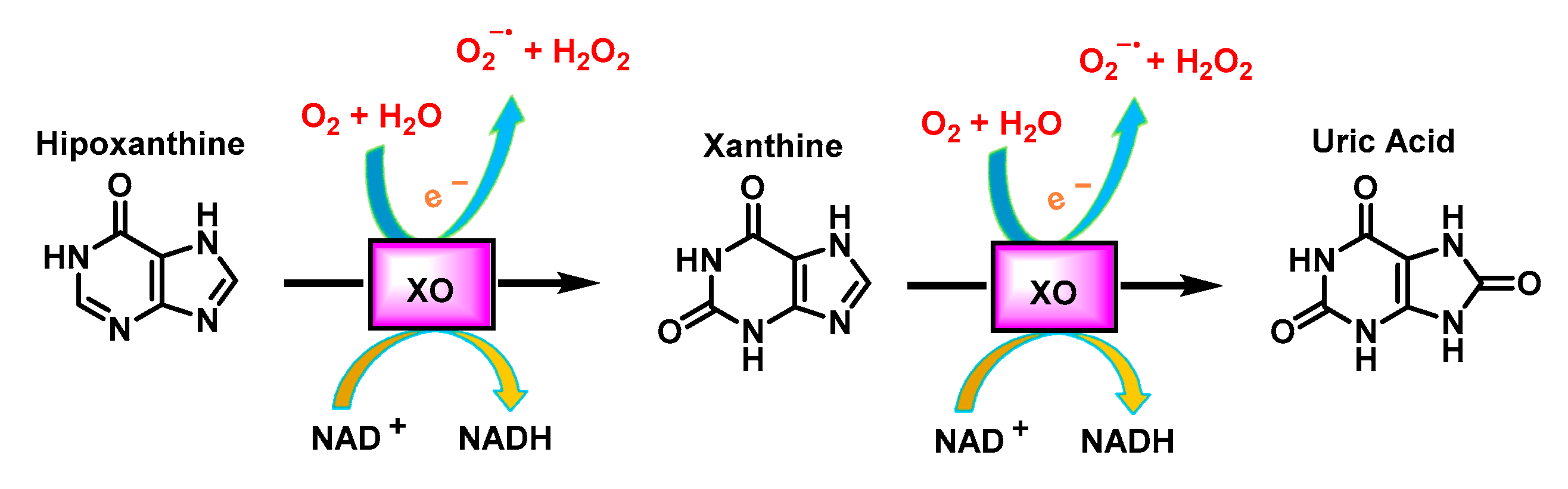
3.5. Xanthine Oxidoreductase
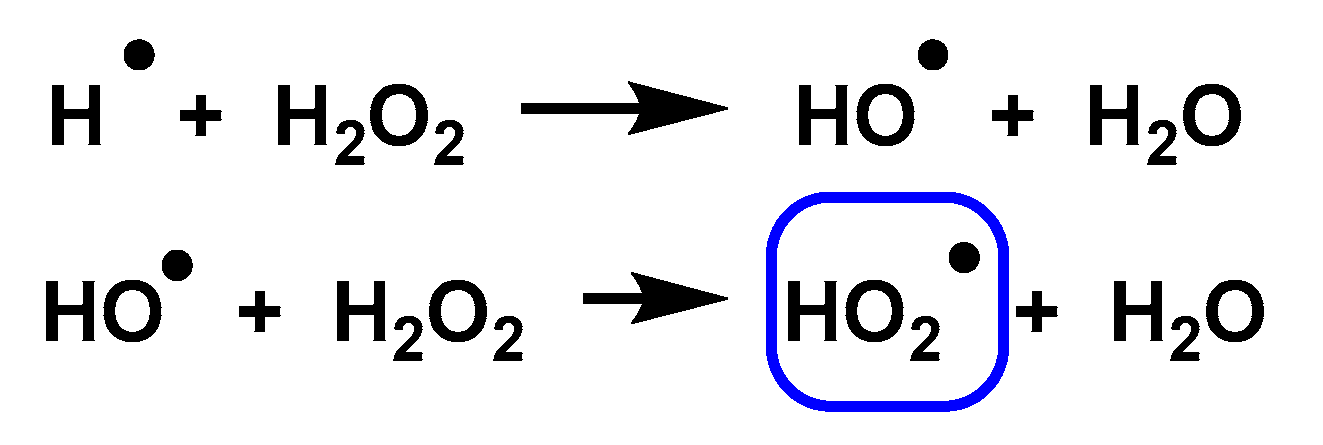
3.6. Non-Enzymatic Production of Superoxide
3.7. Non-Biochemical Sources
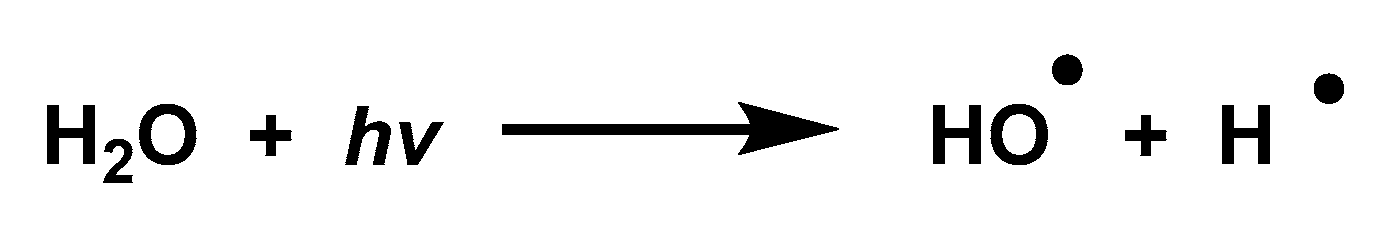
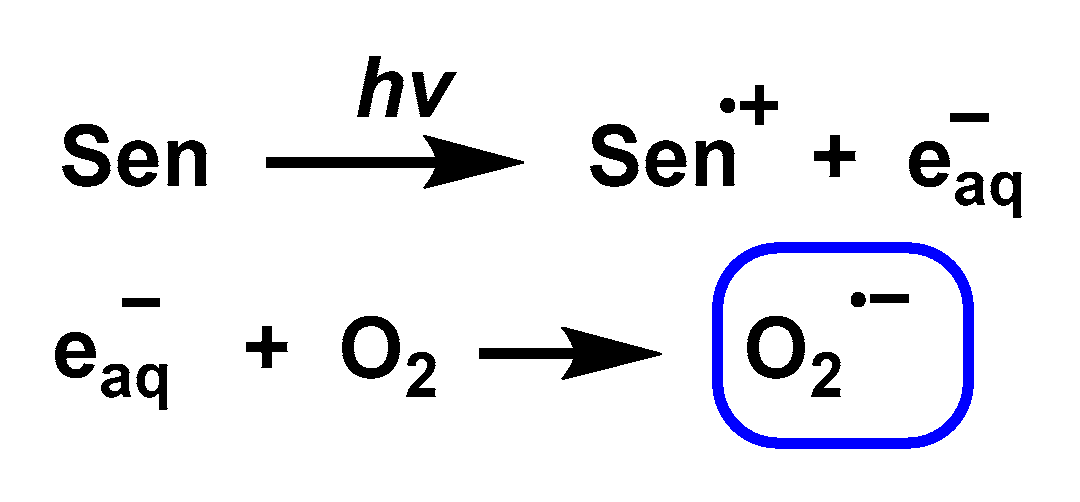
3.8. Photolysis
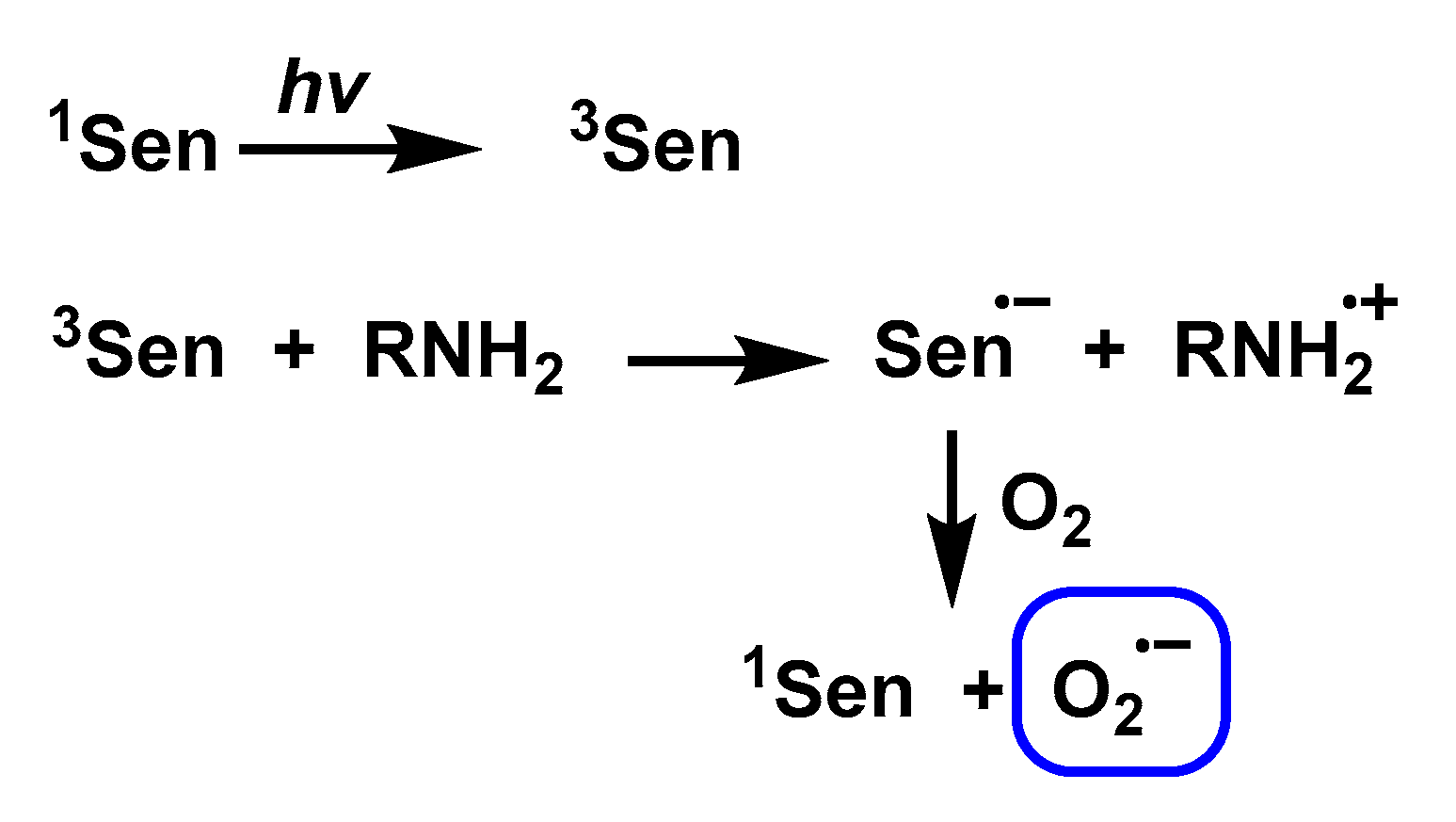
3.9. Photochemical and Photocatalytic Sources
3.10. Chemical Pathway
- Synthesis of superoxide salts of alkali metals, such as potassium and sodium, and alkali earth metals, such as strontium and barium.
- Solvation of these salts in appropriate media to release O2•−.
3.11. Electrochemical
4. Reactions of Superoxide Anion
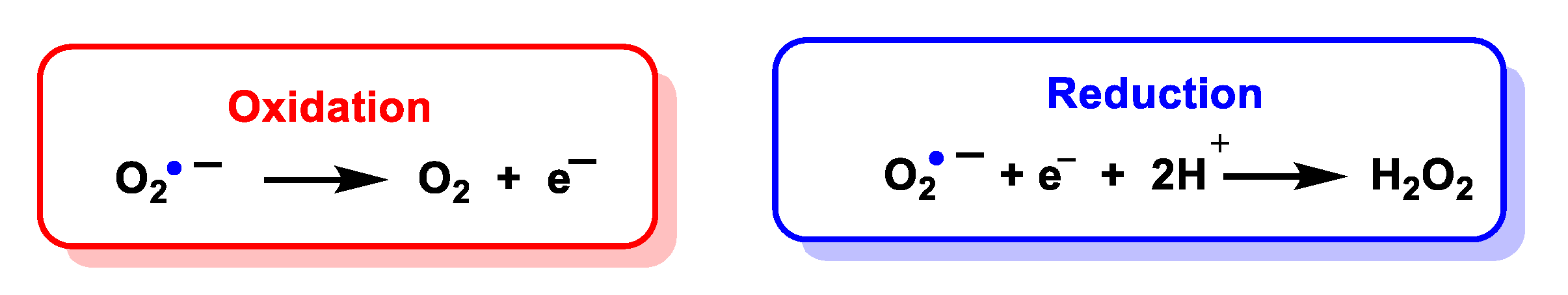
4.1. Dismutation of Superoxide to Hydrogen Peroxide
4.1.1. Non-Enzymatic Spontaneous Dismutation
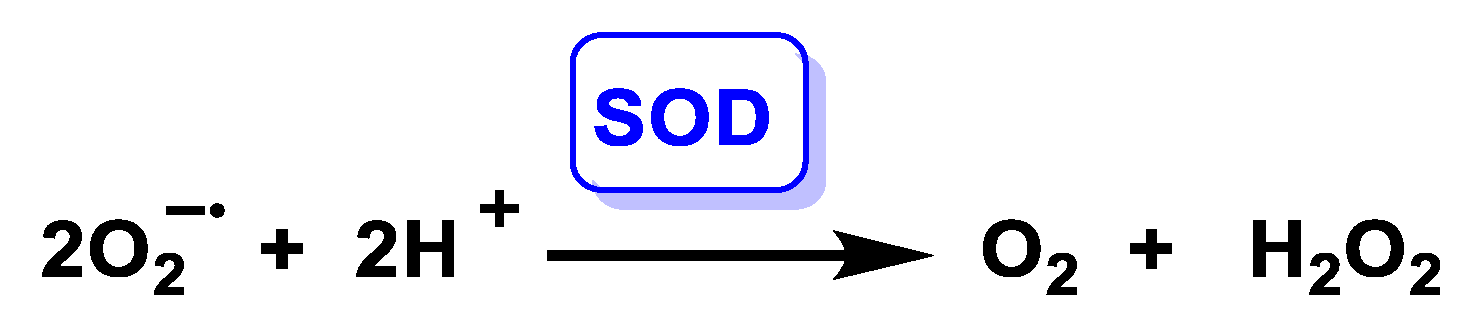
4.1.2. Enzymatically Catalysed Dismutation
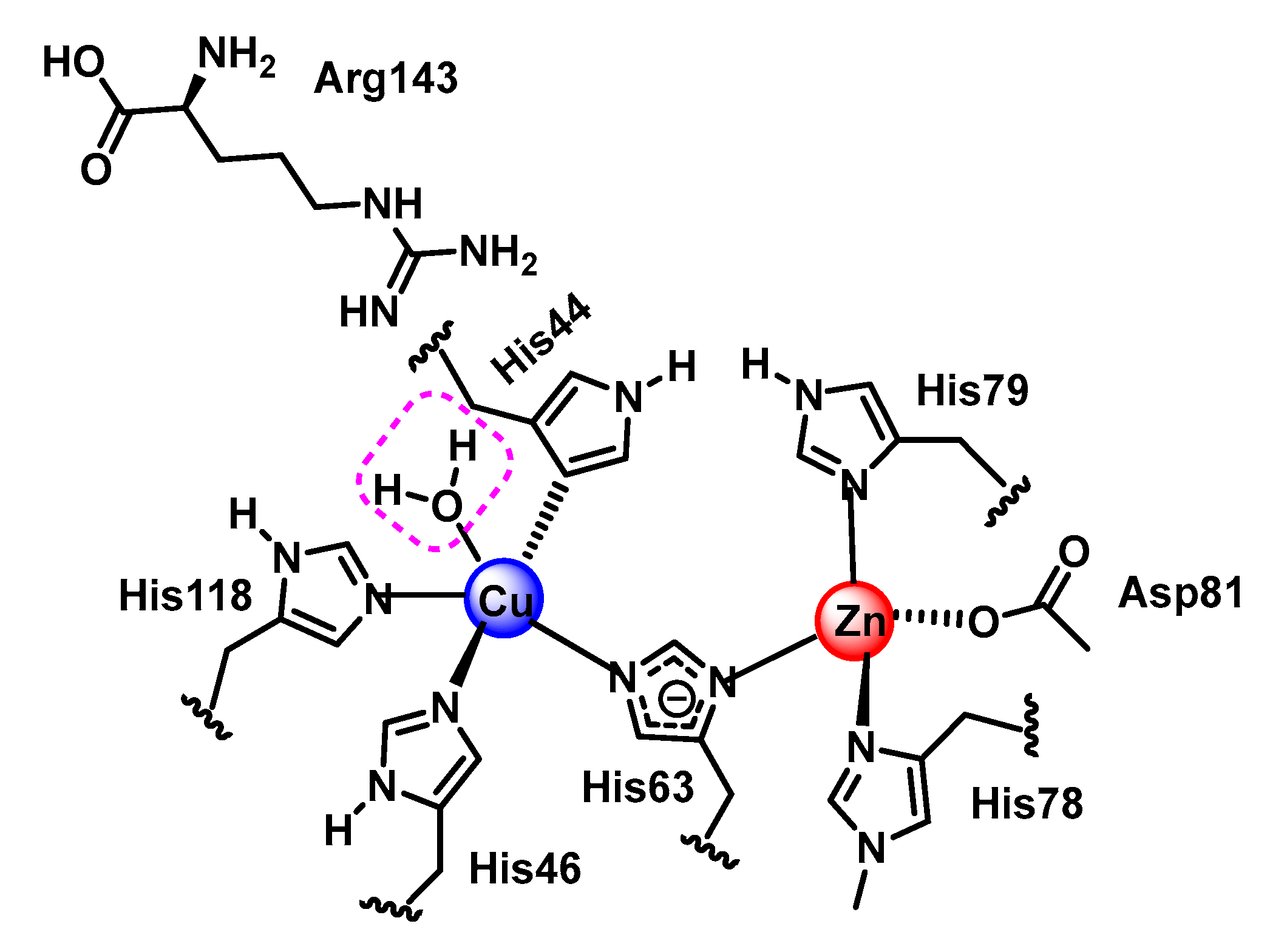
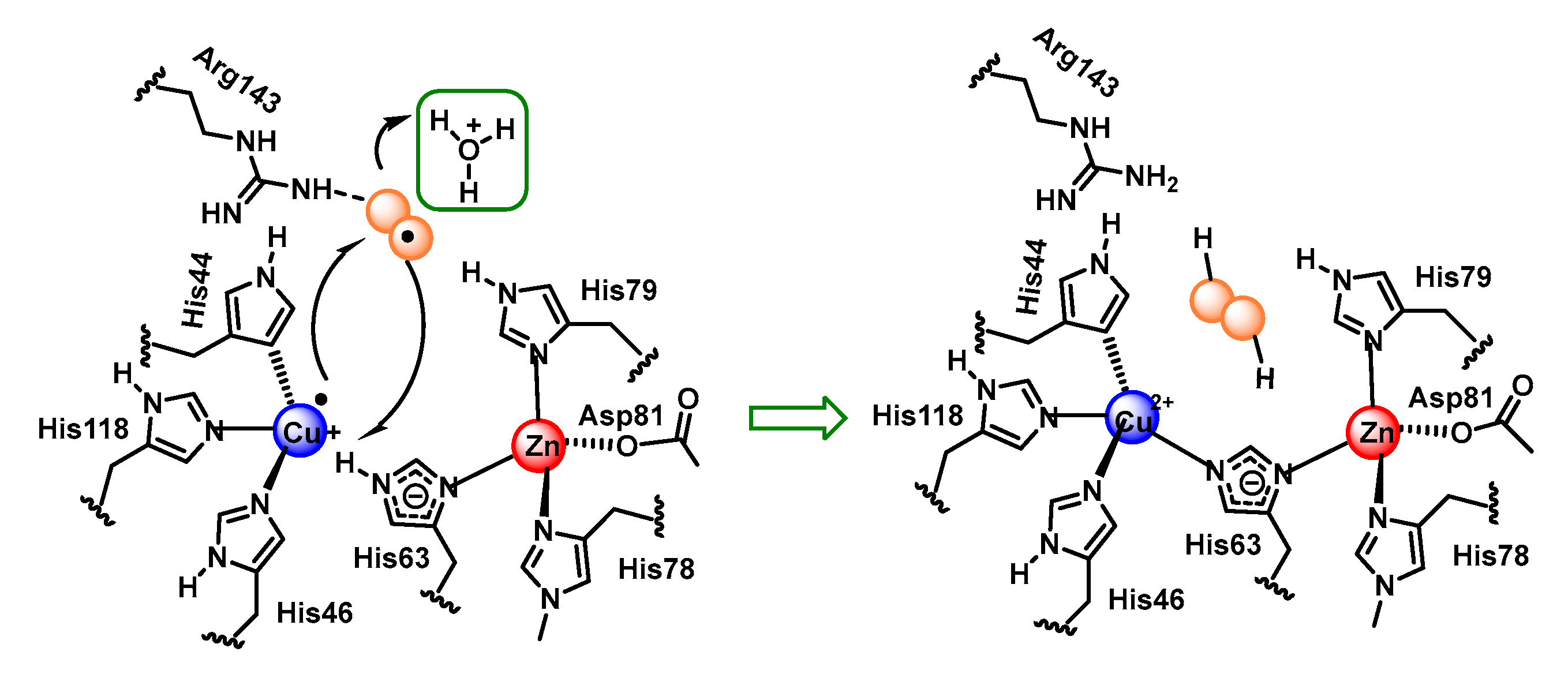
4.1.3. Copper, Zinc-Superoxide Dismutase (Cu,Zn-SOD)
4.1.4. Manganese Superoxide Dismutase Mn-SOD
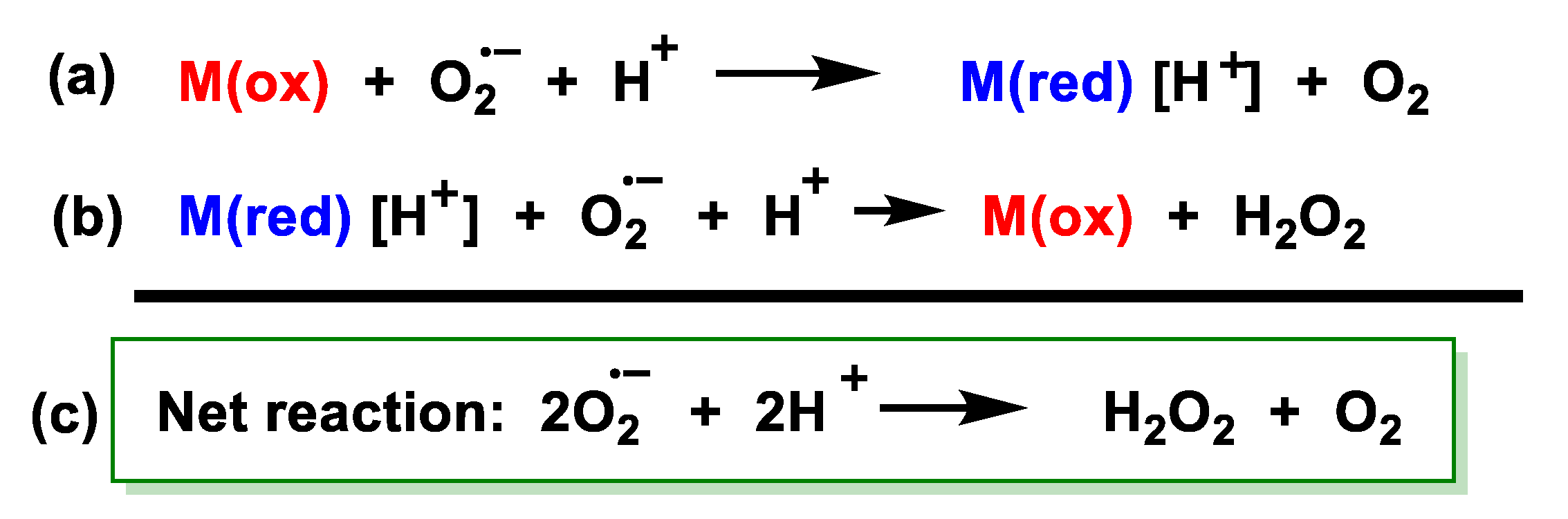
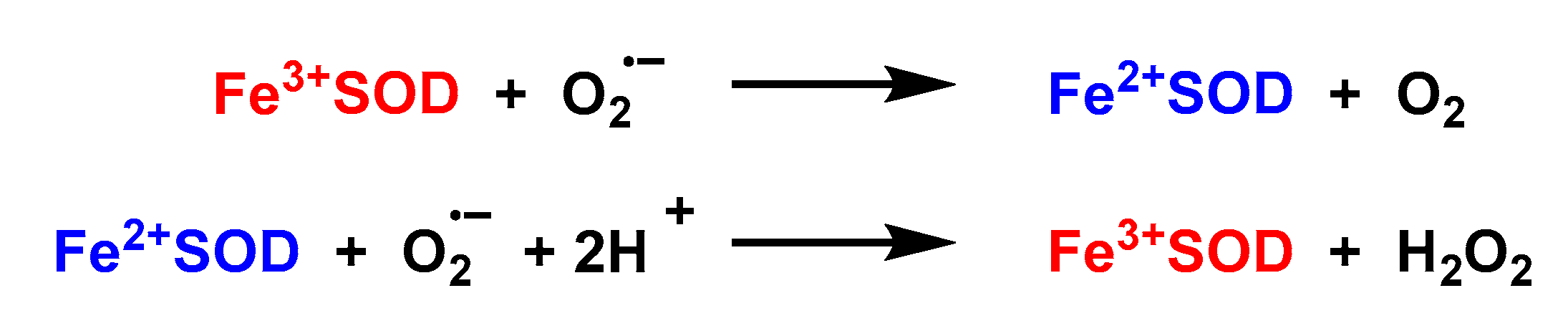
4.1.5. Iron Superoxide Dismutase Fe-SOD
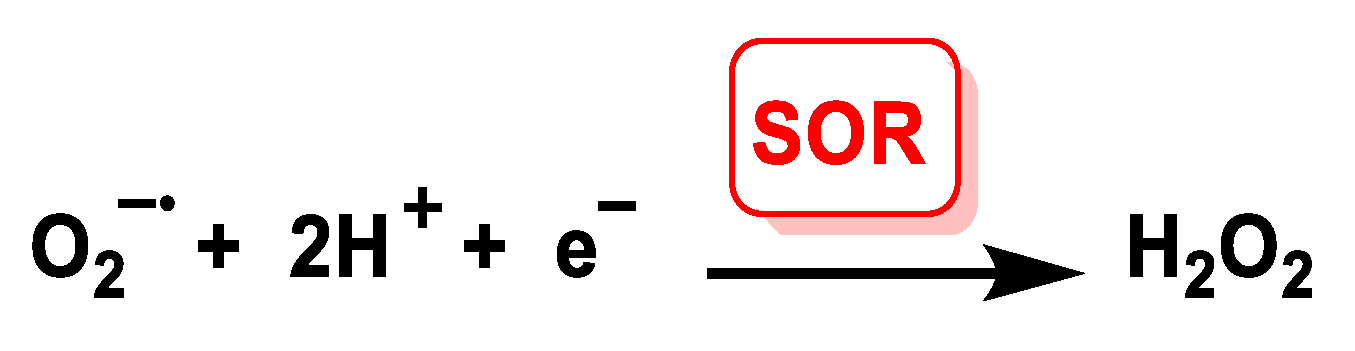
4.1.6. Iron Superoxide Reductase Fe-SOR
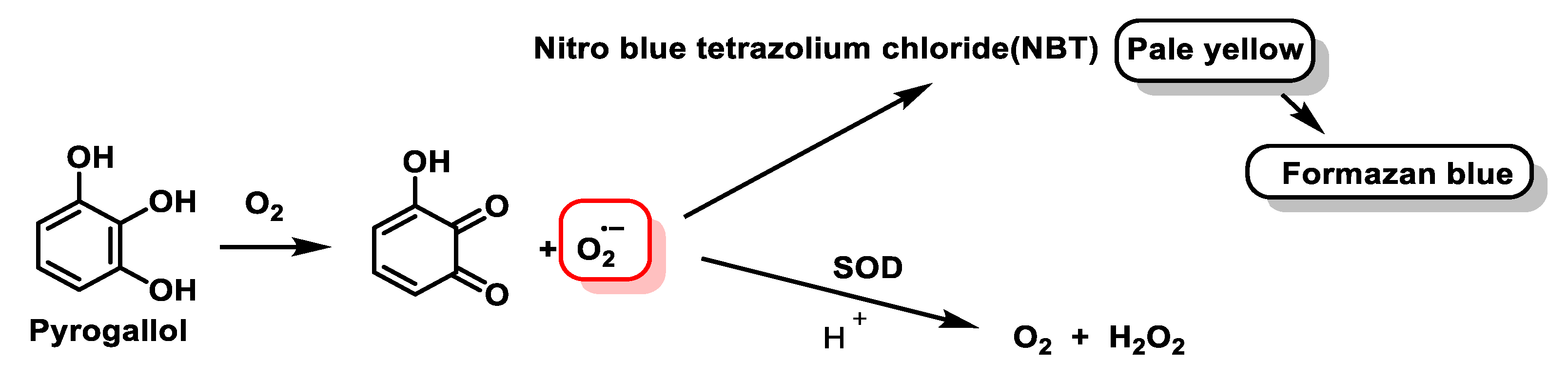
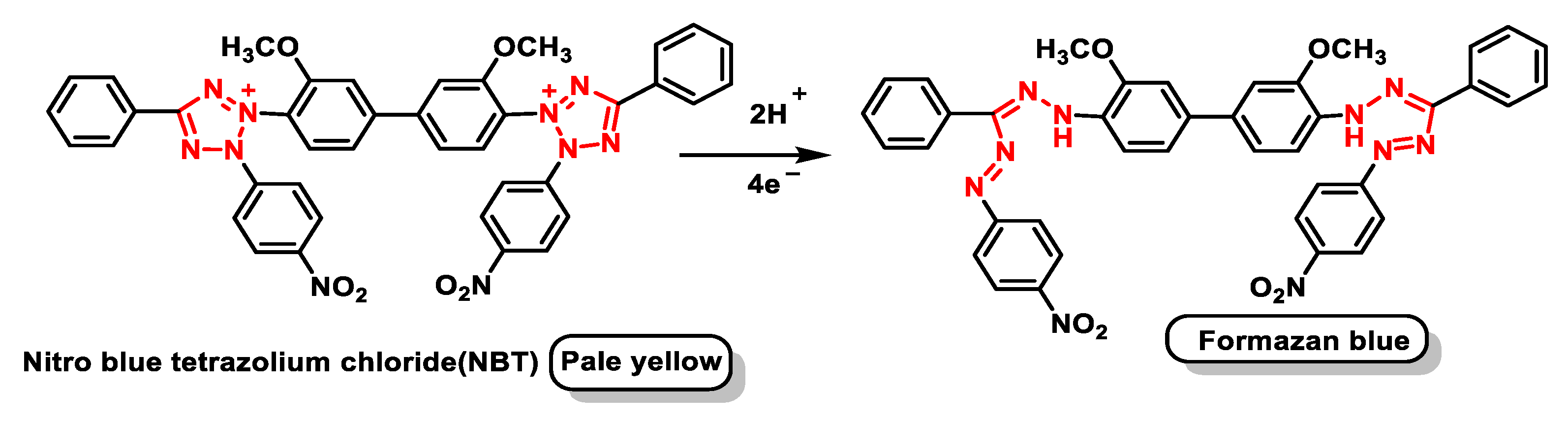
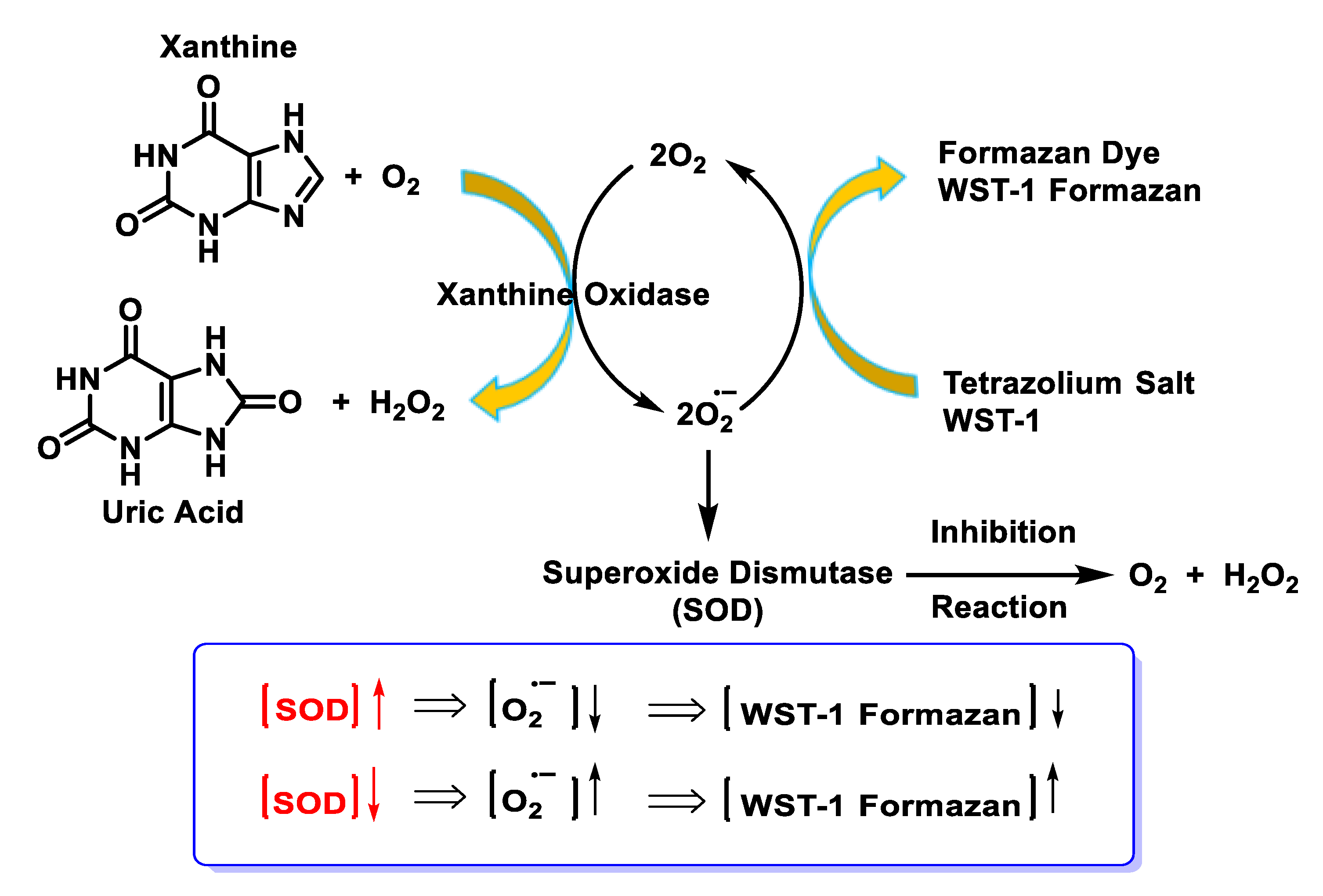
4.1.7. Analytical Determination of Superoxide Dismutase Activity
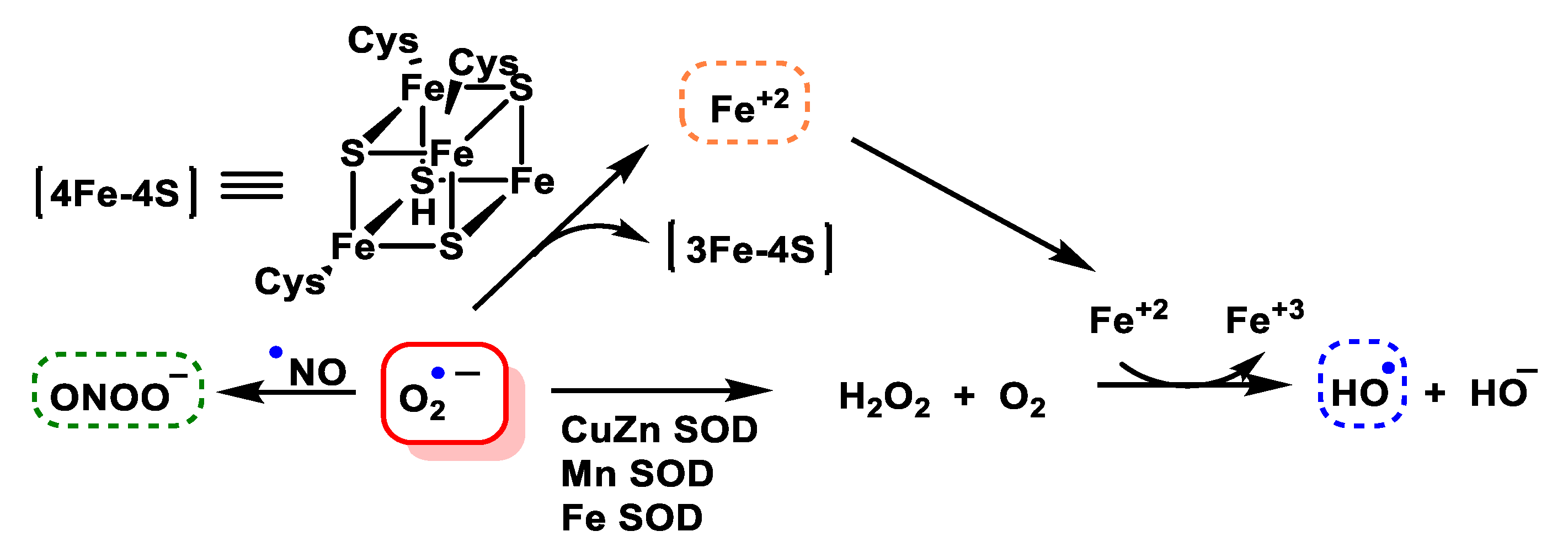
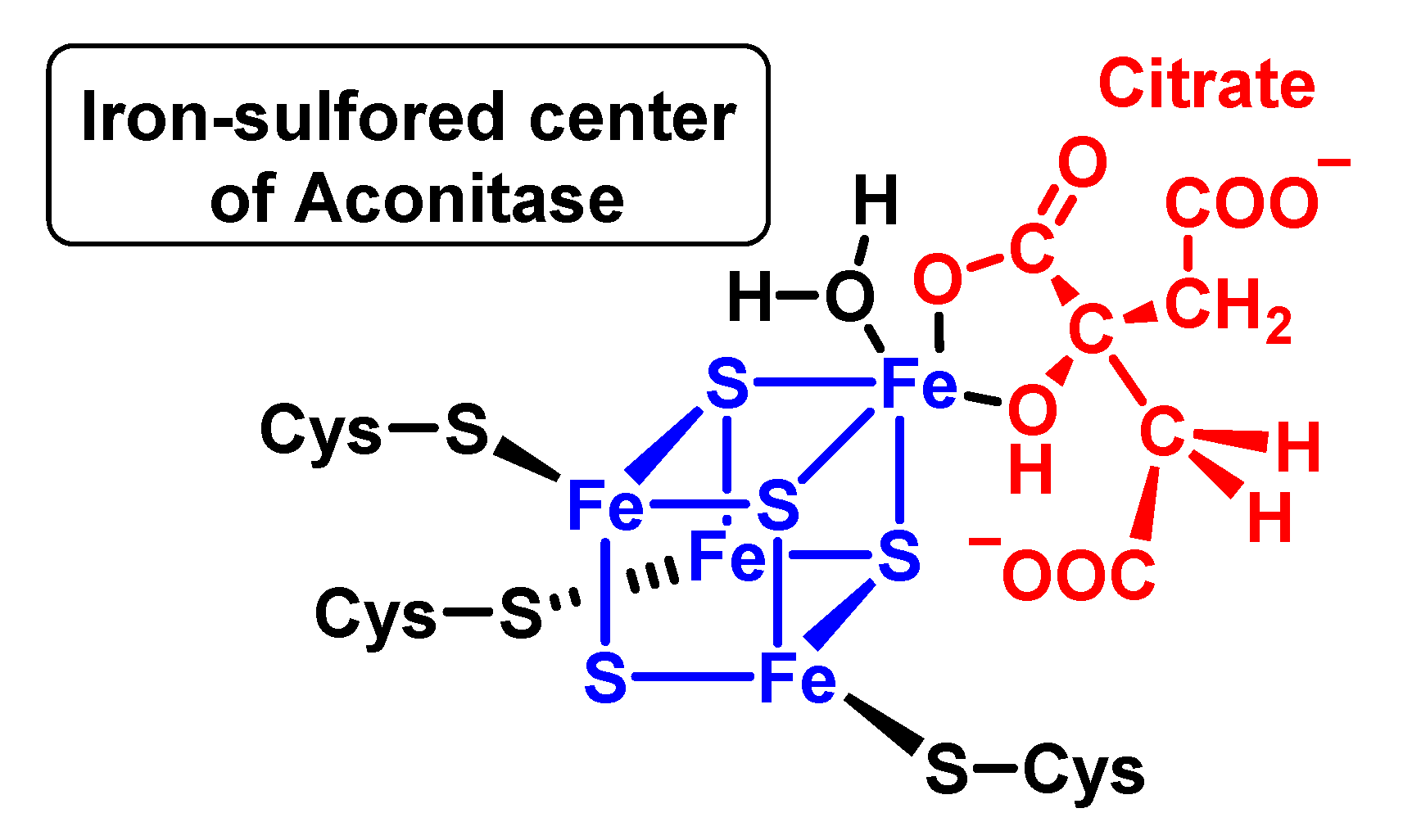
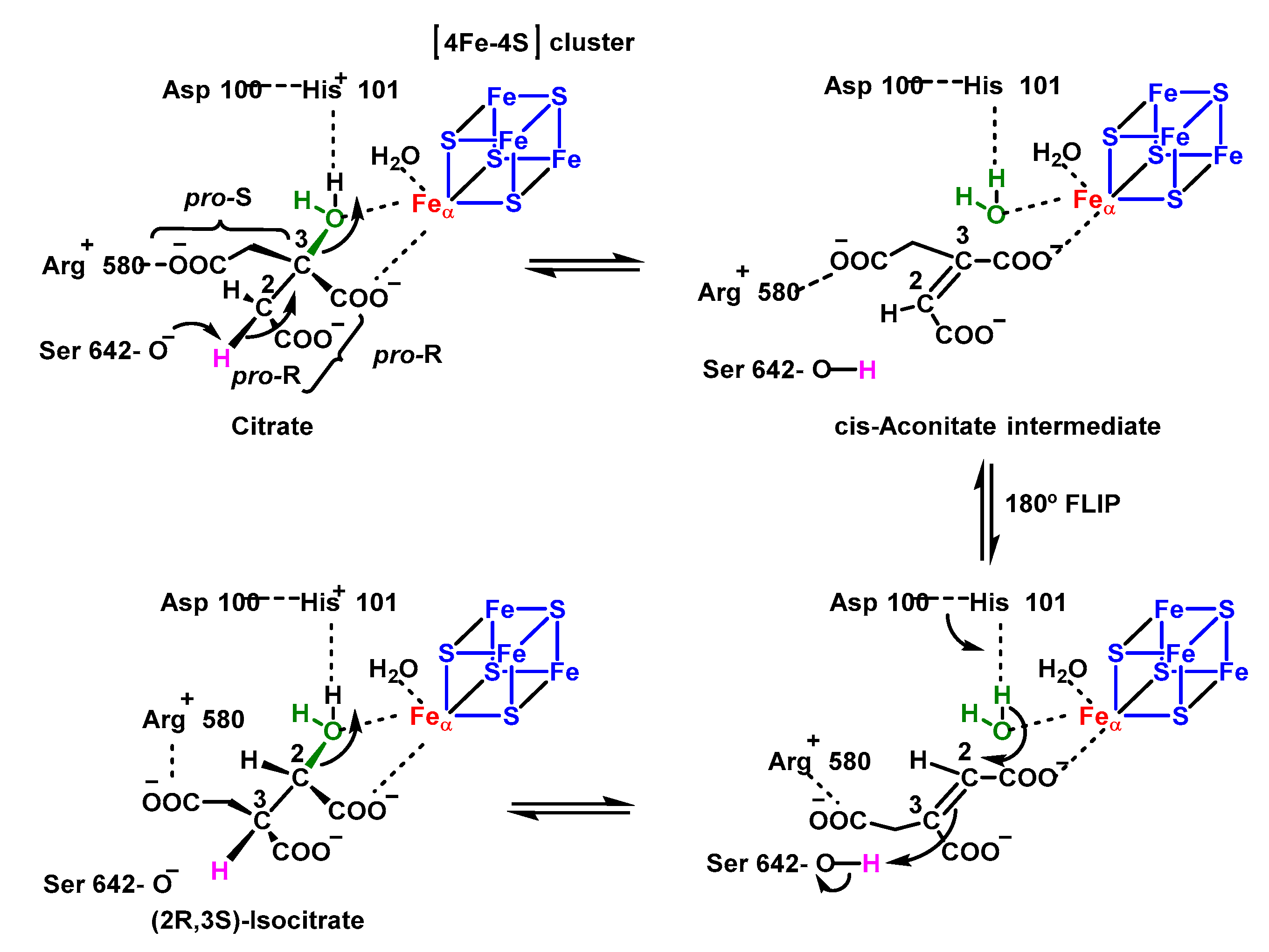
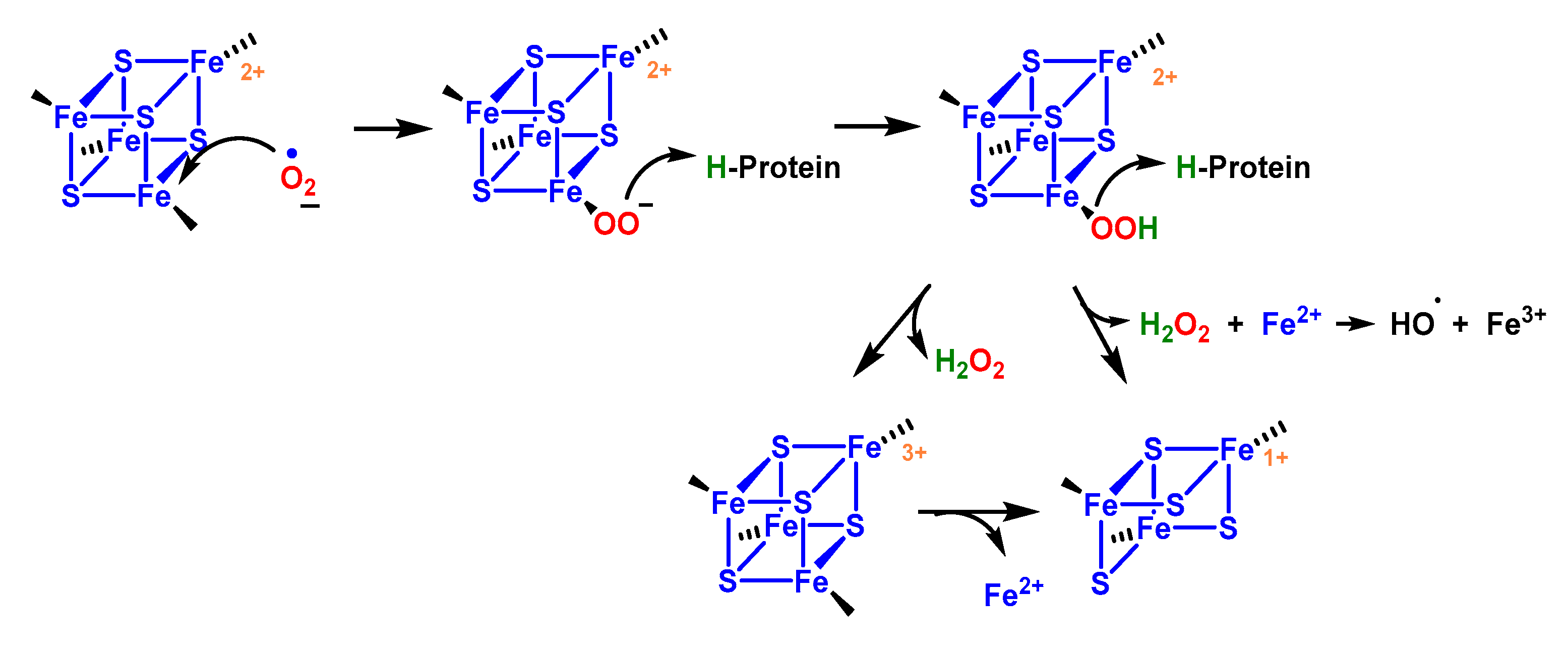
4.1.8. Reaction with Iron–Sulphur [Fe–S] Cluster
4.1.9. Conversion of Nitric Oxide to Peroxynitrite
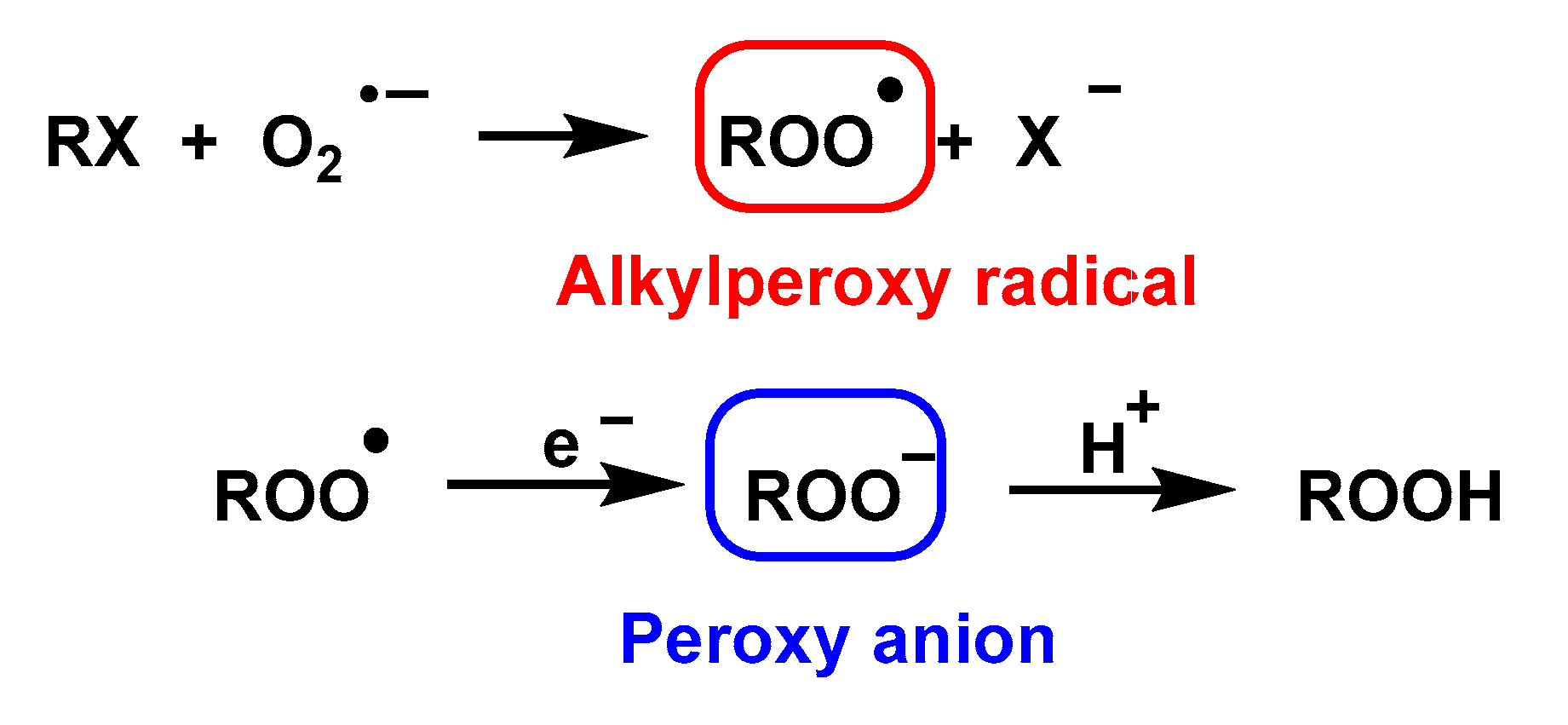
4.1.10. Nucleophilic Substitution Reaction
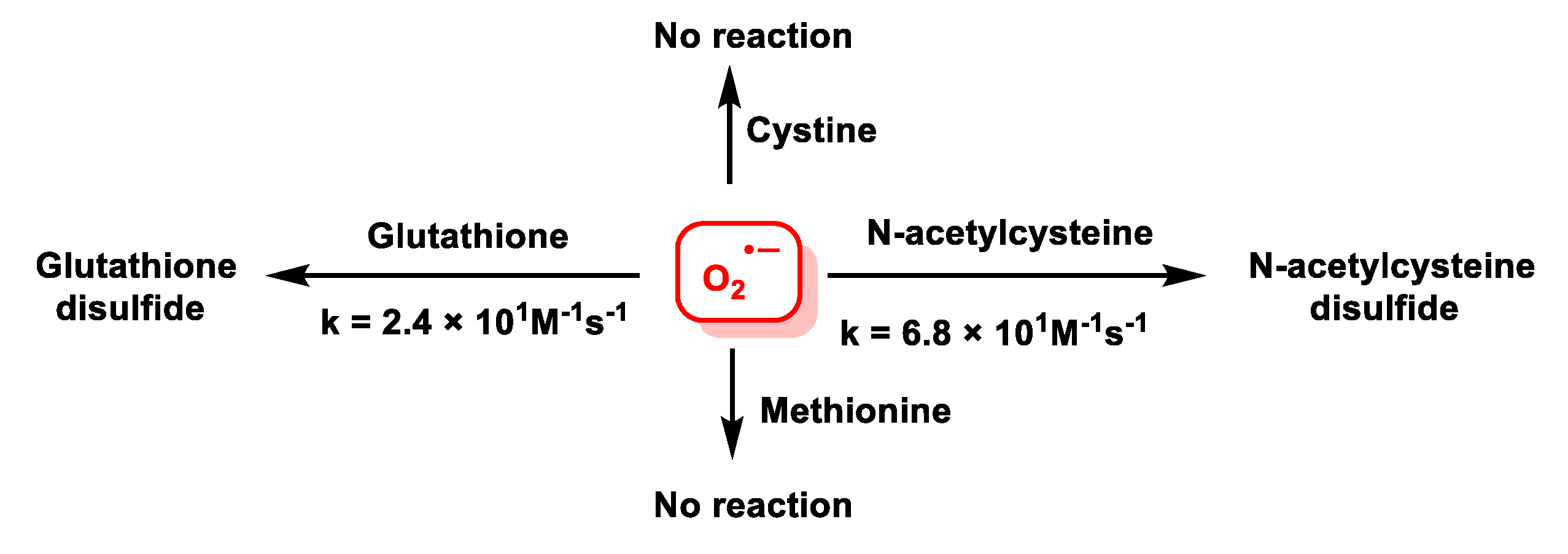
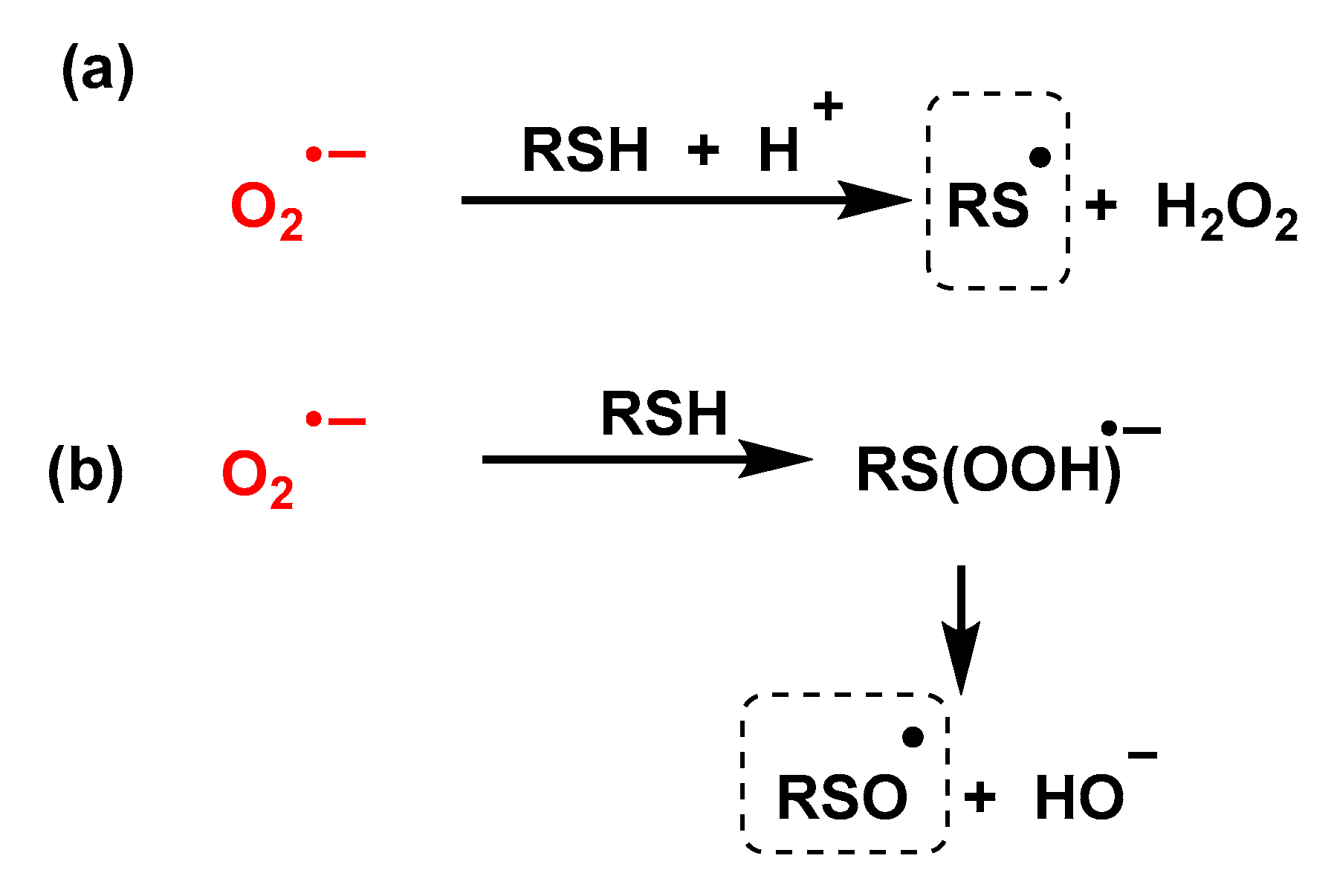
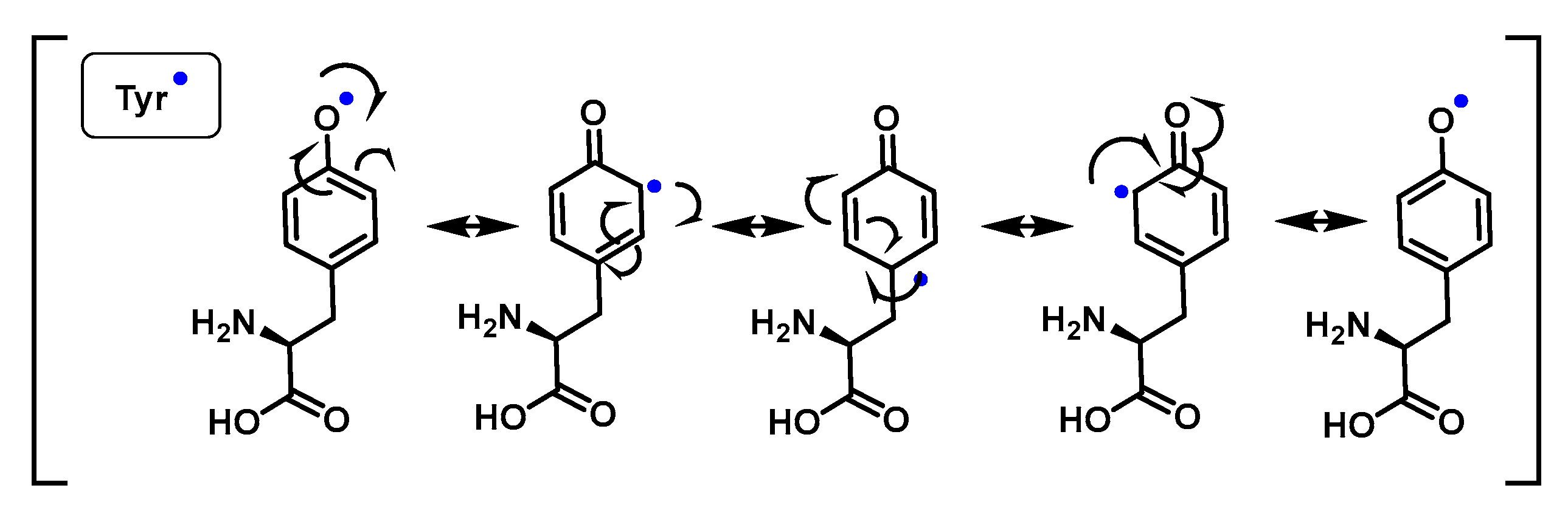
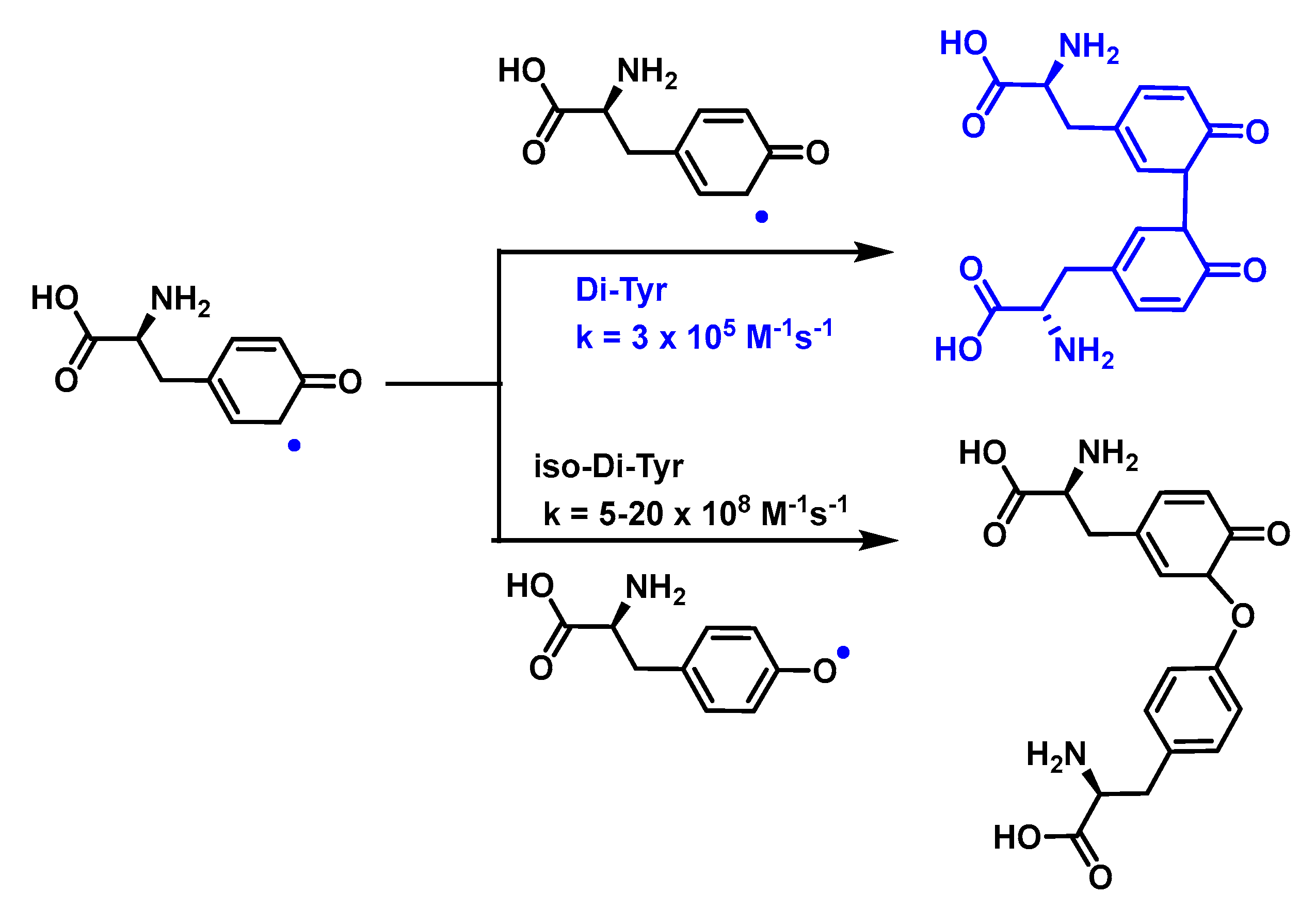
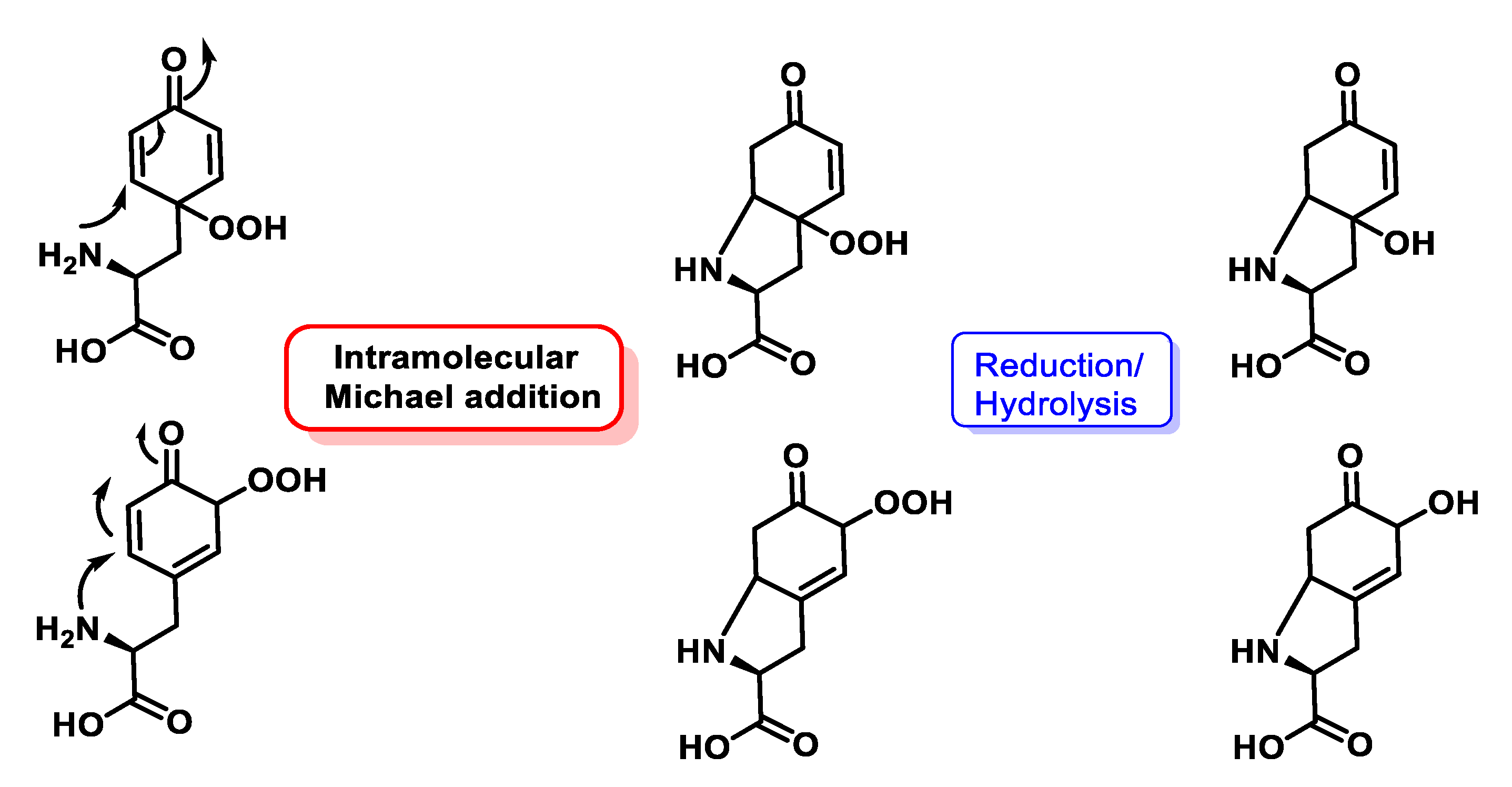
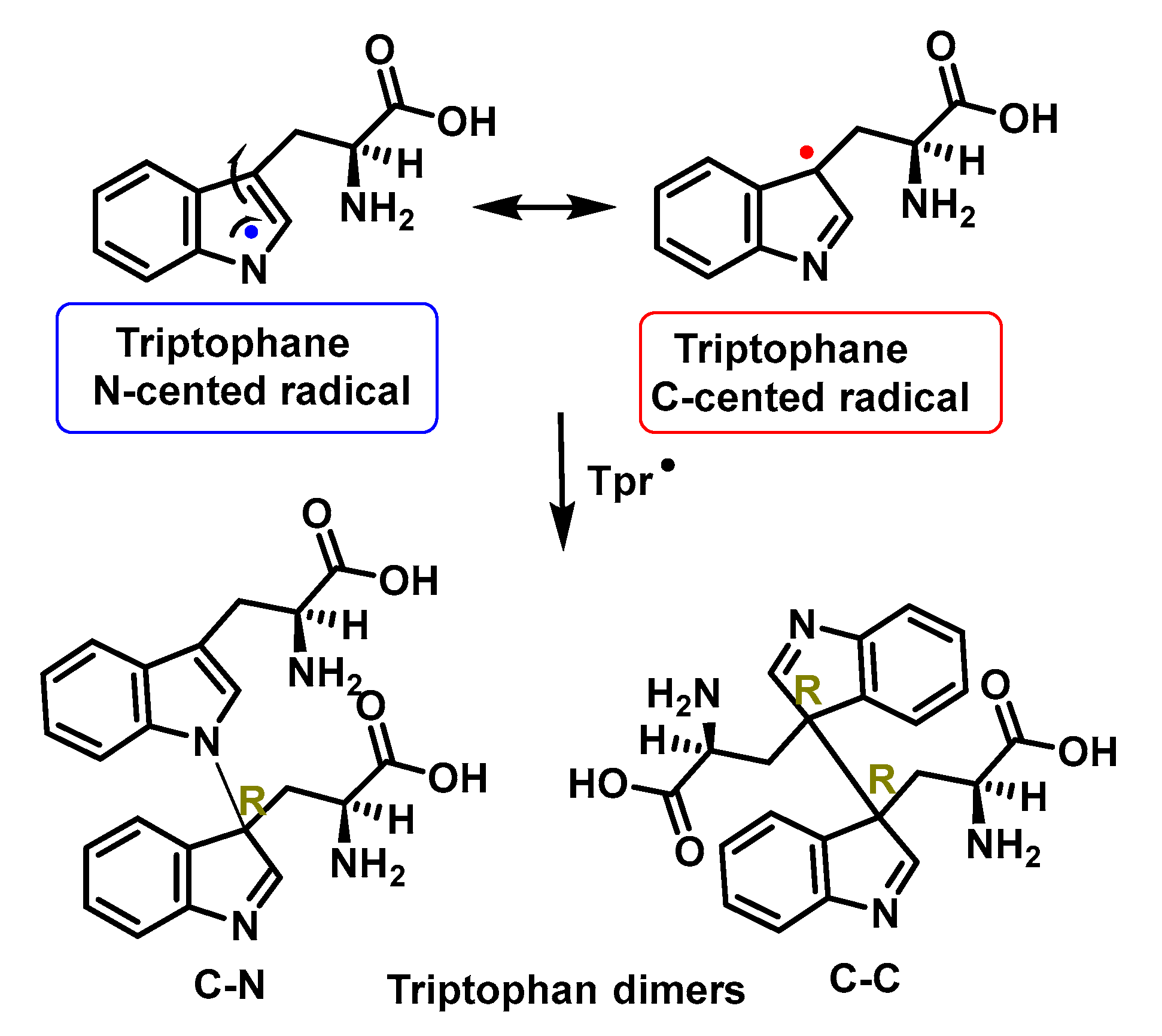
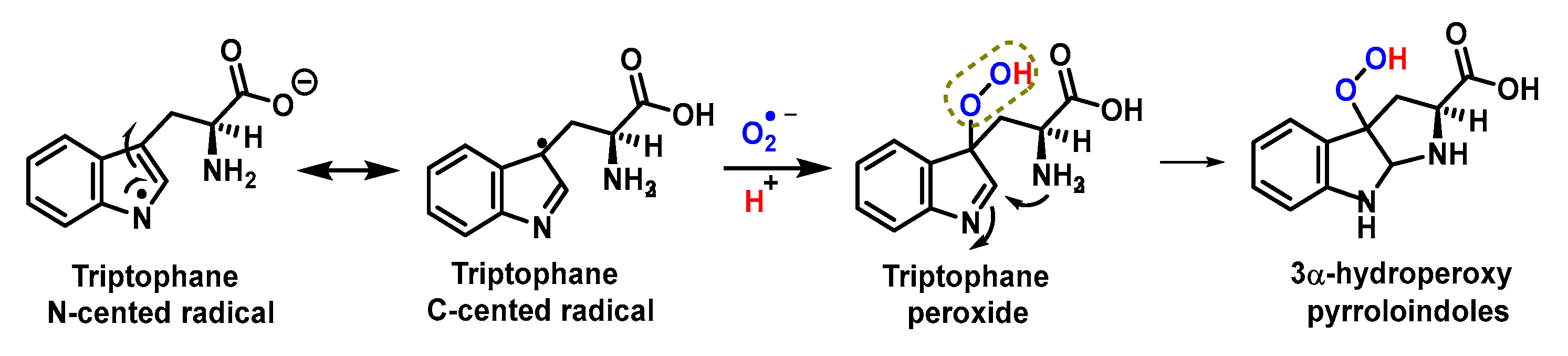
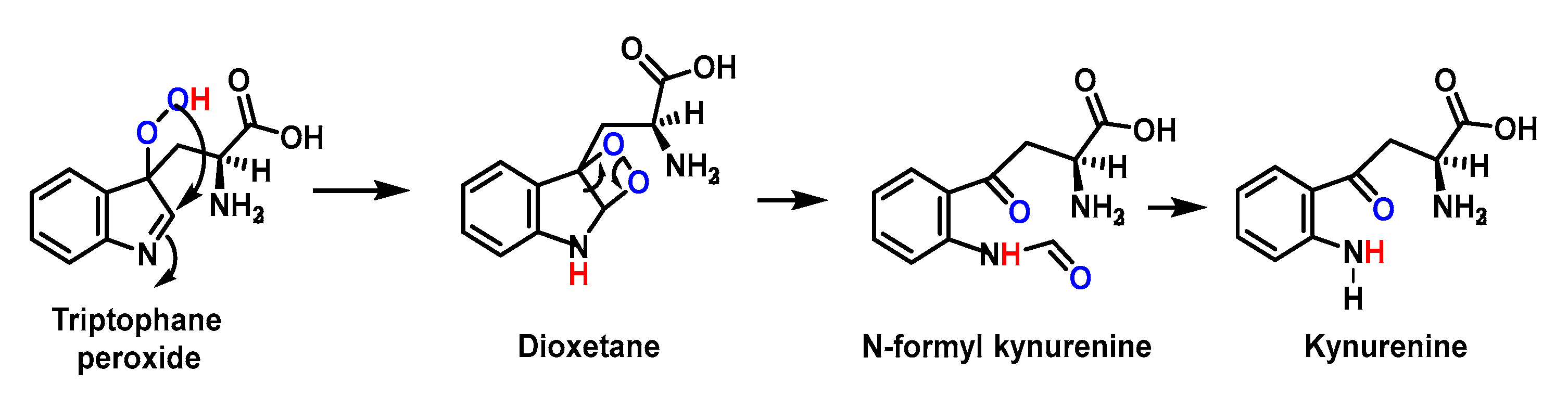
4.1.11. Reactions of Superoxide with Amino Acids
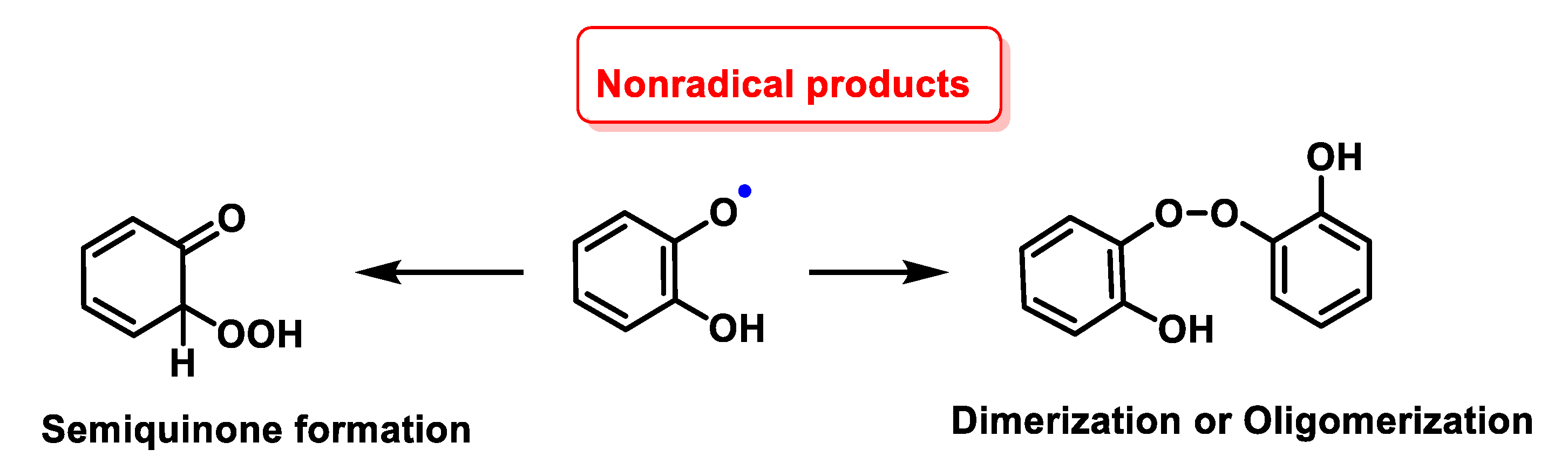
4.1.12. Radical–Radical Reactions of Superoxide
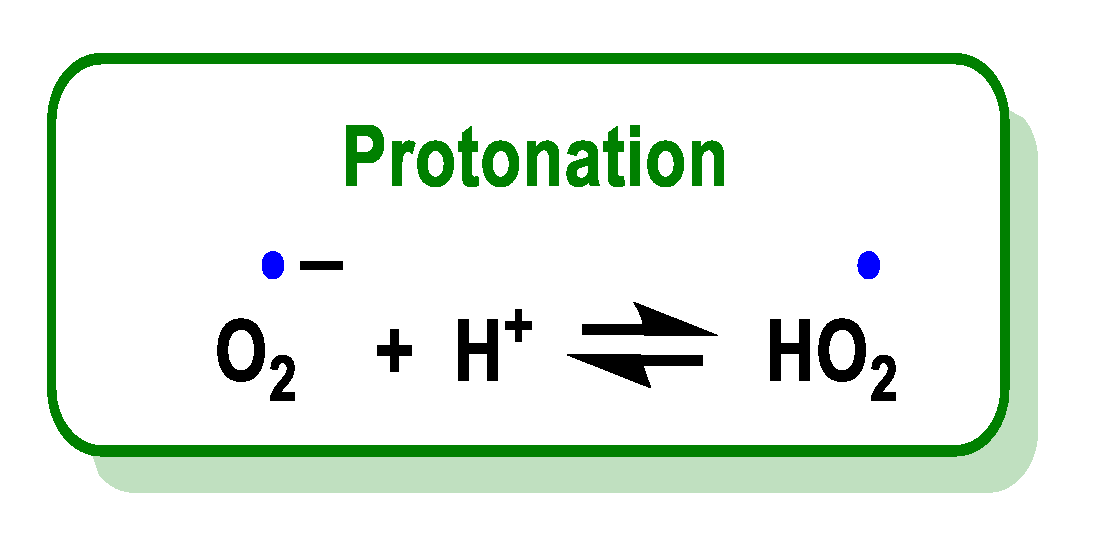
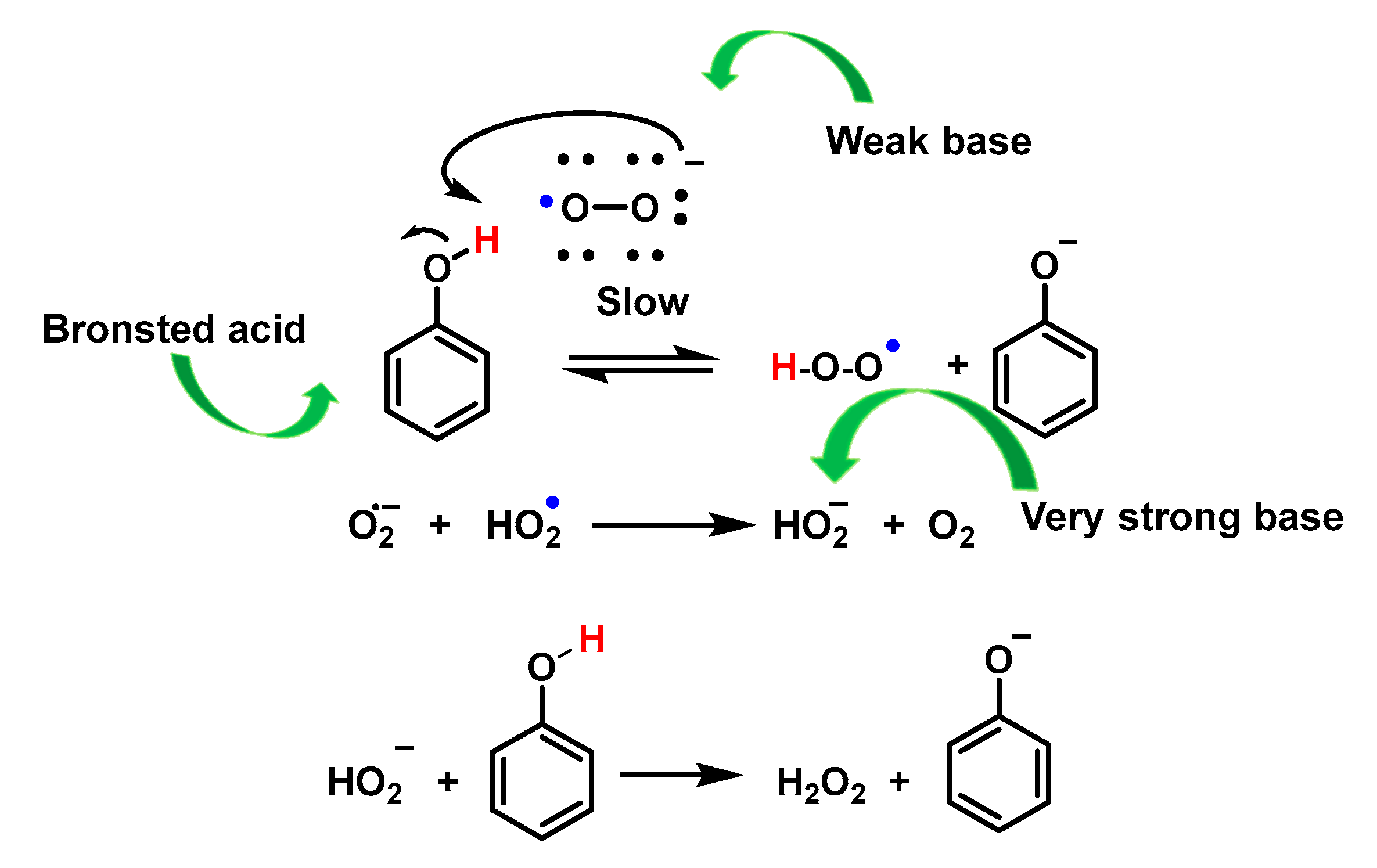
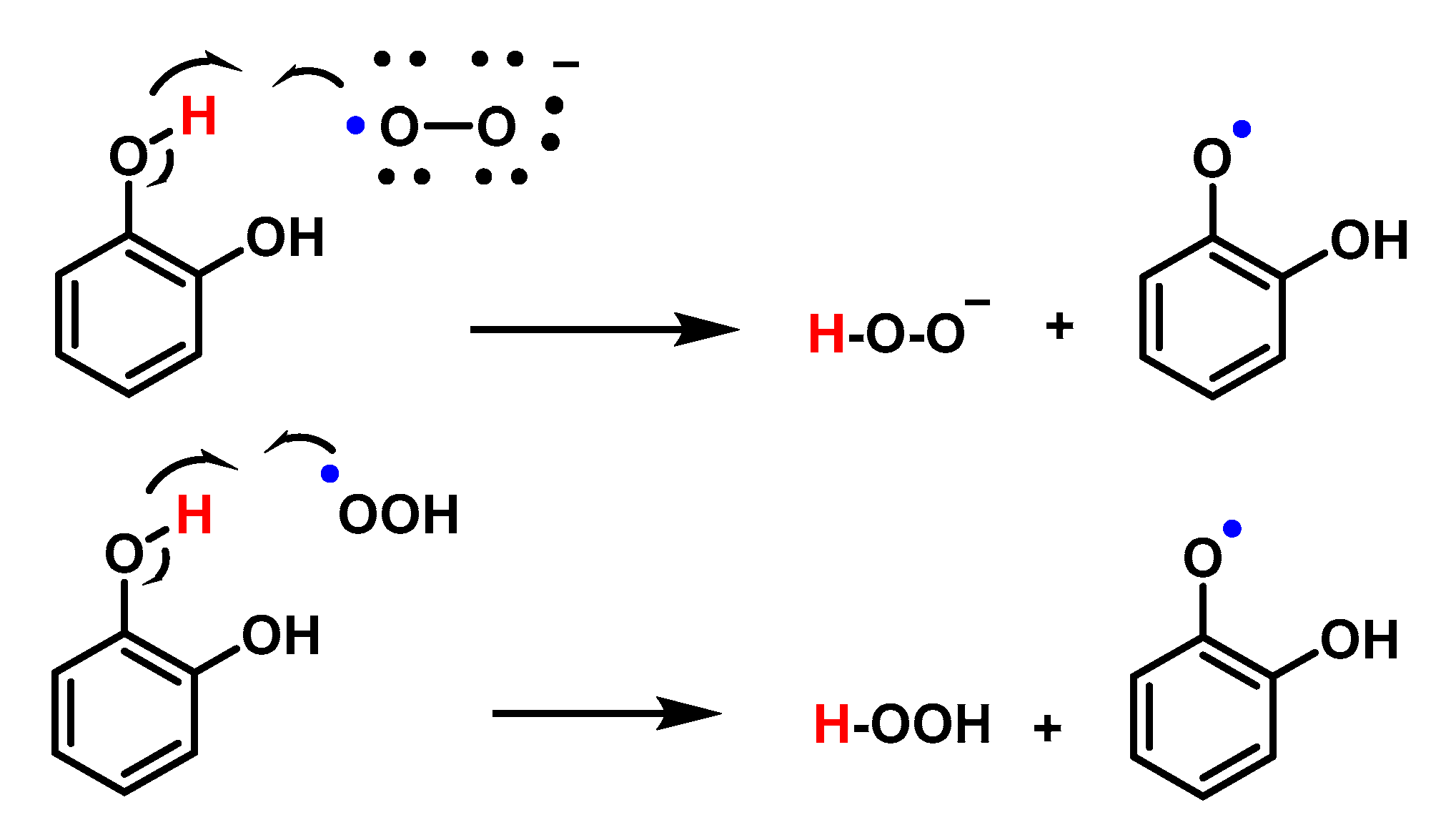
4.1.13. Proton–Radical Transfer
5. Detection of Superoxide Anion
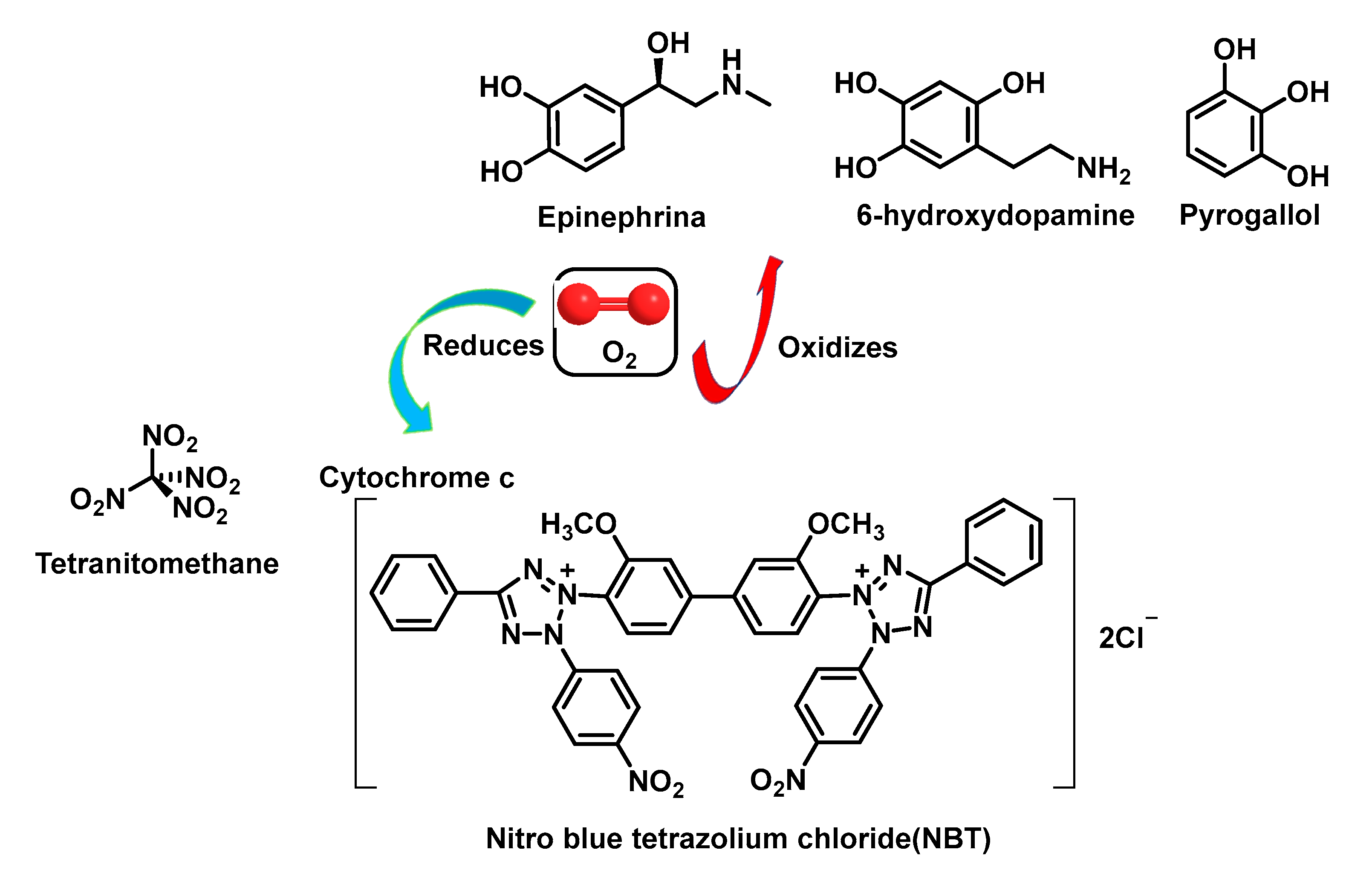
5.1. Detection of Superoxide by Cytochrome C
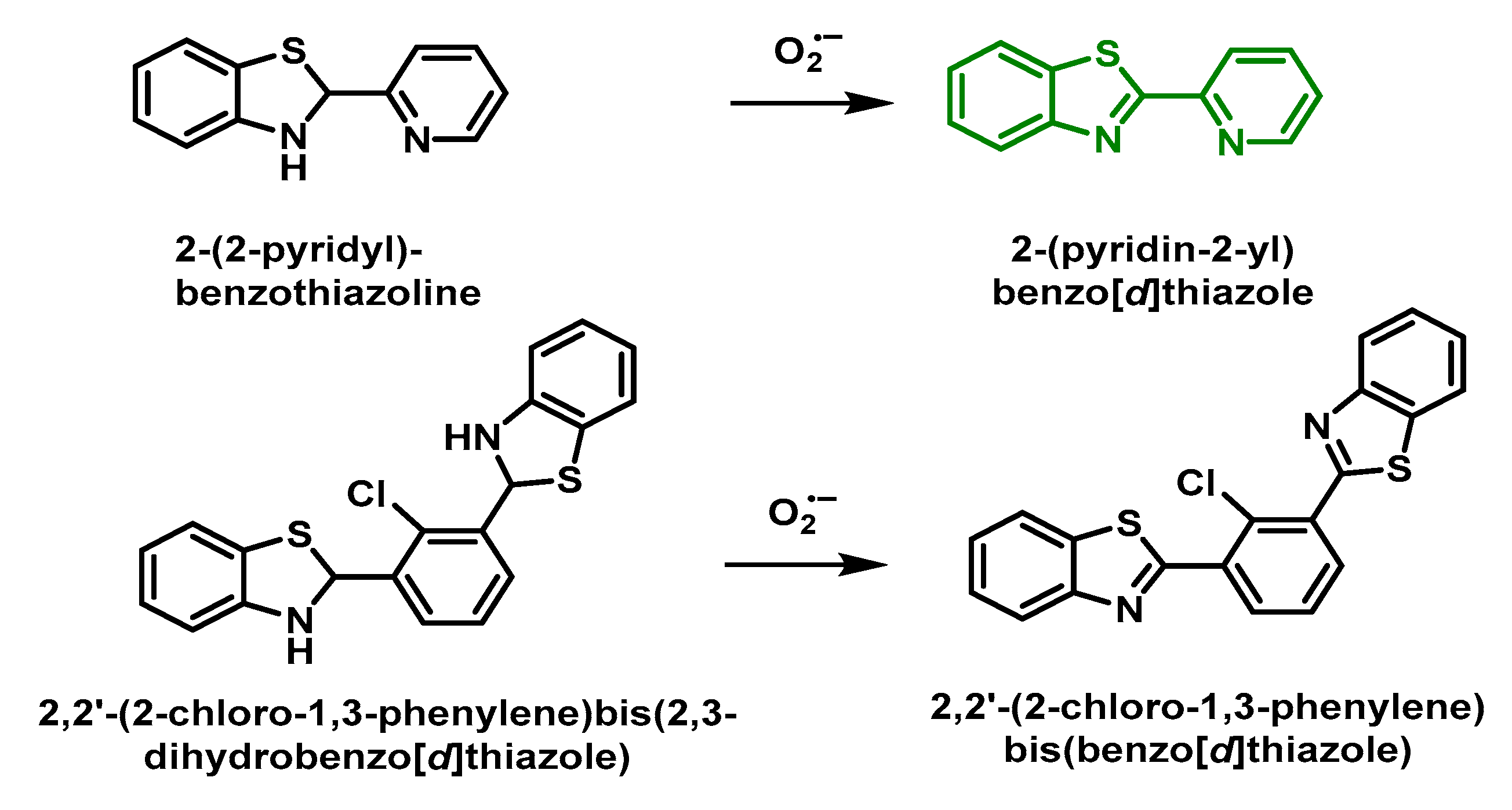
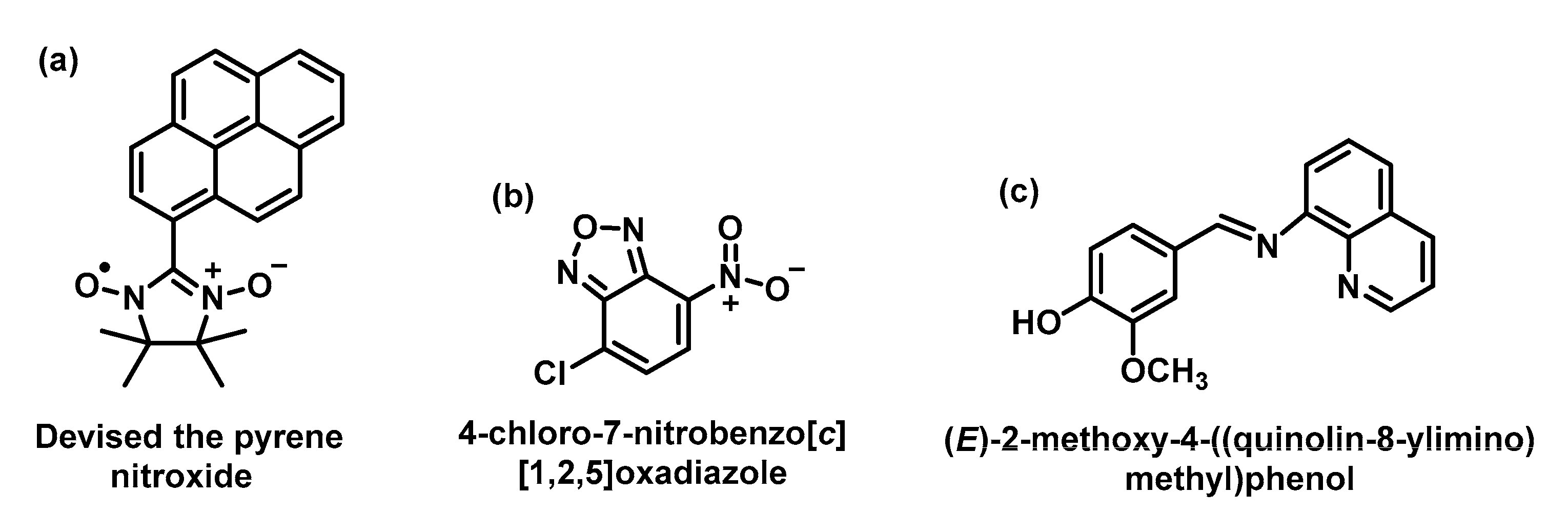
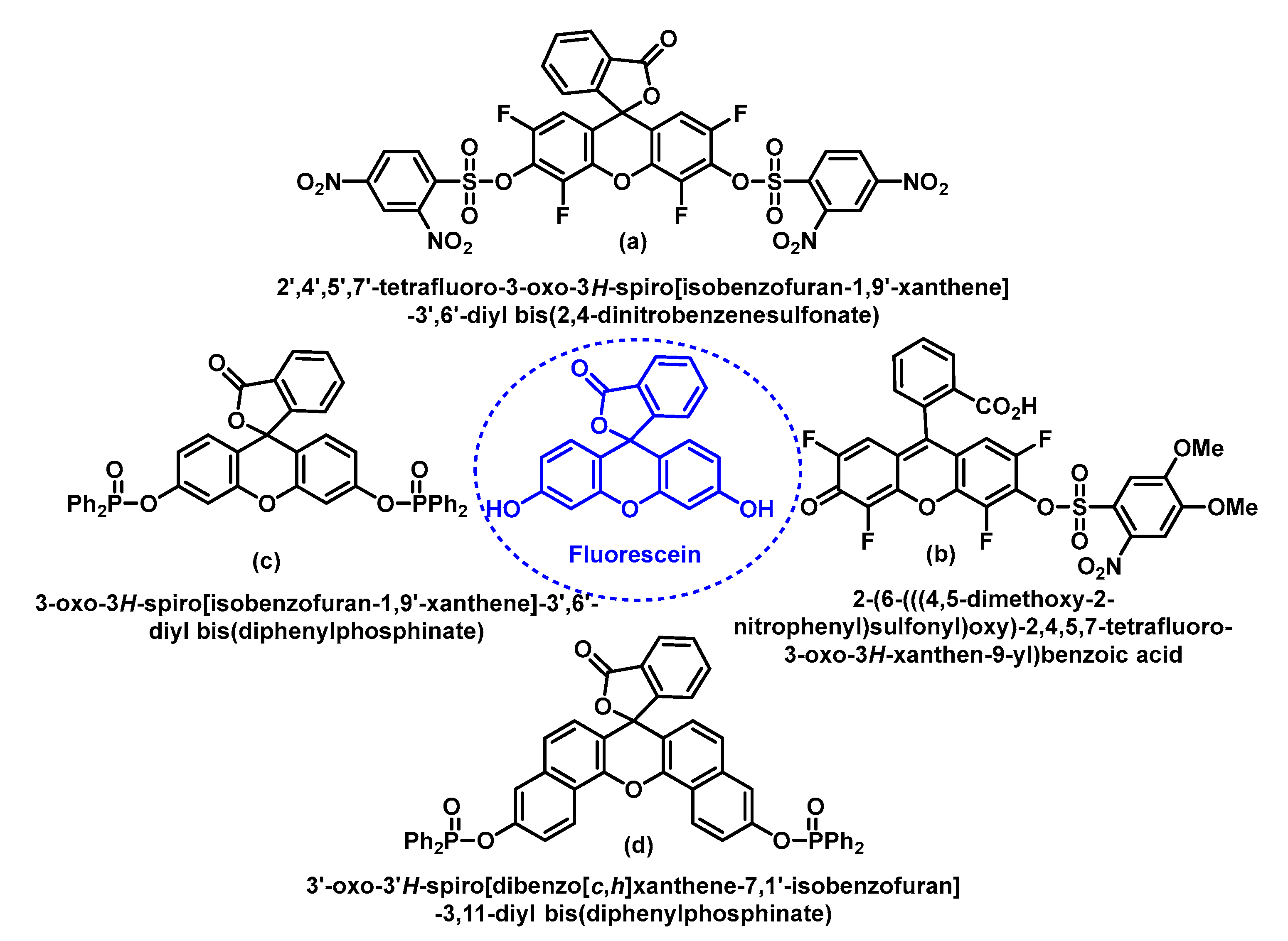
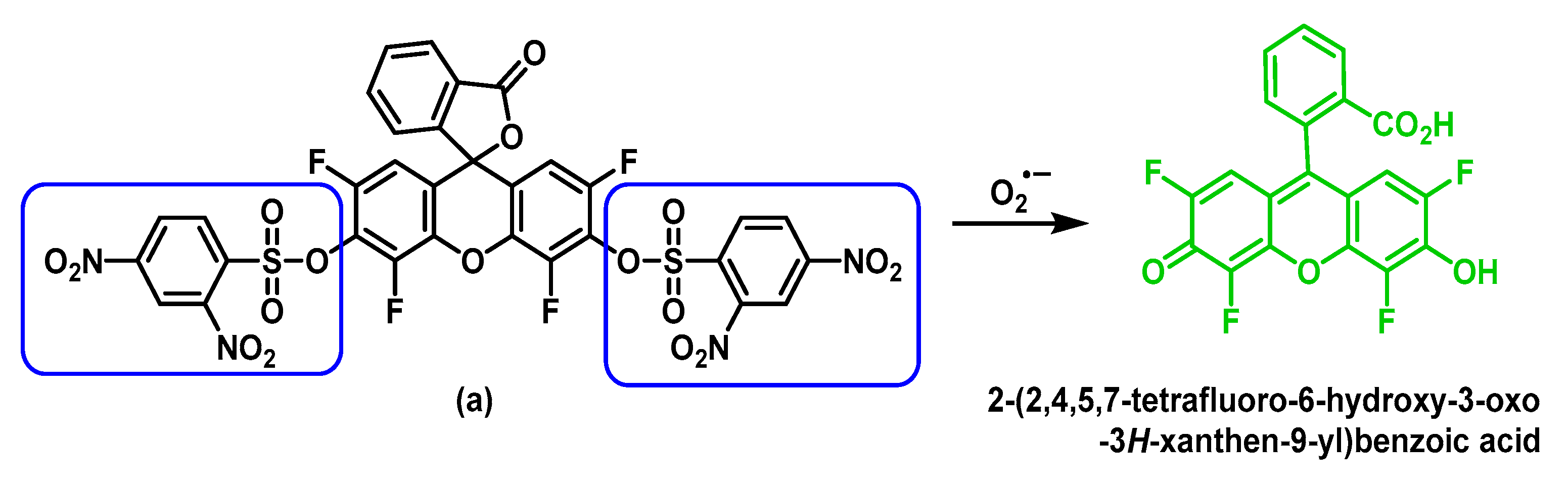
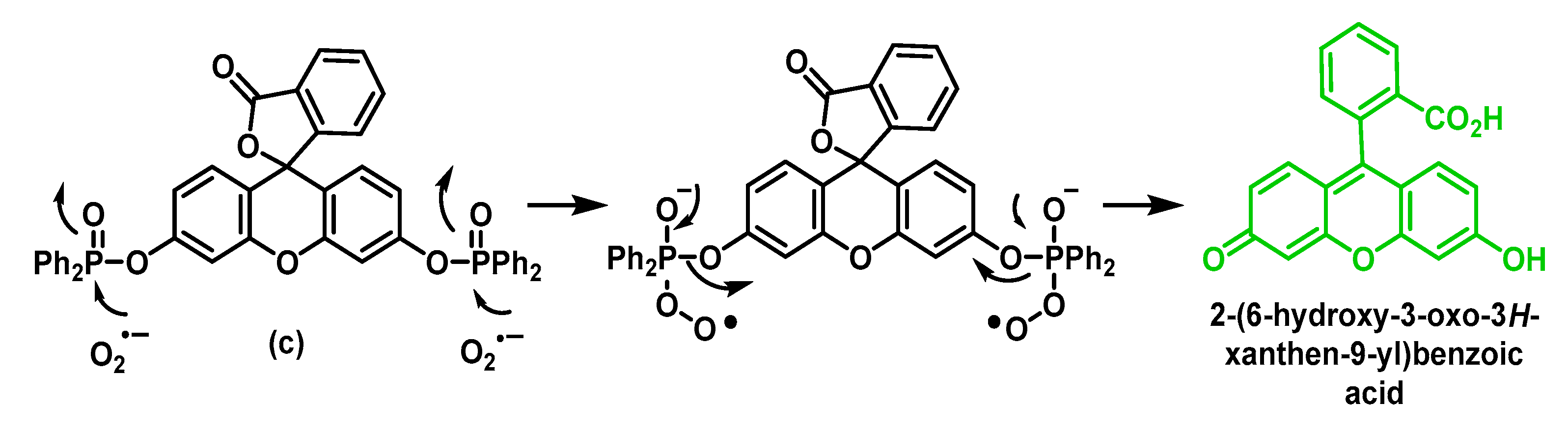
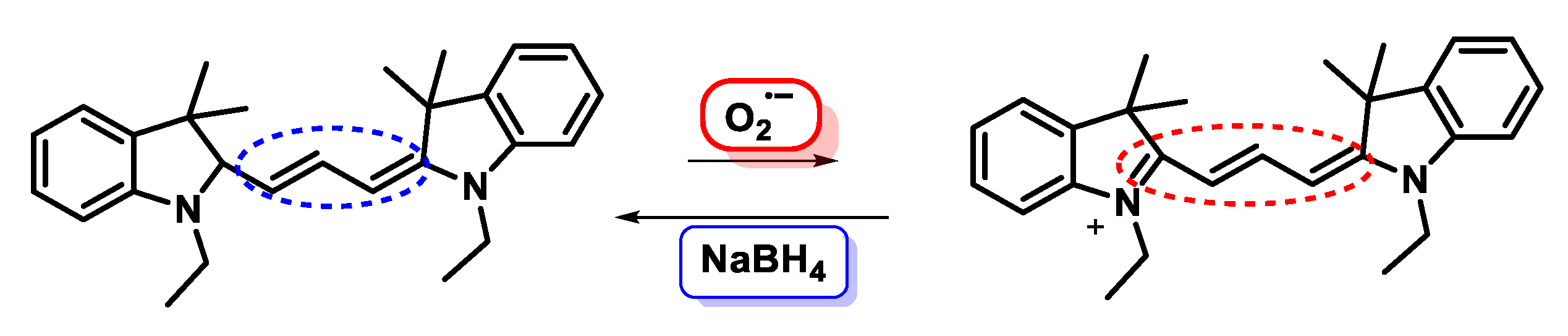
5.2. Fluorescent Probes
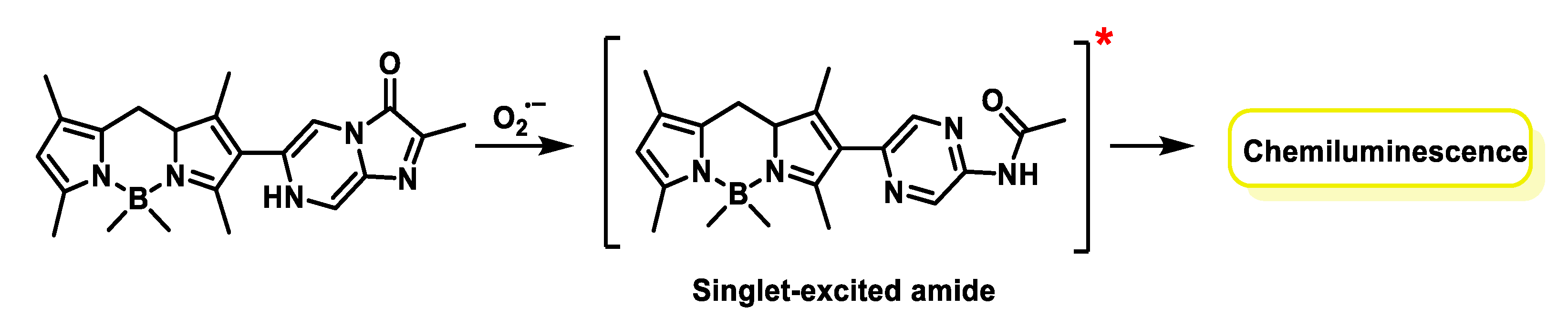
5.3. Chemiluminescent Probes
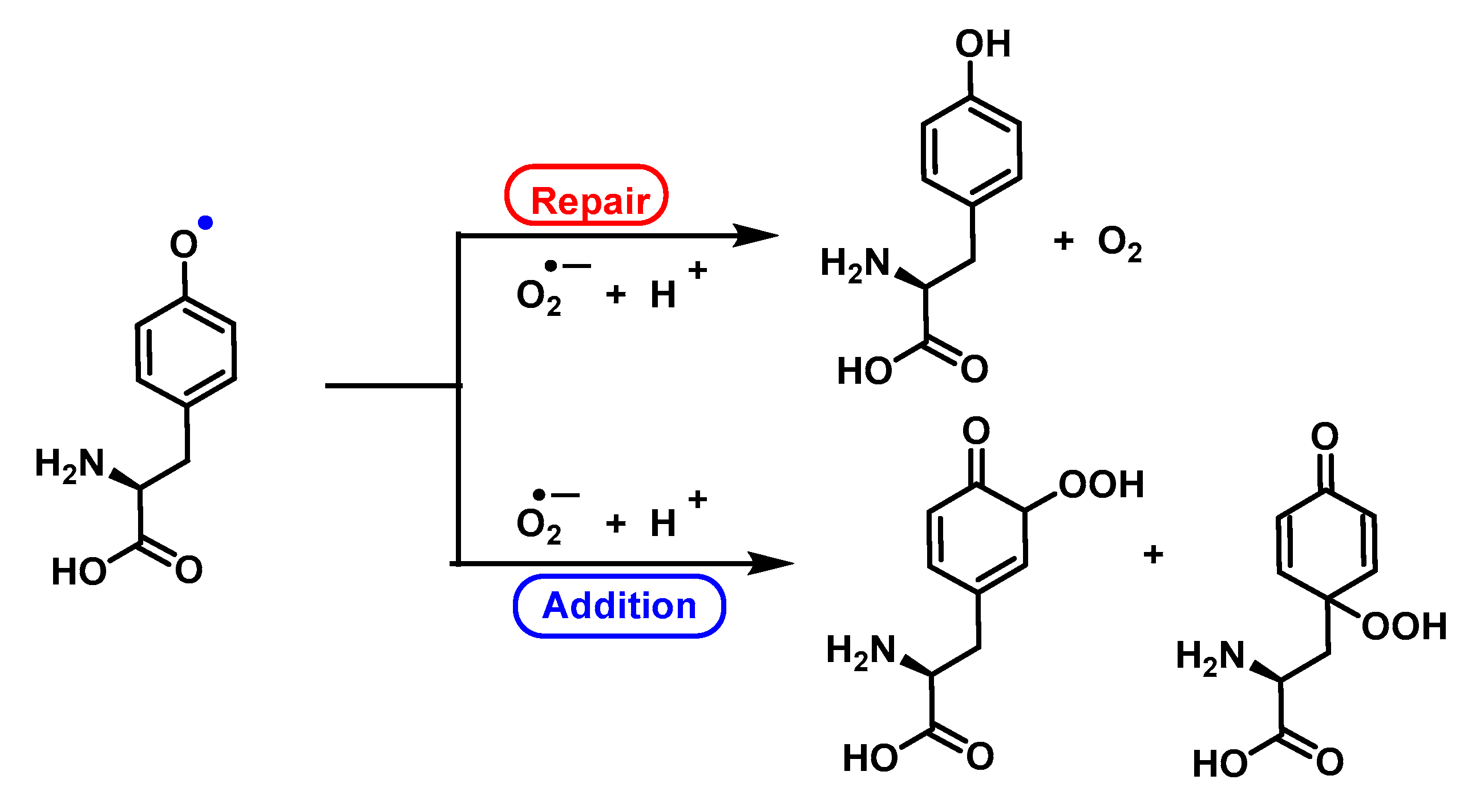
6. Can Superoxide Anion Repair Oxidative Damage?
7. Superoxide Anion in the Antimicrobial Innate Immunity
8. Macrophages, Neutrophils and Superoxide Anion
8.1. Macrophages
8.2. Neutrophils
- (i)
- The production of O2•−. Neutrophils possess the enzyme NOX which, when activated, produces superoxide anion, with strong antimicrobial properties. Superoxide can be released outside the cell, or inside the cell (in the phagosome);
- (ii)
- The coordinated release of proteolytic and antimicrobial granule content. The release of the contents of the primary and secondary granules has important antimicrobial significance. The granules contain MPO, lactoferrin, lysosomes and NGAL [218]. The enzyme myeloperoxidase MPO forms hypochlorous acid HOCl [176] after reaction of chloride anion with hydrogen peroxide. HOCl oxidises tyrosine residues to form the tyrosyl radical [128].
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Corrales, L.C.; Muñoz Ariza, M.M. Estrés oxidativo: Origen, evolución y consecuencias de la toxicidad del oxígeno. Nova 2012, 10, 213–225. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Vona, R.; Pallotta, L.; Cappelletti, M.; Severi, C.; Matarrese, P. The impact of oxidative stress in human pathology: Focus on gastrointestinal disorders. Antioxidants 2021, 10, 201. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, G.; Fasciolo, G.; Venditti, P. The ambiguous aspects of oxygen. Oxygen 2022, 2, 382–409. [Google Scholar] [CrossRef]
- Lozada, S.M.; García, L. Estrés oxidativo y antioxidantes: Cómo mantener el equilibrio. Rev. Asoc. Colomb. Dermatol. Cirugía Derm. 2009, 17, 172–179. [Google Scholar]
- Kohen, R.; Nyska, A. Invited review: Oxidation of biological systems: Oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol. Pathol. 2002, 30, 620–650. [Google Scholar] [CrossRef] [Green Version]
- Hancock, J.T. Oxygen Is Instrumental for Biological Signaling: An Overview. Oxygen 2021, 1, 3–15. [Google Scholar] [CrossRef]
- Turrens, J.F.; Boveris, A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem. J. 1980, 191, 421–427. [Google Scholar] [CrossRef]
- Li, R.; Jia, Z.; Trush, M.A. Defining ROS in biology and medicine. React. Oxyg. Species 2016, 1, 9. [Google Scholar] [CrossRef] [Green Version]
- Storey, K.B. Oxidative stress: Animal adaptations in nature. Braz. J. Med. Biol. Res. 1996, 29, 1715–1733. [Google Scholar]
- Fridovich, I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 1995, 64, 97–112. [Google Scholar] [CrossRef]
- Brglez Mojzer, E.; Knez Hrnčič, M.; Škerget, M.; Knez, Ž.; Bren, U. Polyphenols: Extraction methods, antioxidative action, bioavailability and anticarcinogenic effects. Molecules 2016, 21, 901. [Google Scholar] [CrossRef]
- Williams, R.J.; Spencer, J.P.; Rice-Evans, C. Flavonoids: Antioxidants or signalling molecules? Free Radic. Biol. Med. 2004, 36, 838–849. [Google Scholar] [CrossRef]
- Dong, Z.; Surh, Y.-J. Dietary Modulation of Cell Signaling Pathways; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Wardman, P. Reduction potentials of one-electron couples involving free radicals in aqueous solution. J. Phys. Chem. Ref. Data 1989, 18, 1637–1755. [Google Scholar] [CrossRef] [Green Version]
- Narayana, P.; Suryanarayana, D.; Kevan, L. Electron spin-echo studies of the solvation structure of superoxide ion (O2-) in water. J. Am. Chem. Soc. 1982, 104, 3552–3555. [Google Scholar] [CrossRef]
- Afanas’ ev, I.B. Superoxide Ion: Chemistry and Biological Implications; CRC Press: Boca Raton, FL, USA, 1991; Volume 2. [Google Scholar]
