1. Introduction
The balance between phosphorylation and dephosphorylation is essential for the maintenance of cellular homeostasis in eukaryotes. Phosphorylation is regulated by the antagonistic activity of protein kinases and phosphatases in a highly complex and, usually, conserved manner. The orchestrated interplay between these two enzyme groups can fine-tune and regulate complicated cellular events, such as cell cycle progression. Although the role of kinases governing cell division is well established, we know much less about the function and substrates of the protein phosphatases.
The evolutionarily conserved protein phosphatase 5 (PP5) belongs to the family of Ser/Thr phosphoprotein phosphatases (PPP: includes PP1, PP2A, PP2B, PP4, PP6 and PP7) [
1,
2,
3,
4]. PP5 functions as a monomer and does not employ conventional, non-covalently bound regulatory subunits for substrate recognition or for control of its catalytic activity. All of these features are encoded by its primary amino acid sequence [
5,
6]. Three tetratricopeptide (TPR) motifs are located at the N-terminal region of PP5 (
Figure S1), which fold back onto the C-terminal catalytic (phosphatase) domain, keeping the basal activity of PP5 low [
7]. This auto-inhibition can be released by the binding of fatty acids or the Hsp90 chaperone to the TPR motifs, as well as by phosphorylation of the conserved Thr362 (in human PP5) [
8,
9,
10,
11]. It has been reported that the TPR motifs alone, as well as the catalytic domain itself, are capable of substrate binding [
12].
PP5 regulates diverse cellular processes, including cell growth and differentiation [
13], DNA damage checkpoints and repair [
14,
15,
16], p53 activity [
17], stress response [
18], circadian rhythm [
19] and receptor signaling [
5]. Impaired function of PP5 has been associated with different diseases, such as diabetes, obesity and cancer, therefore PP5 has recently become a potential therapeutic target (reviewed in [
6]). Its function in cell division has also been proposed, due to the presence of the TPR motifs. Ollendorff and Donoghue have reported that PP5 localizes to the mitotic spindle in Cos-1 mammalian cells via its TPR motifs, and physically interacts with subunits of the APC/C ubiquitin ligase required for proper metaphase/anaphase transition during mitosis [
20]. The same research suggested centrosomal localization of PP5, which was also observed in hTERT-RPE1 mammalian cells [
21]. RNAi screen in the Glover lab has revealed that the knock-down of
Drosophila pp5 (
PpD3) creates a mild centrosome number phenotype [
22]. These observations suggest that PP5 might also play a role in centrosome biogenesis that is governed by Polo-like kinase 4 (Plk4). Interestingly, in a high-throughput protein-trap screen, Plk4 was co-purified with the affinity-tagged PP5 in
Drosophila embryos [
23], which has never been further investigated.
Plk4 is a conserved oncogenic kinase that is often referred to as the master regulator of centriole duplication [
24,
25,
26,
27]. Centrioles are the main structural components of centrosomes, which become the major microtubule organizing centers in mitotic cells in animals. Therefore, their duplication must be carried out in a semi-conservative manner with extremely high fidelity, once every cell cycle [
28,
29]. Not surprisingly, the misregulation of Plk4 leads to serious mitotic defects, including chromosome segregation and centrosome number alterations, common hallmarks of cancer cells. These changes can promote tumorigenesis, making Plk4 a highly studied target in cancer diagnostics and therapy (reviewed in [
30]). Comprehensive studies have revealed that the expression, localization, activity and half-life of Plk4 are tightly regulated at pre- and post-transcriptional, as well as post-translational, levels (reviewed in [
30]). The mature homo-dimeric Plk4 kinase is primarily regulated by self-phosphorylation and SCF-mediated ubiquitination in a complex manner. Phosphatases might also be involved in Plk4 regulation [
31], however, it is unknown exactly how these enzymes (and which enzymes besides PP2A) fine-tune the function of Plk4.
In this study, we demonstrate that protein phosphatase 5 interacts with and dephosphorylates Plk4 in Drosophila melanogaster and human cells. We show that PP5 co-localizes with Plk4 to the centrosomes in fruit fly embryos and cultured cells, and speculate that PP5 might be involved in the precise phospho-regulation of Plk4.
3. Discussion
Interestingly, the first discovered phosphatase, the PR enzyme, was published in 1945 by the Cori couple, 10 years before its opposing enzyme, the first kinase, phosphorylase b kinase, was discovered. Nevertheless, phosphatases have been much less studied over the decades for historical and technical reasons [
41]. They had long been considered housekeeping enzymes without any explicit regulatory function. This eventually turned out to be incorrect as they are highly specific to different substrates, including kinases [
31]. Since many kinases are encoded by oncogenes [
30], it is not surprising that the counteracting enzymes are becoming recognized as potential targets for anticancer therapy [
6].
The evolutionarily conserved PP5 phosphatase governs several vital cellular functions, including gene expression, DNA damage response, circadian clock, receptor signaling and apoptosis (reviewed in [
5,
6]). Moreover, its role in cell division has also been reported [
20,
22]. Therefore, we aimed to identify novel mitotic substrates of this enzyme and found that PP5 interacts with the Polo-like kinase 4, Plk4. Plk4 is an oncogenic kinase [
30] and the master regulator of centriole duplication [
24,
25,
26,
27]. The maturation, activity, stability and localization of the Plk4 protein is regulated primarily by phosphorylation. Following synthesis, the enzyme has a low basic activity, which increases upon homo-dimerization of Plk4 and trans-auto-phosphorylation of the kinase domain [
42,
43]. Self-phosphorylation of the DRE element leads to Slimb/SCF-mediated proteasomal degradation of the kinase [
33,
44], which is reversed in mitosis by the phosphatase PP2A-Twins to stabilize Plk4 and promote centrosome duplication in the next S phase [
31]. Interestingly, while other kinases also contribute to Plk4 regulation [
30], the roles of other phosphatases have not yet been found.
