


Early Cell Cultures from Prostate Cancer Tissue Express Tissue Specific Epithelial and Cancer Markers

Abstract
:1. Introduction
2. Results
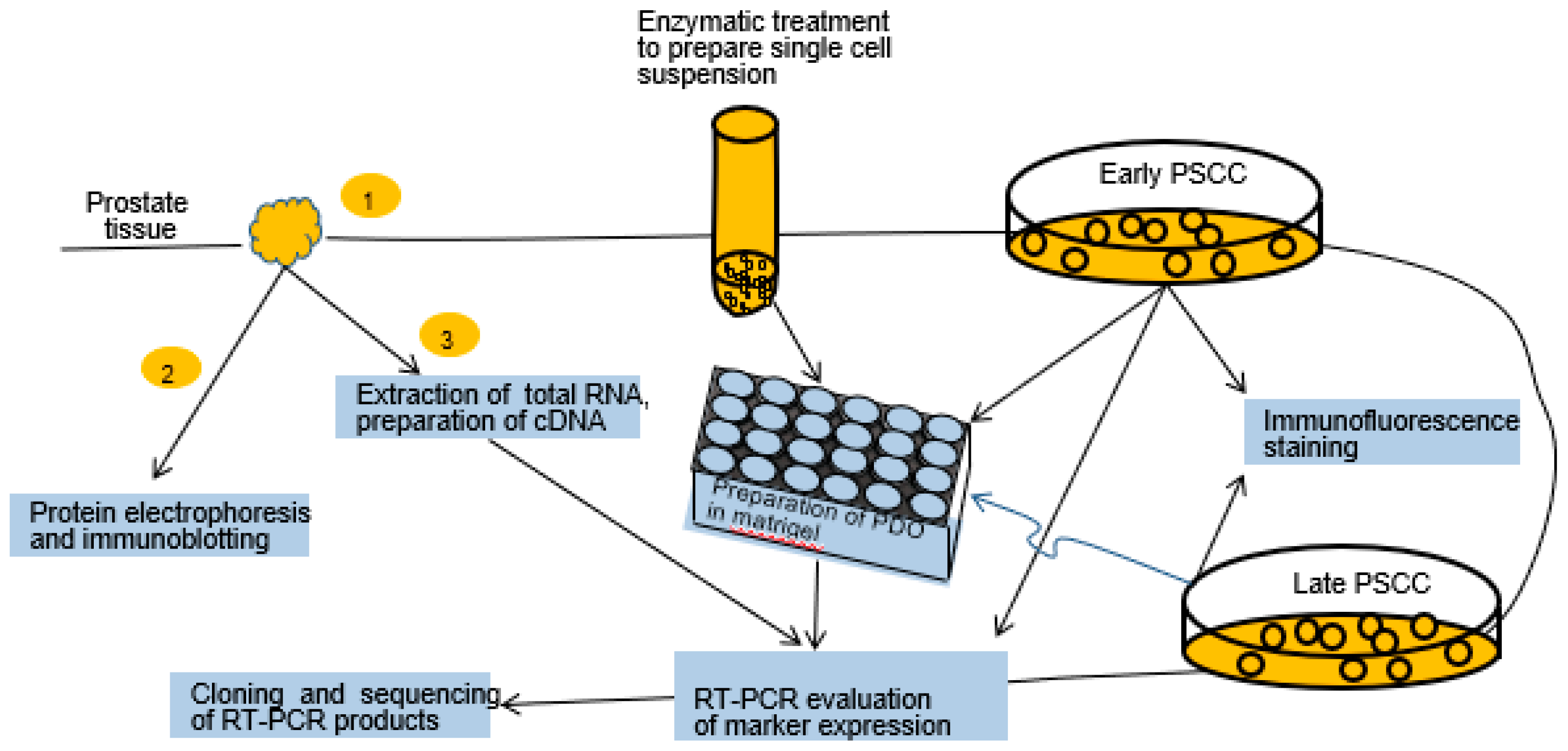
2.1. Experimental Design
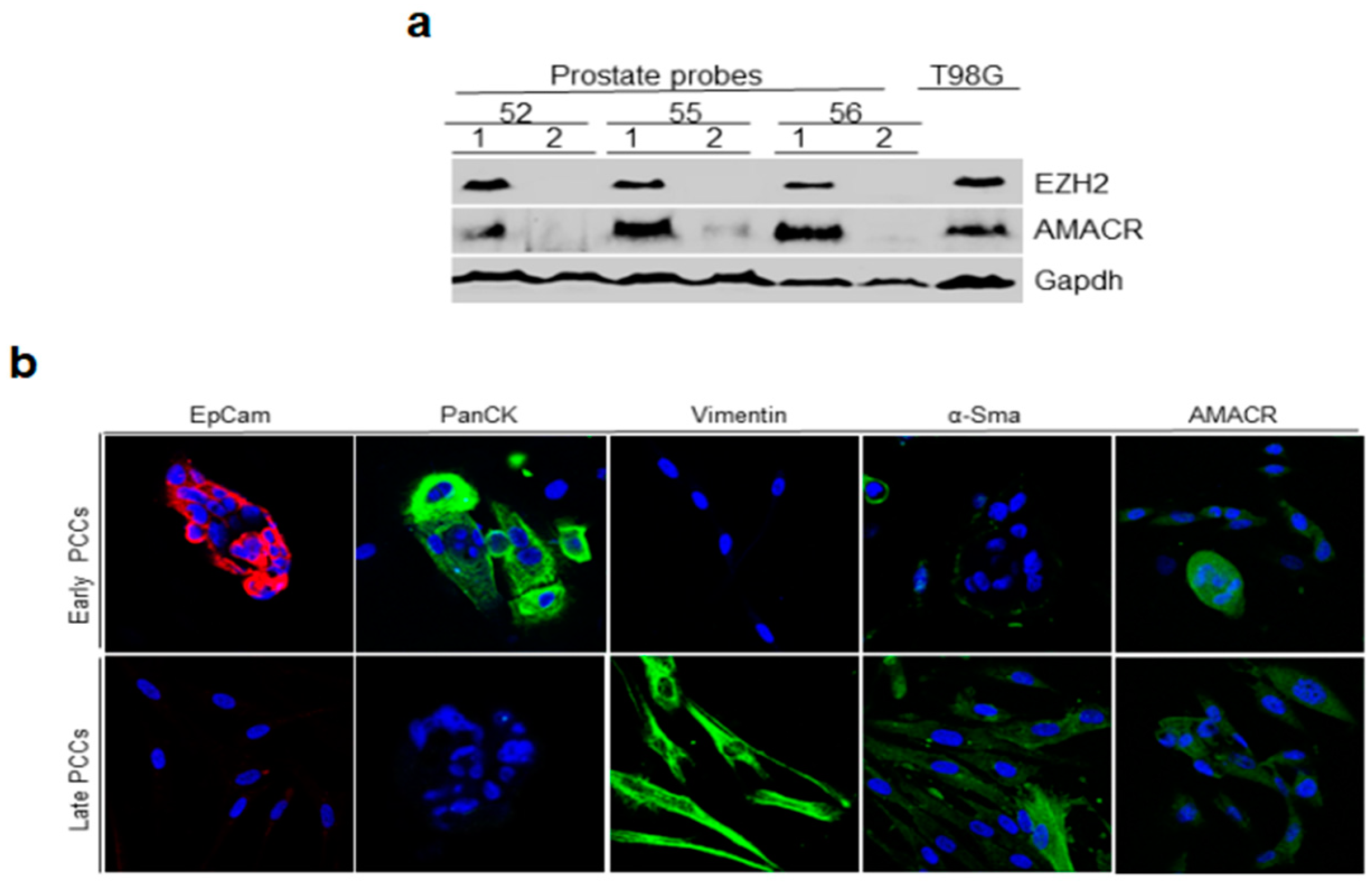
2.2. Immunoblotting Analysis of Prostate Tissue for