- Aikens, J.; Dix, T. Perhydroxyl radical (HOO.) initiated lipid peroxidation. The role of fatty acid hydroperoxides. J. Biol. Chem. 1991, 266, 15091–15098. [Google Scholar] [CrossRef]
- Bielski, B.; Arudi, R.L.; Sutherland, M.W. A study of the reactivity of HO2/O2-with unsaturated fatty acids. J. Biol. Chem. 1983, 258, 4759–4761. [Google Scholar] [CrossRef]
- Bielski, B.H.; Cabelli, D.E.; Arudi, R.L.; Ross, A.B. Reactivity of HO2/O− 2 radicals in aqueous solution. J. Phys. Chem. Ref. Data 1985, 14, 1041–1100. [Google Scholar] [CrossRef]
- Liochev, S.I.; Fridovich, I. Superoxide and iron: Partners in crime. IUBMB life 1999, 48, 157–161. [Google Scholar] [CrossRef]
- Huie, R.E.; Padmaja, S. The reaction of NO with superoxide. Free. Radic. Res. Commun. 1993, 18, 195–199. [Google Scholar] [CrossRef]
- Turrens, J.F. Superoxide production by the mitochondrial respiratory chain. Biosci. Rep. 1997, 17, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Hayyan, M.; Hashim, M.A.; AlNashef, I.M. Superoxide ion: Generation and chemical implications. Chem. Rev. 2016, 116, 3029–3085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boveris, A.; Cadenas, E.; Stoppani, A. Role of ubiquinone in the mitochondrial generation of hydrogen peroxide. Biochem. J. 1976, 156, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.K.; Lu, J.; Bai, Y. Mitochondrial respiratory complex I: Structure, function and implication in human diseases. Curr. Med. Chem. 2009, 16, 1266–1277. [Google Scholar] [CrossRef] [Green Version]
- Lezza, A.; Boffoli, D.; Scacco, S.; Cantatore, P.; Gadaleta, M. Correlation between mitochondrial DNA 4977-bp deletion and respiratory chain enzyme activities in aging human skeletal muscles. Biochem. Biophys. Res. Commun. 1994, 205, 772–779. [Google Scholar] [CrossRef]
- Babior, B.M. NADPH oxidase. Curr. Opin. Immunol. 2004, 16, 42–47. [Google Scholar] [CrossRef]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Babior, B.M.; Kipnes, R.S.; Curnutte, J.T. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J. Clin. Investig. 1973, 52, 741–744. [Google Scholar] [CrossRef] [Green Version]
- Babior, B.M.; Kipnes, R.S. Superoxide-forming enzyme from human neutrophils: Evidence for a flavin requirement. Blood 1977, 50, 517–524. [Google Scholar] [CrossRef] [Green Version]
- Nordzieke, D.E.; Medraño-Fernandez, I. The plasma membrane: A platform for intra-and intercellular redox signaling. Antioxidants 2018, 7, 168. [Google Scholar] [CrossRef]
- Wolin, M.S. Interactions of oxidants with vascular signaling systems. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1430–1442. [Google Scholar] [CrossRef] [Green Version]
- Genestra, M. Oxyl radicals, redox-sensitive signalling cascades and antioxidants. Cell. Signal. 2007, 19, 1807–1819. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, S.; Khan, M.Y.; Rafi, Z.; Khan, H.; Siddiqui, Z.; Rehman, S.; Shahab, U.; Khan, M.S.; Saeed, M.; Alouffi, S. Oxidation, glycation and glycoxidation—The vicious cycle and lung cancer. Semin. Cancer Biol. 2018, 49, 29–36. [Google Scholar] [CrossRef]
- Mujtaba, S.F.; Masih, A.P.; Alqasmi, I.; Alsulimani, A.; Khan, F.H.; Haque, S. Oxidative-Stress-Induced Cellular Toxicity and Glycoxidation of Biomolecules by Cosmetic Products under Sunlight Exposure. Antioxidants 2021, 10, 1008. [Google Scholar] [CrossRef]
- Bielski, B.H.; Richter, H.W. A study of the superoxide radical chemistry by stopped-flow radiolysis and radiation induced oxygen consumption. J. Am. Chem. Soc. 1977, 99, 3019–3023. [Google Scholar] [CrossRef]
- Zhang, D.; Yan, S.; Song, W. Photochemically induced formation of reactive oxygen species (ROS) from effluent organic matter. Environ. Sci. Technol. 2014, 48, 12645–12653. [Google Scholar] [CrossRef]
- Maurette, M.T.; Oliveros, E.; Infelta, P.P.; Ramsteiner, K.; Braun, A.M. Singlet oxygen and superoxide: Experimental differentiation and analysis. Helv. Chim. Acta 1983, 66, 722–733. [Google Scholar] [CrossRef]
- Yamakoshi, Y.; Sueyoshi, S.; Fukuhara, K.; Miyata, N.; Masumizu, T.; Kohno, M. •OH and O2•- Generation in Aqueous C60 and C70 Solutions by Photoirradiation: An EPR Study. J. Am. Chem. Soc. 1998, 120, 12363–12364. [Google Scholar] [CrossRef]
- Garg, S.; Rose, A.L.; Waite, T.D. Production of reactive oxygen species on photolysis of dilute aqueous quinone solutions. Photochem. Photobiol. 2007, 83, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Salthammer, T.; Fuhrmann, F. Photocatalytic surface reactions on indoor wall paint. Environ. Sci. Technol. 2007, 41, 6573–6578. [Google Scholar] [CrossRef] [PubMed]
- Linsebigler, A.L.; Lu, G.; Yates, J.T., Jr. Photocatalysis on TiO2 surfaces: Principles, mechanisms, and selected results. Chem. Rev. 1995, 95, 735–758. [Google Scholar] [CrossRef]
- Hoffmann, M.R.; Martin, S.T.; Choi, W.; Bahnemann, D.W. Environmental applications of semiconductor photocatalysis. Chem. Rev. 1995, 95, 69–96. [Google Scholar] [CrossRef]
- Thompson, T.L.; Yates, J.T. Surface science studies of the photoactivation of TiO2 new photochemical processes. Chem. Rev. 2006, 106, 4428–4453. [Google Scholar] [CrossRef]
- Goto, H.; Hanada, Y.; Ohno, T.; Matsumura, M. Quantitative analysis of superoxide ion and hydrogen peroxide produced from molecular oxygen on photoirradiated TiO2 particles. J. Catal. 2004, 225, 223–229. [Google Scholar] [CrossRef]
- Ding, X.; Zhao, K.; Zhang, L. Enhanced photocatalytic removal of sodium pentachlorophenate with self-doped Bi2WO6 under visible light by generating more superoxide ions. Environ. Sci. Technol. 2014, 48, 5823–5831. [Google Scholar] [CrossRef]
- Merritt, M.V.; Sawyer, D.T. Electrochemical studies of the reactivity of superoxide ion with several alkyl halides in dimethyl sulfoxide. J. Org. Chem. 1970, 35, 2157–2159. [Google Scholar] [CrossRef]
- Johnson, R.A.; Nidy, E.G.; Merritt, M.V. Superoxide chemistry. Reactions of superoxide with alkyl halides and alkyl sulfonate esters. J. Am. Chem. Soc. 1978, 100, 7960–7966. [Google Scholar] [CrossRef]
- Moorcroft, M.J.; Hahn, C.E.; Compton, R.G. Electrochemical studies of the anaesthetic agent enflurane (2-chloro-1, 1, 2-trifluoroethyl difluoromethyl ether) in the presence of oxygen: Reaction with electrogenerated superoxide. J. Electroanal. Chem. 2003, 541, 117–131. [Google Scholar] [CrossRef]