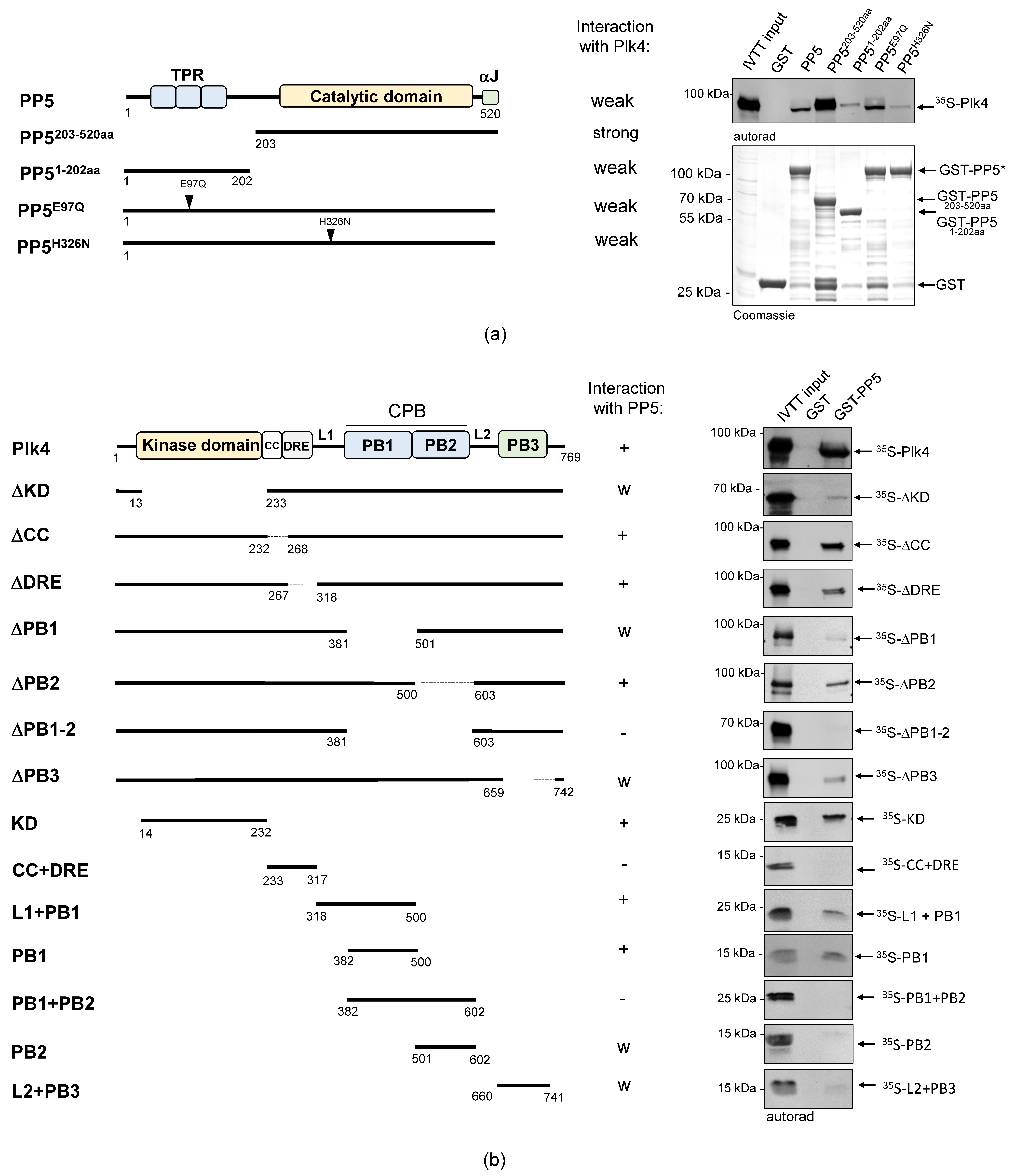
We discovered that PP5 specifically interacts with Plk4 in fruit flies and human cells. We presented that the phosphatase domain of PP5 physically binds to the kinase domain and PB1 domains of Plk4 in
Drosophila. We proved that Plk4 is dephosphorylated by PP5 in vitro and in vivo, and provided evidence that PP5 interacts and co-localizes with Plk4 at the centrosomes in
Drosophila. We observed that the activity of Plk4 was not altered upon PP5 dephosphorylation, and the depletion of PP5 caused only a mild reduction in the centrosome number in cultured cells. However, we also found that the PP5-regulated dephosphorylation of Plk4 affects the interaction of Plk4 with various centrosomal proteins, including its scaffold protein, Asl, which preferentially binds to the dephosphorylated form of the kinase. Similarly, Plk4 binding to Sas4, the centriolar partner of Asl [
36], requires dephosphorylated Plk4. In contrast, Ana1, the key protein in centriole-to-centrosome conversion [
45] interacts exclusively with fully phosphorylated Plk4. Although we do not understand the mechanistic details of this regulation, it may be important for the centrosomal recruitment or maintenance of Plk4.
Considering the fact that PP5 is an evolutionarily conserved enzyme that regulates key determinants, such as tumor suppressor p53 [
17], checkpoint kinases ATM, ATR [
14,
15,
16] and Chk1 [
46] or subunits of APC/C [
20], we were surprised to see that its deletion from the
Drosophila genome did not cause severe mitotic abnormalities or lethality. Indeed, the knock-out of PP5 in mice produced viable animals, too, with increased sensitivity to UV light [
46]. This suggests that PP5 is not essential, but rather acts as a pleiotropic modulator of cellular processes in animals. PP5 is unique among the PPP family of Ser/Thr phosphatases, because it acts as a monomer and its low basal activity is self-regulated by its own TPR motifs [
7], which are released by the molecular chaperone Hsp90 [
12,
47]. It has been shown that PP5 is a co-chaperone of Hsp90 and, together with Cdc37, forms a heteromeric complex [
10] involved in the regulation of oncogenic kinases [
11]. The González laboratory has proved that Hsp90 is a core centrosomal protein in
Drosophila [
48]. The above findings led us to speculate that the Hsp90-Cdc37-PP5 complex might be involved in the phospho-regulation of Plk4, which is not a vital modification. This may happen under special conditions or in defined cell cycle stages to ensure proper homeostasis of Plk4 at the centrosomes. Deciphering and answering this question promises to be an exciting future challenge.