Cancer Markers and Immunofluorescence Staining of Early and Late PCCs for Epithelial and Cancer Markers
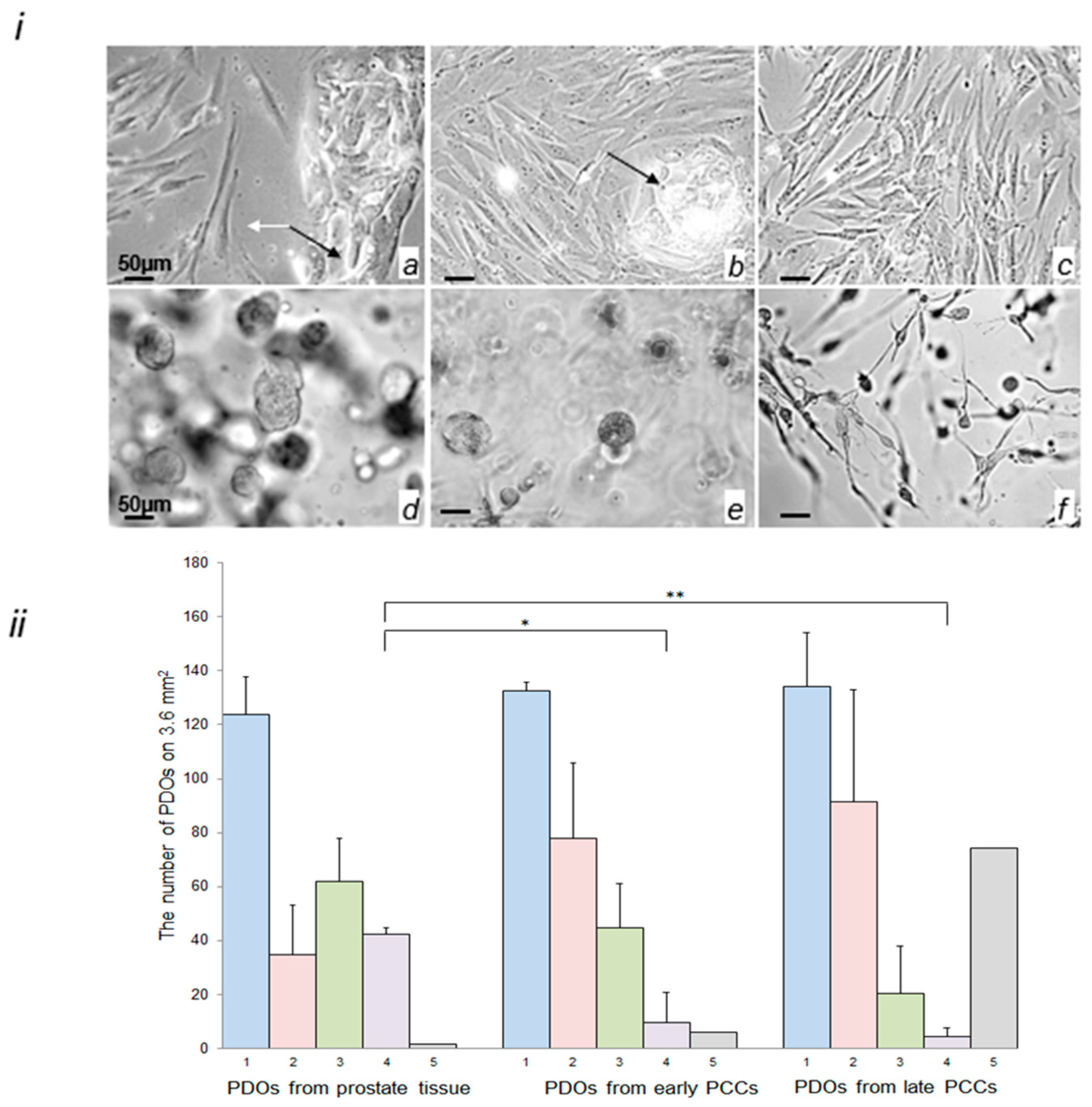