- McElroy, A.; Hashman, J. Synthesis of tetramethylammonium superoxide. Inorg. Chem. 1964, 3, 1798–1799. [Google Scholar] [CrossRef]
- Peters, J.W.; Foote, C.S. Chemistry of superoxide ion. II. Reaction with hydroperoxides. J. Am. Chem. Soc. 1976, 98, 873–875. [Google Scholar] [CrossRef]
- Chern, C.-I.; DiCosimo, R.; De Jesus, R.; San Filippo, J., Jr. A study of superoxide reactivity. Reaction of potassium superoxide with alkyl halides and tosylates. J. Am. Chem. Soc. 1978, 100, 7317–7327. [Google Scholar] [CrossRef]
- Matsumoto, F.; Okajima, T.; Uesugi, S.; Koura, N.; Ohsaka, T. Electrogeneration of superoxide ion and its mechanism at thiol-modified Au electrodes in alkaline aqueous solution. Electrochemistry 2003, 71, 266–273. [Google Scholar] [CrossRef] [Green Version]
- Vasudevan, D.; Wendt, H. Electroreduction of oxygen in aprotic media. J. Electroanal. Chem. 1995, 392, 69–74. [Google Scholar] [CrossRef]
- Islam, M.M.; Saha, M.S.; Okajima, T.; Ohsaka, T. Current oscillatory phenomena based on electrogenerated superoxide ion at the HMDE in dimethylsulfoxide. J. Electroanal. Chem. 2005, 577, 145–154. [Google Scholar] [CrossRef]
- Ortiz, M.; Nunez-Vergara, L.; Squella, J. Voltammetric determination of the heterogeneous charge transfer rate constant for superoxide formation at a glassy carbon electrode in aprotic medium. J. Electroanal. Chem. 2003, 549, 157–160. [Google Scholar] [CrossRef]
- AlNashef, I.M.; Leonard, M.L.; Matthews, M.A.; Weidner, J.W. Superoxide electrochemistry in an ionic liquid. Ind. Eng. Chem. Res. 2002, 41, 4475–4478. [Google Scholar] [CrossRef]
- Guo, Z.; Lin, X. Kinetic studies of dioxygen and superoxide ion in acetonitrile at gold electrodes using ultrafast cyclic voltammetry. J. Electroanal. Chem. 2005, 576, 95–103. [Google Scholar] [CrossRef]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Imlay, J.A. Cellular defenses against superoxide and hydrogen peroxide. Annu. Rev. Biochem. 2008, 77, 755–776. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, D.T. Oxygen Chemistry; Oxford University Press: Oxford, UK, 1991; Volume 26. [Google Scholar]
- Islam, M.M.; Imase, T.; Okajima, T.; Takahashi, M.; Niikura, Y.; Kawashima, N.; Nakamura, Y.; Ohsaka, T. Stability of superoxide ion in imidazolium cation-based room-temperature ionic liquids. J. Phys. Chem. A 2009, 113, 912–916. [Google Scholar] [CrossRef] [PubMed]
- Huynh, M.H.V.; Meyer, T.J. Proton-coupled electron transfer. Chem. Rev. 2007, 107, 5004–5064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, J.; Narita, S.; Gieseg, S.; Gebicki, S.; Gebicki, J.; Dean, R. Long-lived reactive species on free-radical-damaged proteins. Biochem. J. 1992, 282, 621–624. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Y.; Abreu, I.A.; Cabelli, D.E.; Maroney, M.J.; Miller, A.-F.; Teixeira, M.; Valentine, J.S. Superoxide dismutases and superoxide reductases. Chem. Rev. 2014, 114, 3854–3918. [Google Scholar] [CrossRef]
- Fridovich, I. Superoxide dismutases. Adv. Enzymol. Relat. Areas Mol. Biol. 1986, 58, 61–97. [Google Scholar]
- Robinson, B.H. Human complex I deficiency: Clinical spectrum and involvement of oxygen free radicals in the pathogenicity of the defect. Biochim. Biophys. Acta (BBA) Bioenerg. 1998, 1364, 271–286. [Google Scholar] [CrossRef] [Green Version]
- Weisiger, R.A.; Fridovich, I. Superoxide dismutase: Organelle specificity. J. Biol. Chem. 1973, 248, 3582–3592. [Google Scholar] [CrossRef]
- Marklund, S.L. Extracellular superoxide dismutase and other superoxide dismutase isoenzymes in tissues from nine mammalian species. Biochem. J. 1984, 222, 649–655. [Google Scholar] [CrossRef] [Green Version]
- Cudd, A.; Fridovich, I. Electrostatic interactions in the reaction mechanism of bovine erythrocyte superoxide dismutase. J. Biol. Chem. 1982, 257, 11443–11447. [Google Scholar] [CrossRef]
- Fisher, C.L.; Cabelli, D.E.; Tainer, J.A.; Hallewell, R.A.; Getzoff, E.D. The role of arginine 143 in the electrostatics and mechanism of Cu, Zn superoxide dismutase: Computational and experimental evaluation by mutational analysis. Proteins Struct. Funct. Bioinform. 1994, 19, 24–34. [Google Scholar] [CrossRef]
- Getzoff, E.D.; Tainer, J.A.; Weiner, P.K.; Kollman, P.A.; Richardson, J.S.; Richardson, D.C. Electrostatic recognition between superoxide and copper, zinc superoxide dismutase. Nature 1983, 306, 287–290. [Google Scholar] [CrossRef]
- Getzoff, E.D.; Cabelli, D.E.; Fisher, C.L.; Parge, H.E.; Viezzoli, M.S.; Banci, L.; Hallewell, R.A. Faster superoxide dismutase mutants designed by enhancing electrostatic guidance. Nature 1992, 358, 347–351. [Google Scholar] [CrossRef]
- Ellerby, L.M.; Cabelli, D.E.; Graden, J.A.; Valentine, J.S. Copper–zinc superoxide dismutase: Why not pH-dependent? J. Am. Chem. Soc. 1996, 118, 6556–6561. [Google Scholar] [CrossRef]
- Hough, M.A.; Hasnain, S.S. Structure of fully reduced bovine copper zinc superoxide dismutase at 1.15 Å. Structure 2003, 11, 937–946. [Google Scholar] [CrossRef]
- Borgstahl, G.E.; Parge, H.E.; Hickey, M.J.; Beyer, W.F., Jr.; Hallewell, R.A.; Tainer, J.A. The structure of human mitochondrial manganese superoxide dismutase reveals a novel tetrameric interface of two 4-helix bundles. Cell 1992, 71, 107–118. [Google Scholar] [CrossRef]
- Dennis, R.J.; Micossi, E.; McCarthy, J.; Moe, E.; Gordon, E.J.; Kozielski-Stuhrmann, S.; Leonard, G.A.; McSweeney, S. Structure of the manganese superoxide dismutase from Deinococcus radiodurans in two crystal forms. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 325–329. [Google Scholar] [CrossRef] [Green Version]
- Edwards, R.A.; Baker, H.M.; Whittaker, M.M.; Whittaker, J.W.; Jameson, G.B.; Baker, E.N. Crystal structure of Escherichia coli manganese superoxide dismutase at 2.1-Å resolution. JBIC J. Biol. Inorg. Chem. 1998, 3, 161–171. [Google Scholar] [CrossRef]
- Sheng, Y.; Stich, T.A.; Barnese, K.; Gralla, E.B.; Cascio, D.; Britt, R.D.; Cabelli, D.E.; Valentine, J.S. Comparison of two yeast MnSODs: Mitochondrial Saccharomyces cerevisiae versus cytosolic Candida albicans. J. Am. Chem. Soc. 2011, 133, 20878–20889. [Google Scholar] [CrossRef] [Green Version]
- Grove, L.E.; Brunold, T.C. Second-sphere tuning of the metal ion reduction potentials in iron and manganese superoxide dismutases. Comments Inorg. Chem. 2008, 29, 134–168. [Google Scholar] [CrossRef]
- Vance, C.K.; Miller, A.-F. Spectroscopic comparisons of the pH dependencies of Fe-substituted (Mn) superoxide dismutase and Fe-superoxide dismutase. Biochemistry 1998, 37, 5518–5527. [Google Scholar] [CrossRef] [PubMed]