4. Materials and Methods
4.1. DNA Constructs
cDNAs of the fruit fly PP5 (Clone ID: GH12714; Flybase ID: CG8402;
PpD3 in
Drosophila melanogaster), CP110 (Clone ID: GH03511; Flybase ID: CG14617), Slimb (Clone ID: LD08669; Flybase ID: CG3412), Spd2 (Clone ID: LD24702; Flybase ID: CG17286), Polo (Clone ID: LD11851; Flybase ID: CG12306), Sas6 (Clone ID: FI21744, Flybase ID: CG15524), Asterless (hereafter Asl, Clone ID: GH02902, Flybase ID: CG2919) and Ana2 (Clone ID: LD22033, Flybase ID: CG8262) were obtained from the
Drosophila Genomics Resource Centre (DGRC, Bloomington, IN, USA). cDNA of human PP5 (hereafter HsPP5, Clone ID: 3459309) and Plk4 (hereafter HsPlk4, Clone ID: 5273226) were obtained from Horizon Discovery Ltd (Cambridge, UK). Gateway entry clones of Ana1, Sas6, Ana2
1−140aa and Ana2
281−420aa, the constitutively active form of the
Drosophila Plk4 (hereafter Plk4-TE; Flybase ID: CG7186, contains the T172E mutation in the T-loop of the kinase) and its kinase dead version (hereafter Plk4-TEKM, kinase dead due to the K43M mutation), the non-degradable Plk4 (hereafter Plk4-ND, non-degradable due to the S293A and T297A mutations in the DRE region) and its kinase dead version (hereafter Plk4-NDKD, non-degradable and kinase dead due to the mutations S293A, T297A and K43M), and MBP-tagged Plk4-TE and Plk4-TEKM were obtained from the Glover lab (Cambridge, UK) [
32,
35]. Gateway entry clones of fruit fly Polo and PP5, as well as human PP5 were generated, according to the manufacturer’s instructions (cat # 11789100, Thermo Fisher Scientific, Waltham, MA, USA). The following destination vectors were used for the LR reaction to generate expression clones: for N-terminal GFP-tagging in
Drosophila and human cells (pAGW (#1071 DGRC, Bloomington, IN, USA) and pcDNA-Dest53 (cat # 12288015, Thermo Fisher Scientific, Waltham, MA, USA), respectively); N-terminal Flag-tagging in
Drosophila cells (pAFW (#1111, DGRC, Bloomington, IN, USA); N-terminal GST-tagging in
E. coli (pDest15, cat # 11802014, Thermo Fisher Scientific, Waltham, MA, USA); and N-terminal His
6-tagging in
E.coli or IVTT (pDest17, cat # 11803012, Thermo Fisher Scientific, Waltham, MA, USA).
Site-directed mutagenesis with the QuikChange II XL Mutagenesis Kit (cat # 200522, Agilent Technologies, Santa Clara, CA, USA) was performed using entry clones, according to the manufacturer’s instruction to generate truncated fruit fly PP5 and Plk4 proteins (PP5
203−520aa, PP5
1−202aa, Plk4-ΔKD, Plk4-ΔCC, Plk4-ΔDRE, Plk4-ΔPB1, Plk4-ΔPB2, Plk4-ΔPB1-2 and Plk4-ΔPB3); Plk4 fragments (KD, CC + DRE, L1 + PB1, PB1, PB1-2, PB2 and L2 + PB3) and amino acid substitutions in PP5 (fruit fly PP5
E97Q (hyperactive) and PP5
H326N (inactive), according to [
7,
49]) (see also
Figure S1). PP5 truncated forms and point mutants were cloned to pDest15 and used in activity assay or GST-IVTT experiments as bait, while Plk4 truncated forms and fragments were cloned into pDest17 and used for
35S-labelling in IVTT reaction. cDNA of human Plk4 (hereafter HsPlk4, Clone ID: 5273226) was subjected to site-directed mutagenesis, as described above, to generate its non-degradable form (by introducing S285A and T289A mutations based on [
33]), which was cloned to pFlag-CMV-4 plasmid (cat # E7158, Merck-Millipore, Burlington, MA, USA) to express Flag-HsPlk4-ND in human cells.
For co-expression in D.Mel-2 cells the Myc-tagged fruit fly PP5
E97Q or PP5
H326N and the GFP-tagged Plk4-ND or NDKD, respectively, were cloned to the Ac5-STABLE2-neo vector (Sutherland Lab, Bilbao, Spain [
50]) by classical and InFusion cloning methods (cat # 102518, Takara Bio, Kusatsu, Siga, Japan). For co-overexpression in
E. coli cells, the MBP-tagged Plk4-TE or TEKM (in pKM596 plasmid [
32]) and His
6-tagged PP5/PP5
H326N, respectively, were used. To avoid replication origin incompatibility, we cloned the His
6-tagged PP5/PP5
H326N to pACYCDuet-1 (cat # 71147-3, Merck-Millipore, Burlington, MA, USA). For the in vivo co-localization assay, we generated two new
Drosophila Gateway vectors suitable for N-terminal tagging of any gene of interest with NYFP or CYFP, respectively, by modifying the original Split-YFP plasmids published earlier [
39]: pMT(Hygro)-NYFP-Myc-GW and pMT(Hygro)-CYFP-HA-GW (
Figure S9). CDS of Sas6 was cloned into the newly generated pMT(Hygro)-mCherry-GW (
Figure S9) Gateway vector to establish stably transfected D.Mel-2 cell lines expressing mCherry-Sas6 in an inducible manner. The three new pMT vectors (sequences and maps are provided in
Figure S9) are regulated by the metallothionein promoter (MT) suitable for copper-induced expression of the fusion proteins and hygromycin-selection in
Drosophila cultured cells.
For IVTT expression, we used Ana1, Sas4 [
32], Cep135 [
45], Polo and Plk4 constructs cloned to pDest17. Asl, Ana2, CP110 were IVTT synthesized directly from T7-promoter-driven cDNA clones obtained from DGRC; Sas6, Spd2 and Slimb were cloned to pHY22 vector [
34].
All DNA constructs were validated by DNA sequencing. Oligonucleotide primers are provided in
Table S1.
4.2. Recombinant Protein Expression and Purification
Recombinant GST, GST-Ana2
1−140, GST-Ana2
281−420 and GST-tagged PP5 and its derivatives were expressed in SixPack
E. coli [
51] and purified following standard procedures. Briefly, bacteria were grown in Terrific broth auto-induction medium (cat # AIMTB0210, Formedium, Hunstanton, UK) for 48 h at 18 °C. Cells were lysed by sonication in 40 mL phosphate buffered saline (PBS) containing 0.2 mg/mL lysozyme and 1 mM PMSF (phenylmethylsulfonyl fluoride, cat # P7626, Sigma-Aldrich, St. Louis, MO, USA). Cleared supernatants were mixed with pre-equilibrated glutathione sepharose 4B resin (cat # 17-0756-01, Cytiva, Washington, WA, USA) and incubated at 4 °C for 1.5 h. Immobilized proteins were either kept on beads and stored at −20 °C in PBS supplemented with 50% glycerol or eluted with 10 mM reduced glutathione dissolved in phosphatase buffer (50 mM Na-HEPES pH 7.5, 100 mM NaCl, 2 mM DTT, 0.05% Triton X-100, 2 mM MgCl
2, 2 mM MnCl
2). Amicon Ultra-0.5 Centrifugal Filter Units (cat # UFC503096, Merck-Millipore, Burlington, MA, USA) were used to concentrate the eluted proteins, which were then supplemented with 50% glycerol and stored at −80 °C. His
6-tagged PP5 was expressed in SixPack and purified on Ni-sepharose 6 Fast Flow resin (cat # 17531802, Cytiva, Washington, WA, USA), according to the manufacturer. Eluted proteins were dialyzed over PBS for 18 h at 4 °C, concentrated and used for mice immunization.
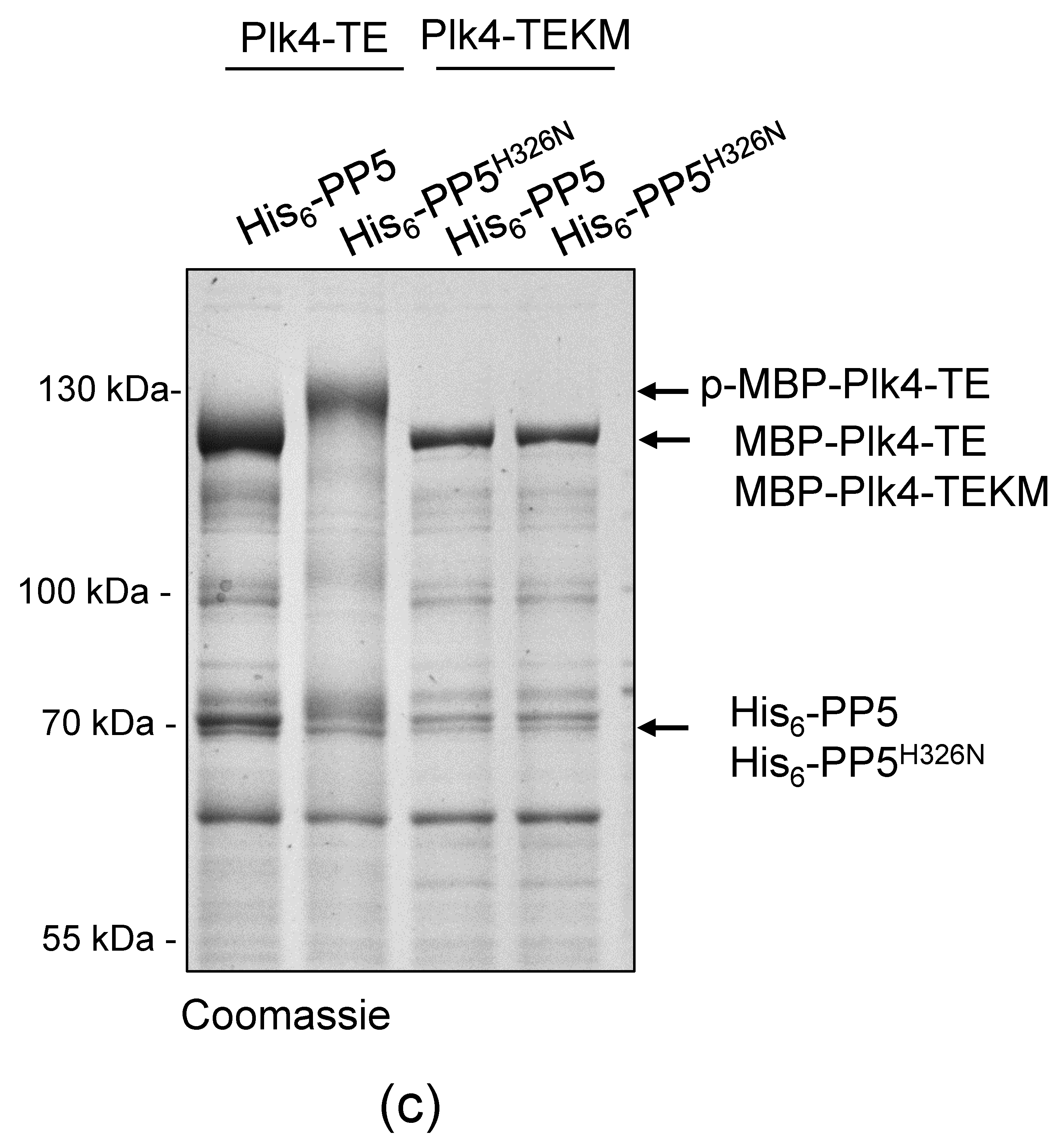
Bacteria expressing MBP-Plk4-TE or MBP-Plk4-TEKM, or co-expressing MBP-Plk4-TE or MBP-Plk4-TEKM with His6-PP5 or His6-PP5H326N, respectively, were grown in LB medium and induced with 1 mM isopropyl 1-thio-β-D-galactopyranoside (cat # R0393, Thermo Fisher Scientific, Waltham, MA, USA) for 2 h at 25 °C, and another 4 h at 30 °C. Cell lysates were prepared by sonication using Branson Sonifier (Branson Ultrasonics Corp., Danbury, CT, USA) in PBS supplemented with 100 mM NaCl, 1 mM PMSF, EDTA-free protease inhibitor cocktail (cat # 11873580001, Roche, Basel, Switzerland), and 0.05% Triton X-100. Cleared supernatants were mixed with pre-equilibrated amylose resin (cat # E8021S, New England Biolabs, Ipswich, MA, USA) and incubated at 4 °C for 1.5 h. Beads were washed with PBS supplemented with 100 mM NaCl and 0.05% Triton X-100 followed by washing with kinase buffer (20 mM Na-HEPES pH 7.5, 100 mM NaCl, 10 mM MgCl2, 10 mM MnCl2, 1 mM DTT). Immobilized MBP-tagged proteins were either kept on beads and stored at −20 °C in DTT-free kinase buffer supplemented with 50% glycerol, or eluted with kinase buffer containing 15 mM D-maltose. Eluted proteins were concentrated, then supplemented with 50% glycerol and stored at −80 °C. For the in vivo phosphatase assay bacteria co-expressing MBP-Plk4-TE or MBP-Plk4-TEKM with His6-PP5 or His6-PP5H326N, respectively, were boiled in Laemmli sample buffer (5 min 100 °C).