- Abreu, I.A.; Cabelli, D.E. Superoxide dismutases—A review of the metal-associated mechanistic variations. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2010, 1804, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Lah, M.S.; Dixon, M.M.; Pattridge, K.A.; Stallings, W.C.; Fee, J.A.; Ludwig, M.L. Structure-function in Escherichia coli iron superoxide dismutase: Comparisons with the manganese enzyme from Thermus thermophilus. Biochemistry 1995, 34, 1646–1660. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, F.; McAdam, M.; Fielden, E.; Roberts, P.; Puget, K.; Michelson, M. A pulse-radiolysis study of the catalytic mechanism of the iron-containing superoxide dismutase from Photobacterium leiognathi. Biochem. J. 1977, 161, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Pinto, A.F.; Rodrigues, J.V.; Teixeira, M. Reductive elimination of superoxide: Structure and mechanism of superoxide reductases. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2010, 1804, 285–297. [Google Scholar] [CrossRef]
- Coelho, A.V.; Matias, P.; Fülöp, V.; Thompson, A.; Gonzalez, A.; Carrondo, M.A. Desulfoferrodoxin structure determined by MAD phasing and refinement to 1.9-Å resolution reveals a unique combination of a tetrahedral FeS4 centre with a square pyramidal FeSN4 centre. JBIC J. Biol. Inorg. Chem. 1997, 2, 680–689. [Google Scholar] [CrossRef]
- Coulter, E.D.; Emerson, J.P.; Kurtz, D.M.; Cabelli, D.E. Superoxide Reactivity of Rubredoxin Oxidoreductase (Desulfoferrodoxin) from Desulfovibrio v ulgaris: A Pulse Radiolysis Study. J. Am. Chem. Soc. 2000, 122, 11555–11556. [Google Scholar] [CrossRef]
- Rodrigues, J.V.; Abreu, I.A.; Cabelli, D.; Teixeira, M. Superoxide reduction mechanism of Archaeoglobus fulgidus one-iron superoxide reductase. Biochemistry 2006, 45, 9266–9278. [Google Scholar] [CrossRef]
- Rodrigues, J.V.; Saraiva, L.M.; Abreu, I.A.; Teixeira, M.; Cabelli, D.E. Superoxide reduction by Archaeoglobus fulgidus desulfoferrodoxin: Comparison with neelaredoxin. JBIC J. Biol. Inorg. Chem. 2007, 12, 248–256. [Google Scholar] [CrossRef]
- Rodrigues, J.V.; Victor, B.L.; Huber, H.; Saraiva, L.M.; Soares, C.M.; Cabelli, D.E.; Teixeira, M. Superoxide reduction by Nanoarchaeum equitans neelaredoxin, an enzyme lacking the highly conserved glutamate iron ligand. JBIC J. Biol. Inorg. Chem. 2008, 13, 219–228. [Google Scholar] [CrossRef]
- Mathé, C.; Mattioli, T.A.; Horner, O.; Lombard, M.; Latour, J.-M.; Fontecave, M.; Nivière, V. Identification of Iron (III) Peroxo Species in the Active Site of the Superoxide Reductase SOR from Desulfoarculus b aarsii. J. Am. Chem. Soc. 2002, 124, 4966–4967. [Google Scholar] [CrossRef]
- Lombard, M.; Houée-Levin, C.; Touati, D.; Fontecave, M.; Nivière, V. Superoxide reductase from Desulfoarculus baarsii: Reaction mechanism and role of glutamate 47 and lysine 48 in catalysis. Biochemistry 2001, 40, 5032–5040. [Google Scholar] [CrossRef]
- Nivière, V.; Asso, M.; Weill, C.O.; Lombard, M.; Guigliarelli, B.; Favaudon, V.; Houée-Levin, C. Superoxide reductase from Desulfoarculus baarsii: Identification of protonation steps in the enzymatic mechanism. Biochemistry 2004, 43, 808–818. [Google Scholar] [CrossRef]
- Chen, L.; Sharma, P.; Le Gall, J.; Mariano, A.M.; Teixeira, M.; Xavier, A.V. A blue non-heme iron protein from Desulfovibrio gigas. Eur. J. Biochem. 1994, 226, 613–618. [Google Scholar] [CrossRef]
- Emerson, J.P.; Coulter, E.D.; Cabelli, D.E.; Phillips, R.S.; Kurtz, D.M. Kinetics and mechanism of superoxide reduction by two-iron superoxide reductase from Desulfovibrio vulgaris. Biochemistry 2002, 41, 4348–4357. [Google Scholar] [CrossRef]
- Beauchamp, C.; Fridovich, I. Superoxide dismutase: Improved assays and an assay applicable to acrylamide gels. Anal. Biochem. 1971, 44, 276–287. [Google Scholar] [CrossRef]
- Misra, H.P.; Fridovich, I. The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J. Biol. Chem. 1972, 247, 3170–3175. [Google Scholar] [CrossRef]
- Greenstock, C.; Miller, R. The oxidation of tiron by superoxide anion. Kinetics of the reaction in aqueous solution and in chloroplasts. Biochim. Biophys. Acta (BBA) Bioenerg. 1975, 396, 11–16. [Google Scholar] [CrossRef]
- Islam, M.N.; Rauf, A.; Fahad, F.I.; Emran, T.B.; Mitra, S.; Olatunde, A.; Shariati, M.A.; Rebezov, M.; Rengasamy, K.R.; Mubarak, M.S. Superoxide dismutase: An updated review on its health benefits and industrial applications. Crit. Rev. Food Sci. Nutr. 2022, 62, 7282–7300. [Google Scholar] [CrossRef]
- Masayasu, M.; Hiroshi, Y. A simplified assay method of superoxide dismutase activity for clinical use. Clin. Chim. Acta 1979, 92, 337–342. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Fridovich, I. Superoxide dismutase: An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef] [PubMed]
- Ukeda, H.; Kawana, D.; Maeda, S.; Sawamura, M. Spectrophotometric Assay for Superoxide Dismutase Based on the Reduction of Highly Water-soluble Tetrazolium Salts by Xanthine-Xanthine Oxidase. Biosci. Biotechnol. Biochem. 1999, 63, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Flint, D.H.; Tuminello, J.; Emptage, M. The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J. Biol. Chem. 1993, 268, 22369–22376. [Google Scholar] [CrossRef] [PubMed]
- Castro, L.A.; Robalinho, R.L.; Cayota, A.; Meneghini, R.; Radi, R. Nitric oxide and peroxynitrite-dependent aconitase inactivation and iron-regulatory protein-1 activation in mammalian fibroblasts. Arch. Biochem. Biophys. 1998, 359, 215–224. [Google Scholar] [CrossRef]
- Drugge, U.; Holmberg, M.; Holmgren, G.; Almay, B.; Linderholm, H. Hereditary myopathy with lactic acidosis, succinate dehydrogenase and aconitase deficiency in northern Sweden: A genealogical study. J. Med. Genet. 1995, 32, 344–347. [Google Scholar] [CrossRef] [Green Version]
- Sadat, R.; Barca, E.; Masand, R.; Donti, T.R.; Naini, A.; Darryl, C.; DiMauro, S.; Hanchard, N.A.; Graham, B.H. Functional cellular analyses reveal energy metabolism defect and mitochondrial DNA depletion in a case of mitochondrial aconitase deficiency. Mol. Genet. Metab. 2016, 118, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Beinert, H.; Kennedy, M.C.; Stout, C.D. Aconitase as iron–sulfur protein, enzyme, and iron-regulatory protein. Chem. Rev. 1996, 96, 2335–2374. [Google Scholar] [CrossRef]