4.3. IVTT and In Vitro Binding Assay
For testing the in vitro physical protein-protein interactions, we produced the
35S-methionine-labeled candidate binding partners in a coupled in vitro transcriptional/translational system (IVTT). The detailed protocol of this method is provided in [
34]. Briefly, bait proteins were immobilized either on glutathione sepharose 4B beads (GST, GST-PP5, GST-PP5
H326N, GST-PP5
1−202aa, GST-PP5
203−520aa, GST-PP5
E97Q, GST-Ana2
281−420aa and GST-Ana2
1−140aa) or amylose resin (MBP or MBP-Plk4-TE), and incubated with
35S-labelled putative partners. Protein samples were subjected to SDS-PAGE followed by Coomassie brilliant blue-staining. Gels were scanned, dried and the specific protein-protein interactions were detected by autoradiography.
4.4. Drosophila and Human Cultured Cell Lines—Transfection and Maintenance
D.Mel-2 cells (Schneider’s Drosophila Line 2 [D. Mel. (2), SL2] (ATCC CRL-1963, Manassas, VA, USA) were cultured in Insectagro DS2 Serum-Free medium (cat # 13-402-CV Corning, Corning, NY, USA) supplemented with 2 mM stable L-glutamine (cat # XC-T1755, Biosera, Nuaille, France) and 1× PenStrep (cat # XC-A4122, Biosera, Nuaille, France) at 25 °C. Cells were transfected using Expifectamine Sf reagent (cat # A38915, Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer. Transiently transfected cells were harvested 2 days-post transfections (if metallothionein promoter was used, cells were treated with 0.5 mM CuSO4 for 24 h before harvesting). For the generation of stable transfected D.Mel-2 lines, cells were subjected 3 days-post transfection to antibiotic selection (following standard procedures) for 4 weeks using G418 at 1 mg/mL concentration (cat # A1720-5G, Merck-Millipore, Burlington, MA, USA) or hygromycin B at 300 µg/mL concentration (cat # 25965.03, Serva, Heidelberg, Germany), respectively.
Human HEK293 cells (ATCC CRL-1573, Manassas, VA, USA) were cultured in DMEM containing GlutaMAX™ Supplement (cat # 61965026, Thermo Fisher Scientific, Waltham, MA, USA) and supplemented with 10% FBS (cat # ECS0180L, Euroclone, Pero, Italy), 1x PenStrep and NEAA mixture (cat # BE13-114E, Lonza, Basel, Switzerland) at 37 °C under 5% CO2. Cells were transiently co-transfected using PEI (cat # 408727, Merck-Millipore, Burlington, MA, USA), according to standard procedures, harvested 2 days post-transfection, then processed for co-immunoprecipitation.
4.5. Gene Silencing in Drosophila Cultured Cells
RNAi approach was used to test the specificity of the anti-PP5 polyclonal antibody. dsRNAs targeting the bacterial kanamycin gene (negative control) or the
PpD3 gene encoding PP5 (either the coding sequence (CDS) or 3′-UTR) were designed. dsRNAs were synthetized using the MEGAscript T7 transcription kit (cat # AM1334, Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer. dsRNA-treatment was performed, as described earlier [
32]. Briefly, D.Mel-2 cells were cultured in 6-well plates and transfected with 10 μg dsRNA using TransFast reagent (cat # E2431, Promega, Madison, WI, USA) for 3 days at 25 °C. Oligonucleotide primers used in this study are shown in
Table S1.
4.6. Protein Sample Preparation from the Tissues
D.Mel-2 cells transiently co-transfected with GFP or GFP-PP5 and 3×Flag-Plk4-ND were lysed by 10 × passing the cell suspension through a G25 needle in EB buffer (20 mM Tris-HCl pH 7.6, 150 mM NaCl, 0.5 mM EGTA, 2 mM MgCl2, 0.1% NP40, 5% glycerol, 1 mM DTT, 1 mM PMSF, EDTA-free protease inhibitor cocktail, 0.1 µL/mL benzonase nuclease (cat # 70746-3, Merck-Millipore, Burlington, MA, USA). Lysates were centrifuged (17,000× g, 20 min, 4 °C) and cleared supernatants were used for immunoblotting and co-immunoprecipitations.
Stably transfected D.Mel-2 cells expressing GFP-Plk4-ND or GFP-Plk4-NDKD and Myc-PP5E97Q or Myc-PP5H326N, respectively, were directly lysed in Laemmli sample buffer containing 0.1 µL/mL benzonase nuclease (treated for 5 min at room temperature followed by boiling for 5 min) and used for immunoblotting; or, they were lysed in EB supplemented with PhosSTOP phosphatase inhibitor cocktail (cat # 4906837001, Merck-Millipore, Burlington, MA, USA) by the needle/syringe method, and used for GFP-Trap (cat # gtma, ChromoTek GmbH, Planegg, Germany) purification. The captured kinases were used for the in vitro kinase assay to phosphorylate GST-Ana2281−420aa, in vitro.
HEK293 cells were transiently co-transfected with GFP or GFP-HsPP5 and Flag-HsPlk4-ND, respectively, lysed in EB by passing the cell suspension through a G25 needle. Lysates were centrifuged (17,000× g, 20 min, 4 °C) and cleared supernatants were used for immunoblotting and co-immunoprecipitations.
Total protein extracts were prepared from Drosophila wild type embryos (0–24 h-old), L1 to L3 larvae, white and brown pupae, pharate adult animals, and adults, respectively, by grinding the samples in RIPA buffer (10 µL buffer for 1 mg sample); 50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% NP 40, 0.5% Na-Deoxycholate, 0.1% SDS, 1 × EDTA-free protease inhibitor cocktail supplemented with 0.1 µL/mL benzonase nuclease. Samples were incubated on ice for 20 min, then supplemented with Laemmli sample buffer, boiled for 5 min and subjected to SDS-PAGE and immunoblotting. Drosophila syncytial embryos (0–2 h-old), and dissected ovaries and testes were collected in Laemmli sample buffer, boiled for 5 min, homogenized by glass pestles and boiled for another 5 min. Equal amounts of samples were subjected to SDS-PAGE and immunoblotting.
For antibody validation, we homogenized the pp5 null mutant adult flies in Laemmli sample buffer with a glass pestle and boiled the samples for 5 min before they were subjected to immunoblotting. Cultured cells depleted for the endogenous PP5 by RNAi were harvested 3 days post-transfection with dsRNAs, washed with PBS then boiled in Laemmli sample buffer for 5 min. Equal amounts of protein samples were subjected to immunoblotting.
4.7. Co-Immunoprecipitation
Co-immunoprecipitation from cultured
Drosophila cells was carried out according to [
52]. Briefly, D.Mel-2 cells expressing GFP or GFP-PP5 and 3×Flag-Plk4-ND were harvested 2 days post-transfection, washed in sterile PBS and lysed in EB buffer. Clarified supernatants were used for purification on GFP-Trap magnetic agarose beads (cat # gtma-20, ChromoTek GmbH, Planegg, Germany) at 4 °C for 90 min. Bound proteins were eluted by boiling the beads in Laemmli sample buffer, then fractionated on SDS-PAGE and blotted onto a PVDF membrane. Immunoprecipitation was tested by immunoblotting with anti-GFP and anti-FlagM
2 antibodies, respectively.
Stably transfected D.Mel-2 cells expressing GFP-Plk4-ND or GFP-Plk4-NDKD and Myc-PP5E97Q or Myc-PP5H326N, respectively, were lysed in EB supplemented with PhosSTOP phosphatase inhibitor cocktail. GFP-tagged proteins were captured from clarified supernatants using GFP-Trap magnetic agarose beads and used for an in vitro kinase assay to phosphorylate GST-Ana2281−420aa.
For testing the in vivo interaction between human Plk4 and PP5, HEK293 cells were transiently transfected with GFP or GFP-HsPP5 and Flag-HsPlk4-ND, respectively. Cells were harvested 2 days post-transfection and lysed in EB, as above. GFP or GFP-HsPP5 were captured using GFP-Trap magnetic agarose beads at 4 °C for 90 min and eluted by boiling in Laemmli sample buffer, followed by SDS-PAGE fractionation and immunoblotting with anti-FlagM2 and anti-GFP antibodies.
4.8. In Vitro Kinase and Phosphatase Assay
In vitro kinase assay was carried out according to [
32]. Briefly, 6 μg recombinant purified MBP-Plk4-TE (expressed either alone or in combination with His
6-PP5 or PP5
H326N in
E. coli) was used in 50 μL kinase reaction containing 250 μM ATP and 5 μg GST-Ana2
1−140 aa immobilized on glutathione sepharose 4B beads. The reaction was performed at 30 °C for 1 h, then the beads were washed once with λ-phosphatase buffer and divided into two equal parts: one part was untreated, while the other part was treated with 0.3 μL λ-phosphatase (cat # P0753S, New England Biolabs, Ipswich, MA, USA) for 30 min at 30 °C. The reaction was terminated by boiling in Laemmli sample buffer. Proteins were fractionated on SDS-PAGE and subjected to Coomassie brilliant blue-staining.
The STAN-motif-phosphorylating activity of purified MBP-Plk4-TE (expressed either alone or in combination with His6-PP5 or PP5H326N in E. coli) was tested in vitro in kinase reaction containing 250 μM ATP and 3 μg GST-Ana2281−420aa immobilized on glutathione sepharose 4B beads. The reaction was performed at 30 °C for 1 h, followed by washing the beads with 50 mM Na-HEPES pH 7.5, 150 mm NaCl, 2 mm MgCl2, 1 mm EGTA, 1 mm DTT, 0.1% Triton X-100 and PhosSTOP phosphatase inhibitor cocktail. Then, the beads were incubated with 35S-labeled Sas6 generated in IVTT reaction for 1 h at 4 °C, and finally boiled in Laemmli sample buffer. Protein samples were run on SDS-PAGE, Coomassie brilliant blue-stained gels were dried and subjected to autoradiography.
GFP-Plk4-ND or NDKD co-expressed with Myc-PP5E97Q or PP5H326N, respectively, in D.Mel-2 cells were captured on GFP-Trap magnetic agarose beads as above, and directly used in 50 μL kinase reaction containing 250 μM ATP and 3 μg purified GST-Ana2281−420aa for 1 h at 30 °C. Then, pre-phosphorylated GST-Ana2281-420aa was immobilized on glutathione sepharose 4B beads in the presence of PhosSTOP phosphatase inhibitor cocktail and incubated with 35S-Sas6 for 1 h at 4 °C. Protein samples were subjected to SDS-PAGE followed by Coomassie brilliant blue-staining. Dried gel was subjected to autoradiography.