- Englard, S.; Colowick, S.P. On the mechanism of the aconitase and isocitric dehydrogenase reactions. J. Biol. Chem. 1957, 226, 1047–1058. [Google Scholar] [CrossRef]
- Flint, D.H.; Allen, R.M. Iron–sulfur proteins with nonredox functions. Chem. Rev. 1996, 96, 2315–2334. [Google Scholar] [CrossRef]
- Srinivasan, C.; Liba, A.; Imlay, J.A.; Valentine, J.S.; Gralla, E.B. Yeast lacking superoxide dismutase (s) show elevated levels of “free iron” as measured by whole cell electron paramagnetic resonance. J. Biol. Chem. 2000, 275, 29187–29192. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, J.O.; Weitzberg, E.; Gladwin, M.T. The nitrate–nitrite–nitric oxide pathway in physiology and therapeutics. Nat. Rev. Drug Discov. 2008, 7, 156–167. [Google Scholar] [CrossRef]
- Tejero, J.; Shiva, S.; Gladwin, M.T. Sources of vascular nitric oxide and reactive oxygen species and their regulation. Physiol. Rev. 2019, 99, 311–379. [Google Scholar] [CrossRef]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, S.; Czapski, G.; Lind, J.; Merényi, G. Tyrosine Nitration by Simultaneous Generation of⋅ NO and O⨪ 2 under Physiological Conditions: HOW THE RADICALS DO THE JOB. J. Biol. Chem. 2000, 275, 3031–3036. [Google Scholar] [CrossRef] [Green Version]
- Pérez de la Lastra, J.M.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. The Nitration of Proteins, Lipids and DNA by Peroxynitrite Derivatives-Chemistry Involved and Biological Relevance. Stresses 2022, 2, 53–64. [Google Scholar] [CrossRef]
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA 1990, 87, 1620–1624. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Sueta, G.; Radi, R. Chemical biology of peroxynitrite: Kinetics, diffusion, and radicals. ACS Chem. Biol. 2009, 4, 161–177. [Google Scholar] [CrossRef]
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The chemistry of reactive oxygen species (ROS) revisited: Outlining their role in biological macromolecules (DNA, lipids and proteins) and induced pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Squadrito, G.L.; Pryor, W.A. The formation of peroxynitrite in vivo from nitric oxide and superoxide. Chem.-Biol. Interact. 1995, 96, 203–206. [Google Scholar] [CrossRef]
- Packer, M.A.; Porteous, C.M.; Murphy, M.P. Superoxide production by mitochondria in the presence of nitric oxide forms peroxynitrite. IUBMB Life 1996, 40, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Uchida, K.; Kawakishi, S. Oxidative fragmentation of collagen and prolyl peptide by Cu (II)/H2O2. Conversion of proline residue to 2-pyrrolidone. J. Biol. Chem. 1992, 267, 23646–23651. [Google Scholar] [CrossRef] [PubMed]
- Pryor, W.A.; Squadrito, G.L. The chemistry of peroxynitrite: A product from the reaction of nitric oxide with superoxide. Am. J. Physiol.-Lung Cell. Mol. Physiol. 1995, 268, L699–L722. [Google Scholar] [CrossRef] [PubMed]
- Berlett, B.; Friguet, B.; Yim, M.; Chock, P.; Stadtman, E. Peroxynitrite-mediated nitration of tyrosine residues in Escherichia coli glutamine synthetase mimics adenylylation: Relevance to signal transduction. Proc. Natl. Acad. Sci. USA 1996, 93, 1776–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrés, C.M.C.; Pérez de la Lastra, J.M.; Andrés Juan, C.; Plou, F.J.; Pérez-Lebeña, E. Impact of Reactive Species on Amino Acids—Biological Relevance in Proteins and Induced Pathologies. Int. J. Mol. Sci. 2022, 23, 14049. [Google Scholar] [CrossRef]
- Davico, G.E.; Bierbaum, V.M. Reactivity and secondary kinetic isotope effects in the SN2 reaction mechanism: Dioxygen radical anion and related nucleophiles. J. Am. Chem. Soc. 2000, 122, 1740–1748. [Google Scholar] [CrossRef]
- San Filippo, J., Jr.; Chern, C.-I.; Valentine, J.S. Reaction of superoxide with alkyl halides and tosylates. J. Org. Chem. 1975, 40, 1678–1680. [Google Scholar] [CrossRef]
- Bielski, B.H.; Shiue, G.G. Reaction rates of superoxide radicals with the essential amino acids. Oxyg. Free. Radic. Tissue Damage 1979, 43–56. [Google Scholar] [CrossRef]
- Benrahmoune, M.; Thérond, P.; Abedinzadeh, Z. The reaction of superoxide radical with N-acetylcysteine. Free Radic. Biol. Med. 2000, 29, 775–782. [Google Scholar] [CrossRef]
- Zhang, N.; Schuchmann, H.P.; Von Sonntag, C. The reaction of superoxide radical anion with dithiothreitol: A chain process. J. Phys. Chem. 1991, 95, 4718–4722. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Metodiewa, D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 1999, 27, 322–328. [Google Scholar] [CrossRef]
- Dikalov, S.; Khramtsov, V.; Zimmer, G. Determination of rate constants of the reactions of thiols with superoxide radical by electron paramagnetic resonance: Critical remarks on spectrophotometric approaches. Arch. Biochem. Biophys. 1996, 326, 207–218. [Google Scholar] [CrossRef]
- Jones, C.; Lawrence, A.; Wardman, P.; Burkitt, M. Kinetics of superoxide scavenging by glutathione: An evaluation of its role in the removal of mitochondrial superoxide. Biochem. Soc. Trans. 2003, 31, 1337–1339. [Google Scholar] [CrossRef]
- Feroci, G.; Fini, A. Voltammetric investigation of the interactions between superoxide ion and some sulfur amino acids. Inorg. Chim. Acta 2007, 360, 1023–1031. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Kettle, A.J. Radical–radical reactions of superoxide: A potential route to toxicity. Biochem. Biophys. Res. Commun. 2003, 305, 729–736. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Parsons-Mair, H.N.; Gebicki, S.; Gebicki, J.M.; Davies, M.J. Requirements for superoxide-dependent tyrosine hydroperoxide formation in peptides. Biochem. J. 2004, 381, 241–248. [Google Scholar] [CrossRef] [Green Version]
- Möller, M.a.N.; Hatch, D.M.; Kim, H.-Y.H.; Porter, N.A. Superoxide reaction with tyrosyl radicals generates para-hydroperoxy and para-hydroxy derivatives of tyrosine. J. Am. Chem. Soc. 2012, 134, 16773–16780. [Google Scholar] [CrossRef]
- Hunter, E.P.; Desrosiers, M.F.; Simic, M.G. The effect of oxygen, antioxidants, and superoxide radical on tyrosine phenoxyl radical dimerization. Free Radic. Biol. Med. 1989, 6, 581–585. [Google Scholar] [CrossRef]
- Carroll, L.; Pattison, D.I.; Davies, J.B.; Anderson, R.F.; Lopez-Alarcon, C.; Davies, M.J. Formation and detection of oxidant-generated tryptophan dimers in peptides and proteins. Free Radic. Biol. Med. 2017, 113, 132–142. [Google Scholar] [CrossRef]