In vitro phosphatase assay was carried out in 20 μL phosphatase buffer containing 2 μg purified MBP-Plk4-TE and 5 μg purified GST-PP5 or GST-PP5H326N at 30 °C for 1 h; 0.3 μL λ-phosphatase served as positive control and non-treated MBP-Plk4-TE served as negative control. Protein samples were boiled in Laemmli sample buffer, separated by SDS-PAGE and subjected to Coomassie brilliant blue-staining. To test the kinetics of in vitro dephosphorylation, we used 32 μg purified MBP-Plk4-TE in 160 μL phosphatase buffer, and removed 20 µL (for non-treated control). Then we added 5 μg GST-PP5 to the kinase and the reaction was performed at 30 °C. Then, 20 μL aliquots were taken at the indicated time points (5–60 min), supplemented with Laemmli sample buffer, and boiled for 5 min. Then, 4 μg purified kinase was treated with 0.3 μL λ-phosphatase (control treatment) for 60 min at 30 °C. Protein samples were separated on SDS-PAGE and subjected to Coomassie brilliant blue-staining.
For testing the activity of purified GST-PP5 and its derivatives, we used p-nitrophenyl phosphate (pNPP) as a general chromogenic phosphatase substrate. The reaction was performed in 1 mL phosphatase buffer containing 2 μg purified proteins and 2,5 μL pNPP (500 mM, cat # P0757S, New England Biolabs, Ipswich, MA, USA) at 30 °C for 10 min, then the absorbance of the liberated p-nitrophenol (its amount is proportional with the phosphatase activity) was measured at 405 nm using NanoDrop OneC spectrophotometer (cat # ND-ONE-W, Thermo Fisher Scientific, Waltham, MA, USA).
4.9. Centrosome Purification with Sucrose Gradient Ultracentrifugation
0.2 g of wild type (
w1118) 0–2 h-old syncytial
Drosophila embryos were collected, dechorionated and lysed in 1 mL BRB80 buffer (80 mM PIPES, 1 mM EGTA, 1 mM MgCl
2 pH 6.8) using Dounce homogenizer (cat # 357542, VWR, Radnor, PA, USA) according to [
53]. The lysate was clarified by mild centrifugation (1500×
g, 10 min at 4 °C) and filtered through Miracloth (cat # 475855, Merck Millipore, Burlington, MA, USA); 0.4 mL filtrate was loaded onto a 5 mL sucrose gradient (25–70
w/
v % linear sucrose gradient in BRB80 buffer supplemented with 100 mM KCl). Centrosomes were separated by ultracentrifugation (131,000×
g in Beckman-Coulter (Brea, CA, USA) MLS-50 rotor for 90 min at 4 °C), fractions (0.2 mL each) were collected from the bottom of the tube and subjected to SDS-PAGE analysis. Immunoblotting was performed by using anti-γTubulin and anti-Asl as centrosome fraction markers (centrosomes enriched around 55–60% of sucrose) and anti-αTubulin as cytoplasmic fractions (around 25–33% of sucrose) marker. The co-migration and centrosomal co-localization of Plk4 and PP5 were also tested.
4.10. Antibodies
The anti-PP5 polyclonal antibody was generated in-house by immunizing BALB/c mice with purified His
6-PP5 following standard procedures. The specificity of the antibody was confirmed by immunoblotting (
Figure S5). The following antibodies were used in immunoblot (IB) or immunofluorescence (IF) experiments: mouse anti-PP5 (IB: 1:10,000), rabbit anti-Asl (IB: 1:2000, IF: 1:300 [
36]), rat anti-Sas6 (IF: 1:500 [
32]), sheep anti-Plk4 (IB: 1: 2000 [
32], anti-γTubulin (IB: 1: 10,000, cat # T6557, Merck Millipore, Burlington, MA, USA), anti-αTubulin (IB: 1: 10,000, cat # T6199, Merck Millipore, Burlington, MA, USA), anti-GFP (IB: 1: 1000, cat # 11814460001, Roche, Basel, Switzerland), anti-Actin (IB: 1:2000, cat # A4700, Merck Millipore, Burlington, MA, USA), anti-FlagM
2 (IB: 1:10,000, cat # F1804, Merck Millipore, Burlington, MA, USA), goat anti-mouse IgG conjugated to horseradish peroxidase (1:10,000, cat # P044701-2 Dako, Glostrup, Denmark), goat anti-rabbit IgG conjugated to horseradish peroxidase (1:10,000, cat # P044801-2, Dako, Glostrup, Denmark), goat anti-sheep IgG conjugated to horseradish peroxidase (1: 10,000, cat # 31480, Thermo Fisher Scientific, Waltham, MA, USA), donkey anti-Rabbit IgG Alexa Fluor 647 (IF: 1:300, cat # A31573, Thermo Fisher Scientific, Waltham, MA, USA), and donkey anti-Rat IgG DyLight 650 (IF: 1:500, cat # SA5-10029, Thermo Fisher Scientific, Waltham, MA, USA).
4.11. Microscopy
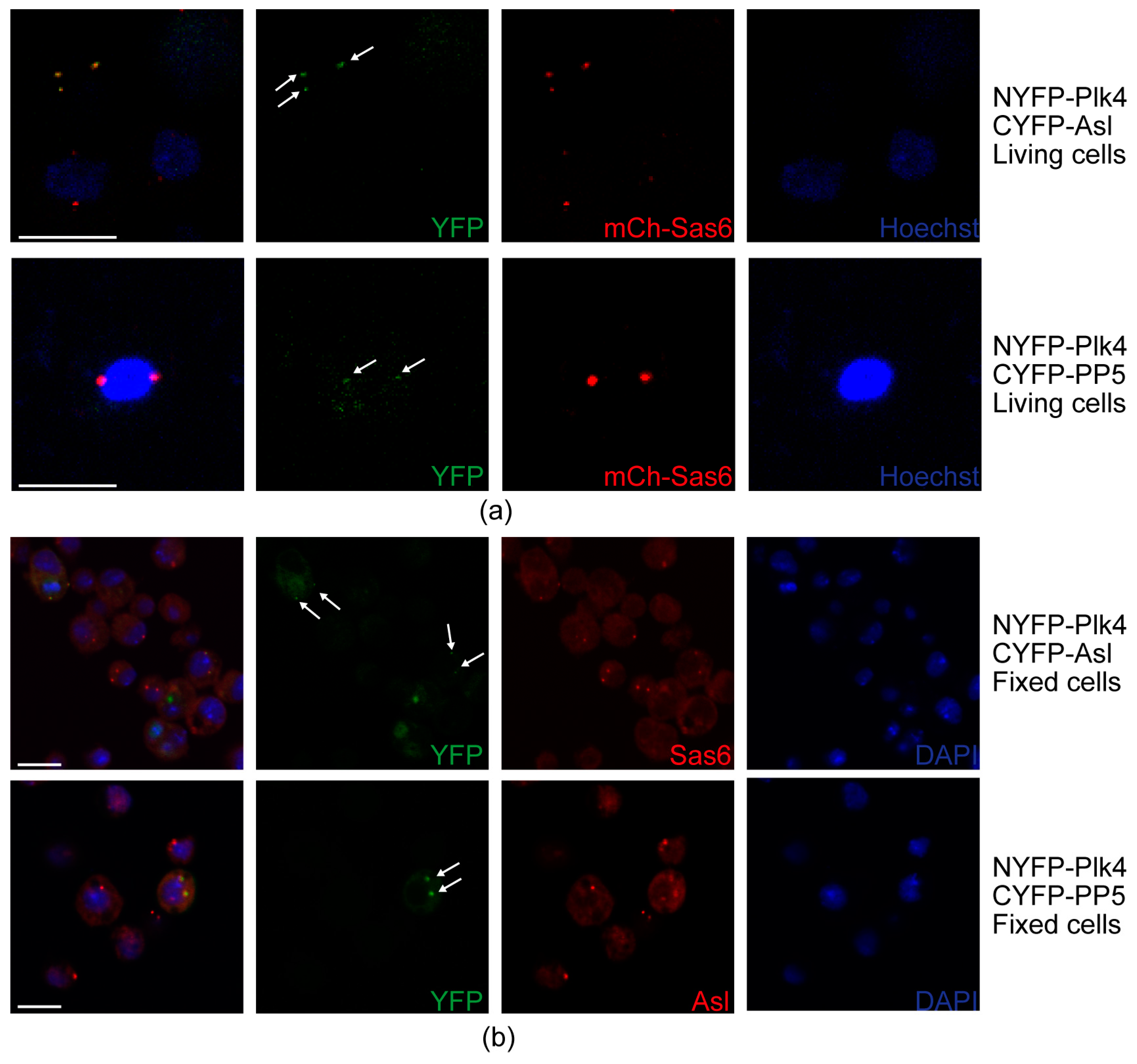
NYFP-Plk4 and CYFP (negative control), CYFP-PP5 and NYFP (negative control), NYFP-Plk4 and CYFP-Asl (positive control) or NYFP-Plk4 and CYFP-PP5, respectively, were transiently co-expressed in D.Mel-2 cells expressing the mCherry-Sas6 centrosomal marker protein under the control of MT promoter in glass bottom dishes (cat # P35G-0-14-C, Mattek Corporation, Ashland, MA, USA). Transgene expression was induced by adding 0.5 mM CuSO4 to the medium for 24 h. Then, 0.33 µg/mL Hoechst 33342 (cat # 62249, Thermo Fisher Scientific, Waltham, MA, USA) was added to the medium for 30 min to visualize DNA. Then the media was removed and cells were kept in mounting medium (cat # 50001, Ibidi, Gräfelfing, Germany) during microscopy. Images from live cells were taken with a Zeiss LSM 800 confocal microscope (Carl Zeiss, Jena, Germany) using an 63× OIL objective. YFP fluorescence signal of the samples was detected on the GFP channel with the same settings. mCherry signal was detected on the mCherry channel with the same settings.
D.Mel-2 cells co-expressing NYFP-Plk4 and CYFP (negative control), CYFP-PP5 and NYFP (negative control), NYFP-Plk4 and CYFP-Asl (positive control) or NYFP-Plk4 and CYFP-PP5, respectively, were collected, fixed and stained with DAPI, anti-Sas6 or anti-Asl antibodies according to [
32]. Experiments were repeated four times. Centrosomal localization of the YFP signal was counted in
n = 100 transfected cells. Images were taken using Olympus Fluoview Fv10i Confocal microscope (Olympus Corp., Tokyo, Japan).
Preparation of testis, fixation and staining with DAPI and Phalloidin were performed as described earlier [
54]. Samples were mounted using SlowFade Gold antifade reagent (cat # S36967, Thermo Fisher Scientific, Waltham, MA, USA) and images were taken using Olympus Fluoview Fv10i Confocal microscope.
4.12. Fly Stocks, Mutants and Fertility Test
Drosophila melanogaster flies were crossed and maintained on standard cornmeal agar medium at 25 °C. CRISPR/Cas9-mediated gene deletion was performed to generate the
pp5Δ7/1/2 null mutant (hereafter
pp5Δ) following standard procedures [
55]. Two guide RNAs (
Table S1) were designed to target the 5′-end of the second exon and 3′-UTR region of the PP5-encoding
PpD3 gene (FlyBase ID: CG8402). The deletion removed a 1936 bp-long sequence from the gene, which was validated by DNA sequencing (breakpoints: 3R:9748626 and 3R:9750562). The fertility of the animals was tested by crossing the appropriate genotype of individual females (
n = 8) with three wild type (wt) or
pp5/Df(3R)ED5454 males. Experiments were repeated three times and the significance was determined by Welch’s test. Testes were analyzed in flies expressing the basal body marker GFP-PACT in wild type or
pp5/Df(3R)ED5454 hemizygotic genetic background. Stock used in this study:
w1118 (BDSC ID: 5905)—isogenic wild type (wt) control stock.
Df(3R)ED5454 (BDSC ID: 86547)—isogenic deficiency stock with chromosomal deletion over PpD3.
Df(3R)ED5428 (BDSC ID: 9227)—isogenic deficiency stock with chromosomal deletion over pPD3.
w; +; pp5Δ7/1/2/TM6bTbHu—PP5 null mutant stock.
w; PlpPACT.Ubi-p63E.GFP—expressing GFP-PACT, a basal body marker in testis [
56].





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