- Sormacheva, E.D.; Sherin, P.S.; Tsentalovich, Y.P. Dimerization and oxidation of tryptophan in UV-A photolysis sensitized by kynurenic acid. Free Radic. Biol. Med. 2017, 113, 372–384. [Google Scholar] [CrossRef]
- Carroll, L.; Pattison, D.I.; Davies, J.B.; Anderson, R.F.; Lopez-Alarcon, C.; Davies, M.J. Superoxide radicals react with peptide-derived tryptophan radicals with very high rate constants to give hydroperoxides as major products. Free Radic. Biol. Med. 2018, 118, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Ehrenshaft, M.; Deterding, L.J.; Mason, R.P. Tripping up Trp: Modification of protein tryptophan residues by reactive oxygen species, modes of detection, and biological consequences. Free Radic. Biol. Med. 2015, 89, 220–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, X.; Jin, F.; Jin, H.; von Sonntag, C. Reaction of the superoxide radical with the N-centered radical derived from N-acetyltryptophan methyl ester. J. Chem. Soc. Perkin Trans. 2 1998, 259–264. [Google Scholar] [CrossRef]
- Wrona, M.Z.; Dryhurst, G. Oxidation of serotonin by superoxide radical: Implications to neurodegenerative brain disorders. Chem. Res. Toxicol. 1998, 11, 639–650. [Google Scholar] [CrossRef]
- de Oliveira Silva, S.; Ximenes, V.F.; Catalani, L.H.; Campa, A. Myeloperoxidase-catalyzed oxidation of melatonin by activated neutrophils. Biochem. Biophys. Res. Commun. 2000, 279, 657–662. [Google Scholar] [CrossRef]
- Santus, R.; Patterson, L.K.; Bazin, M. The diffusion-controlled reaction of semioxidized tryptophan with the superoxide radical anion. Free Radic. Biol. Med. 1995, 19, 837–842. [Google Scholar] [CrossRef]
- Santus, R.; Patterson, L.; Bazin, M.; Maziere, J.; Morliere, P. Intra and intermolecular charge effects on the reaction of the superoxide radical anion with semi-oxidized tryptophan in peptides and N-acetyl tryptophan. Free Radic. Res. 1998, 29, 409–419. [Google Scholar] [CrossRef]
- Santus, R.; Patterson, L.K.; Hug, G.L.; Bazin, M.; Mazière, J.-C.; Morlière, P. Interactions of superoxide anion with enzyme radicals: Kinetics of reaction with lysozyme tryptophan radicals and corresponding effects on tyrosine electron transfer. Free Radic. Res. 2000, 33, 383–391. [Google Scholar] [CrossRef]
- Zhao, H.; Kalivendi, S.; Zhang, H.; Joseph, J.; Nithipatikom, K.; Vásquez-Vivar, J.; Kalyanaraman, B. Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: Potential implications in intracellular fluorescence detection of superoxide. Free Radic. Biol. Med. 2003, 34, 1359–1368. [Google Scholar] [CrossRef]
- Yamazaki, T.; Kawai, C.; Yamauchi, A.; Kuribayashi, F. A highly sensitive chemiluminescence assay for superoxide detection and chronic granulomatous disease diagnosis. Trop. Med. Health 2011, 39, 41–45. [Google Scholar] [CrossRef] [Green Version]
- Nauseef, W.M. Detection of superoxide anion and hydrogen peroxide production by cellular NADPH oxidases. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2014, 1840, 757–767. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.S.; Berridge, M.V. Superoxide produced by activated neutrophils efficiently reduces the tetrazolium salt, WST-1 to produce a soluble formazan: A simple colorimetric assay for measuring respiratory burst activation and for screening anti-inflammatory agents. J. Immunol. Methods 2000, 238, 59–68. [Google Scholar] [CrossRef]
- Tarpey, M.M.; Wink, D.A.; Grisham, M.B. Methods for detection of reactive metabolites of oxygen and nitrogen: In vitro and in vivo considerations. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R431–R444. [Google Scholar] [CrossRef]
- Chen, X.; Tian, X.; Shin, I.; Yoon, J. Fluorescent and luminescent probes for detection of reactive oxygen and nitrogen species. Chem. Soc. Rev. 2011, 40, 4783–4804. [Google Scholar] [CrossRef]
- Georgiou, C.D.; Papapostolou, I.; Patsoukis, N.; Tsegenidis, T.; Sideris, T. An ultrasensitive fluorescent assay for the in vivo quantification of superoxide radical in organisms. Anal. Biochem. 2005, 347, 144–151. [Google Scholar] [CrossRef]
- Xiao, H.; Zhang, W.; Li, P.; Zhang, W.; Wang, X.; Tang, B. Versatile fluorescent probes for imaging the superoxide anion in living cells and in vivo. Angew. Chem. 2020, 132, 4244–4258. [Google Scholar] [CrossRef]
- Gao, J.J.; Xu, K.H.; Tang, B.; Yin, L.L.; Yang, G.W.; An, L.G. Selective detection of superoxide anion radicals generated from macrophages by using a novel fluorescent probe. FEBS J. 2007, 274, 1725–1733. [Google Scholar] [CrossRef]
- Medvedeva, N.; Martin, V.V.; Weis, A.L.; Likhtenshten, G.I. Dual fluorophore-nitronyl probe for investigation of superoxide dynamics and antioxidant status of biological systems. J. Photochem. Photobiol. A Chem. 2004, 163, 45–51. [Google Scholar] [CrossRef]
- Olojo, R.; Xia, R.; Abramson, J. Spectrophotometric and fluorometric assay of superoxide ion using 4-chloro-7-nitrobenzo-2-oxa-1, 3-diazole. Anal. Biochem. 2005, 339, 338–344. [Google Scholar] [CrossRef]
- Tang, B.; Zhang, L.; Hu, J.-X.; Li, P.; Zhang, H.; Zhao, Y.-X. Indirect determination of superoxide anion radical in the plant of red sage based on vanillin-8-aminoquinoline with fluorescence. Anal. Chim. Acta 2004, 502, 125–131. [Google Scholar] [CrossRef]
- Maeda, H.; Yamamoto, K.; Nomura, Y.; Kohno, I.; Hafsi, L.; Ueda, N.; Yoshida, S.; Fukuda, M.; Fukuyasu, Y.; Yamauchi, Y. A design of fluorescent probes for superoxide based on a nonredox mechanism. J. Am. Chem. Soc. 2005, 127, 68–69. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Yamamoto, K.; Kohno, I.; Hafsi, L.; Itoh, N.; Nakagawa, S.; Kanagawa, N.; Suzuki, K.; Uno, T. Design of a practical fluorescent probe for superoxide based on protection–deprotection chemistry of fluoresceins with benzenesulfonyl protecting groups. Chem. A Eur. J. 2007, 13, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Liu, X.; Tang, B.; Yang, G.; Yang, Y.; An, L. Design of a phosphinate-based fluorescent probe for superoxide detection in mouse peritoneal macrophages. Chem. A Eur. J. 2007, 13, 1411–1416. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Liu, X.; Tang, B. A phosphinate-based red fluorescent probe for imaging the superoxide radical anion generated by RAW264. 7 macrophages. ChemBioChem 2007, 8, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Kundu, K.; Knight, S.F.; Willett, N.; Lee, S.; Taylor, W.R.; Murthy, N. Hydrocyanines: A class of fluorescent sensors that can image reactive oxygen species in cell culture, tissue, and in vivo. Angew. Chem. Int. Ed. 2009, 48, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Rugeles, L.; Galano, A.; Alvarez-Idaboy, J.R. The other side of the superoxide radical anion: Its ability to chemically repair DNA oxidized sites. Chem. Commun. 2018, 54, 13710–13713. [Google Scholar] [CrossRef]
- O’Neill, S.; Brault, J.; Stasia, M.-J.; Knaus, U.G. Genetic disorders coupled to ROS deficiency. Redox Biol. 2015, 6, 135–156. [Google Scholar] [CrossRef] [Green Version]
- Dupré-Crochet, S.; Erard, M.; Nüβe, O. ROS production in phagocytes: Why, when, and where? J. Leukoc. Biol. 2013, 94, 657–670. [Google Scholar] [CrossRef]
- Ostuni, M.A.; Gelinotte, M.; Bizouarn, T.; Baciou, L.; Houée-Levin, C. Targeting NADPH-oxidase by reactive oxygen species reveals an initial sensitive step in the assembly process. Free Radic. Biol. Med. 2010, 49, 900–907. [Google Scholar] [CrossRef]
- Dworakowski, R.; Anilkumar, N.; Zhang, M.; Shah, A. Redox signalling involving NADPH oxidase-derived reactive oxygen species. Biochem. Soc. Trans. 2006, 34 Pt 5, 960–964. [Google Scholar]
- Holmdahl, R.; Sareila, O.; Olsson, L.M.; Bäckdahl, L.; Wing, K. Ncf1 polymorphism reveals oxidative regulation of autoimmune chronic inflammation. Immunol. Rev. 2016, 269, 228–247. [Google Scholar] [CrossRef]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH oxidase activation and bacterial resistance. Front. Cell. Infect. Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef] [Green Version]
- Andrés, C.M.C.; Pérez de la Lastra, J.M.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. Hypochlorous Acid Chemistry in Mammalian Cells—Influence on Infection and Role in Various Pathologies. Int. J. Mol. Sci. 2022, 23, 10735. [Google Scholar] [CrossRef]
- Chen, X.; Lee, K.-A.; Ha, E.-M.; Lee, K.M.; Seo, Y.Y.; Choi, H.K.; Kim, H.N.; Kim, M.J.; Cho, C.-S.; Lee, S.Y. A specific and sensitive method for detection of hypochlorous acid for the imaging of microbe-induced HOCl production. Chem. Commun. 2011, 47, 4373–4375. [Google Scholar] [CrossRef]
- Nauseef, W.M. The phagocyte NOX2 NADPH oxidase in microbial killing and cell signaling. Curr. Opin. Immunol. 2019, 60, 130–140. [Google Scholar] [CrossRef]
- Mantegazza, A.R.; Savina, A.; Vermeulen, M.; Pérez, L.; Geffner, J.; Hermine, O.; Rosenzweig, S.D.; Faure, F.; Amigorena, S. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood 2008, 112, 4712–4722. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, S.M.; Corriden, R.; Nizet, V. How neutrophils meet their end. Trends Immunol. 2020, 41, 531–544. [Google Scholar] [CrossRef]
- Tauber, A.I.; Chernyak, L. Metchnikoff and the Origins of Immunology: From Metaphor to Theory; Oxford University Press on Demand: Oxford, UK, 1991. [Google Scholar]
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar] [CrossRef]
- Mahla, R.S.; Kumar, A.; Tutill, H.J.; Krishnaji, S.T.; Sathyamoorthy, B.; Noursadeghi, M.; Breuer, J.; Pandey, A.K.; Kumar, H. NIX-mediated mitophagy regulate metabolic reprogramming in phagocytic cells during mycobacterial infection. Tuberculosis 2021, 126, 102046. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. New insights into tissue macrophages: From their origin to the development of memory. Immune network 2015, 15, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Janeway, C.A., Jr.; Travers, P.; Walport, M.; Shlomchik, M.J. Principles of innate and adaptive immunity. In Immunobiology: The Immune System in Health and Disease, 5th ed.; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Marciano, B.E.; Spalding, C.; Fitzgerald, A.; Mann, D.; Brown, T.; Osgood, S.; Yockey, L.; Darnell, D.N.; Barnhart, L.; Daub, J. Common severe infections in chronic granulomatous disease. Clin. Infect. Dis. 2015, 60, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Krombach, F.; Münzing, S.; Allmeling, A.-M.; Gerlach, J.T.; Behr, J.; Dörger, M. Cell size of alveolar macrophages: An interspecies comparison. Environ. Health Perspect. 1997, 105, 1261–1263. [Google Scholar] [PubMed] [Green Version]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Chakravarty, S.D.; Ivashkiv, L.B. Regulation of IFN and TLR signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol. Rev. 2008, 226, 41. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.; Keshav, S.; Harris, N.; Gordon, S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: A marker of alternative immunologic macrophage activation. J. Exp. Med. 1992, 176, 287–292. [Google Scholar] [CrossRef] [Green Version]
- Doherty, T.; Kastelein, R.; Menon, S.; Andrade, S.; Coffman, R. Modulation of murine macrophage function by IL-13. J. Immunol. 1993, 151, 7151–7160. [Google Scholar] [CrossRef]
- McWhorter, F.Y.; Davis, C.T.; Liu, W.F. Physical and mechanical regulation of macrophage phenotype and function. Cell. Mol. Life Sci. 2015, 72, 1303–1316. [Google Scholar] [CrossRef] [Green Version]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Corrigendum: Macrophage polarization: Different gene signatures in M1 (LPS+) vs. classically and M2 (LPS–) vs. alternatively activated macrophages. Front. Immunol. 2020, 11, 234. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Pérez, S.; Rius-Pérez, S. Macrophage polarization and reprogramming in acute inflammation: A redox perspective. Antioxidants 2022, 11, 1394. [Google Scholar] [CrossRef]
- Parisi, L.; Gini, E.; Baci, D.; Tremolati, M.; Fanuli, M.; Bassani, B.; Farronato, G.; Bruno, A.; Mortara, L. Macrophage polarization in chronic inflammatory diseases: Killers or builders? J. Immunol. Res. 2018, 2018, 8917804. [Google Scholar] [CrossRef] [Green Version]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The metabolic signature of macrophage responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [Green Version]
- Palma, A.; Jarrah, A.S.; Tieri, P.; Cesareni, G.; Castiglione, F. Gene regulatory network modeling of macrophage differentiation corroborates the continuum hypothesis of polarization states. Front. Physiol. 2018, 9, 1659. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef]
- Cornelissen, R.; Lievense, L.A.; Maat, A.P.; Hendriks, R.W.; Hoogsteden, H.C.; Bogers, A.J.; Hegmans, J.P.; Aerts, J.G. Ratio of intratumoral macrophage phenotypes is a prognostic factor in epithelioid malignant pleural mesothelioma. PLoS ONE 2014, 9, e106742. [Google Scholar] [CrossRef]
- Andrés, C.M.C.; Pérez de la Lastra, J.M.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. The Role of Reactive Species on Innate Immunity. Vaccines 2022, 10, 1735. [Google Scholar] [CrossRef]
- Mitchell, G.; Chen, C.; Portnoy, D.A. Strategies used by bacteria to grow in macrophages. Myeloid Cells Health Dis. A Synth. 2017, 4, 701–725. [Google Scholar]
- Weiss, G.; Schaible, U.E. Macrophage defense mechanisms against intracellular bacteria. Immunol. Rev. 2015, 264, 182–203. [Google Scholar] [CrossRef] [Green Version]
- del Cerro-Vadillo, E.; Madrazo-Toca, F.; Carrasco-Marín, E.; Fernandez-Prieto, L.; Beck, C.; Leyva-Cobián, F.; Saftig, P.; Alvarez-Dominguez, C. Cutting edge: A novel nonoxidative phagosomal mechanism exerted by cathepsin-D controls Listeria monocytogenes intracellular growth. J. Immunol. 2006, 176, 1321–1325. [Google Scholar] [CrossRef] [Green Version]
- Allan, E.R.; Tailor, P.; Balce, D.R.; Pirzadeh, P.; McKenna, N.T.; Renaux, B.; Warren, A.L.; Jirik, F.R.; Yates, R.M. NADPH oxidase modifies patterns of MHC class II–restricted epitopic repertoires through redox control of antigen processing. J. Immunol. 2014, 192, 4989–5001. [Google Scholar] [CrossRef] [Green Version]
- White, P.; Anderson, D. In vivo transplantation of mammalian neural crest cells into chick hosts reveals a new autonomic sublineage restriction. Development 1999, 126, 4351–4363. [Google Scholar] [CrossRef] [PubMed]
- Bainton, D.F.; Ullyot, J.L.; Farquhar, M.G. The development of neutrophilic polymorphonuclear leukocytes in human bone marrow: Origin and content of azurophil and specific granules. J. Exp. Med. 1971, 134, 907–934. [Google Scholar] [CrossRef] [PubMed]
- Rosales, C. Neutrophils at the crossroads of innate and adaptive immunity. J. Leukoc. Biol. 2020, 108, 377–396. [Google Scholar] [CrossRef] [PubMed]
- Rigby, K.M.; DeLeo, F.R. Neutrophils in innate host defense against Staphylococcus aureus infections. Semin. Immunopathol. 2012, 34, 237–259. [Google Scholar] [CrossRef] [Green Version]
- Savill, J.S.; Wyllie, A.H.; Henson, J.E.; Walport, M.J.; Henson, P.M.; Haslett, C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J. Clin. Investig. 1989, 83, 865–875. [Google Scholar] [CrossRef]
- Greenlee-Wacker, M.C. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol. Rev. 2016, 273, 357–370. [Google Scholar] [CrossRef] [Green Version]
- Edwards, S.W.; Moulding, D.A.; Derouet, M.; Moots, R.J. Regulation of neutrophil apoptosis. Chem. Immunol. Allergy 2003, 83, 204–224. [Google Scholar]
- Schmidt, E.P.; Lee, W.L.; Zemans, R.L.; Yamashita, C.; Downey, G.P. On, around, and through: Neutrophil-endothelial interactions in innate immunity. Physiology 2011, 26, 334–347. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.D.; Voyich, J.M.; Burlak, C.; DeLeo, F.R. Neutrophils in the innate immune response. Arch. Immunol. Ther. Exp. Engl. Ed. 2005, 53, 505. [Google Scholar]
- Metzemaekers, M.; Gouwy, M.; Proost, P. Neutrophil chemoattractant receptors in health and disease: Double-edged swords. Cell. Mol. Immunol. 2020, 17, 433–450. [Google Scholar] [CrossRef]
- McPhail, L.C.; Clayton, C.C.; Snyderman, R. The NADPH oxidase of human polymorphonuclear leukocytes. Evidence for regulation by multiple signals. J. Biol. Chem. 1984, 259, 5768–5775. [Google Scholar] [CrossRef]
- Noronha, N.d.C.; Mizukami, A.; Caliári-Oliveira, C.; Cominal, J.G.; Rocha, J.L.M.; Covas, D.T.; Swiech, K.; Malmegrim, K.C. Priming approaches to improve the efficacy of mesenchymal stromal cell-based therapies. Stem Cell Res. Ther. 2019, 10, 131. [Google Scholar] [CrossRef] [Green Version]
- Sheshachalam, A.; Srivastava, N.; Mitchell, T.; Lacy, P.; Eitzen, G. Granule protein processing and regulated secretion in neutrophils. Front. Immunol. 2014, 5, 448. [Google Scholar] [CrossRef] [Green Version]
- Ethuin, F.; Gérard, B.; Benna, J.E.; Boutten, A.; Gougereot-Pocidalo, M.-A.; Jacob, L.; Chollet-Martin, S. Human neutrophils produce interferon gamma upon stimulation by interleukin-12. Lab. Investig. 2004, 84, 1363–1371. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.; Whyte, M.K.; Haslett, C. Inhibition of apoptosis and prolongation of neutrophil functional longevity by inflammatory mediators. J. Leukoc. Biol. 1993, 54, 283–288. [Google Scholar] [CrossRef]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef]
- Megiovanni, A.M.; Sanchez, F.; Robledo-Sarmiento, M.; Morel, C.; Gluckman, J.C.; Boudaly, S. Polymorphonuclear neutrophils deliver activation signals and antigenic molecules to dendritic cells: A new link between leukocytes upstream of T lymphocytes. J. Leukoc. Biol. 2006, 79, 977–988. [Google Scholar] [CrossRef]
- Scott, M.G.; Dullaghan, E.; Mookherjee, N.; Glavas, N.; Waldbrook, M.; Thompson, A.; Wang, A.; Lee, K.; Doria, S.; Hamill, P. An anti-infective peptide that selectively modulates the innate immune response. Nat. Biotechnol. 2007, 25, 465–472. [Google Scholar] [CrossRef]
- Soehnlein, O.; Lindbom, L. Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 2010, 10, 427–439. [Google Scholar] [CrossRef]
- Bronte, V.; Zanovello, P. Regulation of immune responses by L-arginine metabolism. Nat. Rev. Immunol. 2005, 5, 641–654. [Google Scholar] [CrossRef]






































































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrés, C.M.C.; Pérez de la Lastra, J.M.; Andrés Juan, C.; Plou, F.J.; Pérez-Lebeña, E. Superoxide Anion Chemistry—Its Role at the Core of the Innate Immunity. Int. J. Mol. Sci. 2023, 24, 1841. https://doi.org/10.3390/ijms24031841
Andrés CMC, Pérez de la Lastra JM, Andrés Juan C, Plou FJ, Pérez-Lebeña E. Superoxide Anion Chemistry—Its Role at the Core of the Innate Immunity. International Journal of Molecular Sciences. 2023; 24(3):1841. https://doi.org/10.3390/ijms24031841
Chicago/Turabian StyleAndrés, Celia María Curieses, José Manuel Pérez de la Lastra, Celia Andrés Juan, Francisco J. Plou, and Eduardo Pérez-Lebeña. 2023. "Superoxide Anion Chemistry—Its Role at the Core of the Innate Immunity" International Journal of Molecular Sciences 24, no. 3: 1841. https://doi.org/10.3390/ijms24031841
APA StyleAndrés, C. M. C., Pérez de la Lastra, J. M., Andrés Juan, C., Plou, F. J., & Pérez-Lebeña, E. (2023). Superoxide Anion Chemistry—Its Role at the Core of the Innate Immunity. International Journal of Molecular Sciences, 24(3), 1841. https://doi.org/10.3390/ijms24031841