2.3. Morphological Characterization of Cells in the Early and Late PCCs and in PDOs Prepared from Prostate Tissue and PCCs
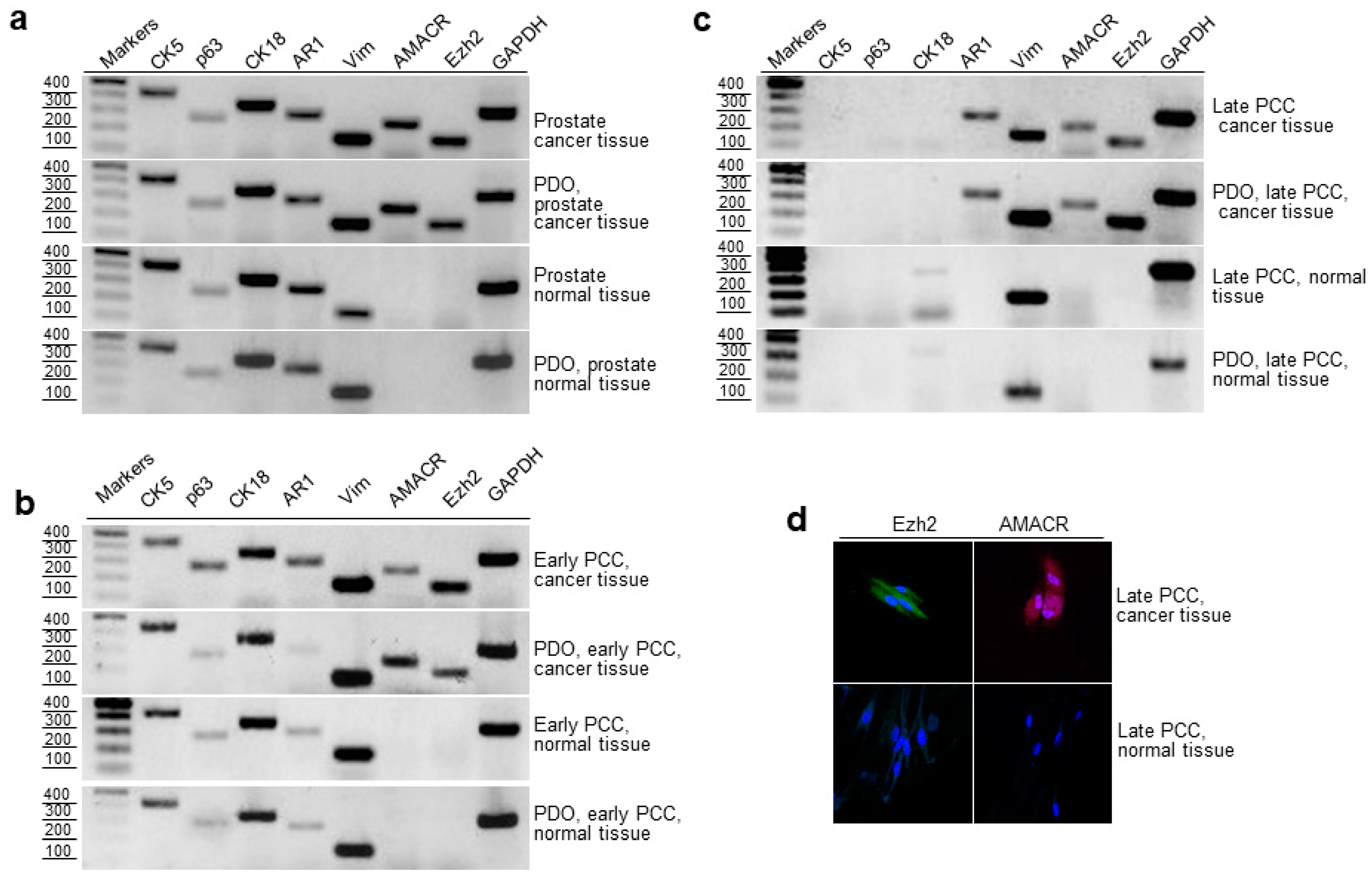
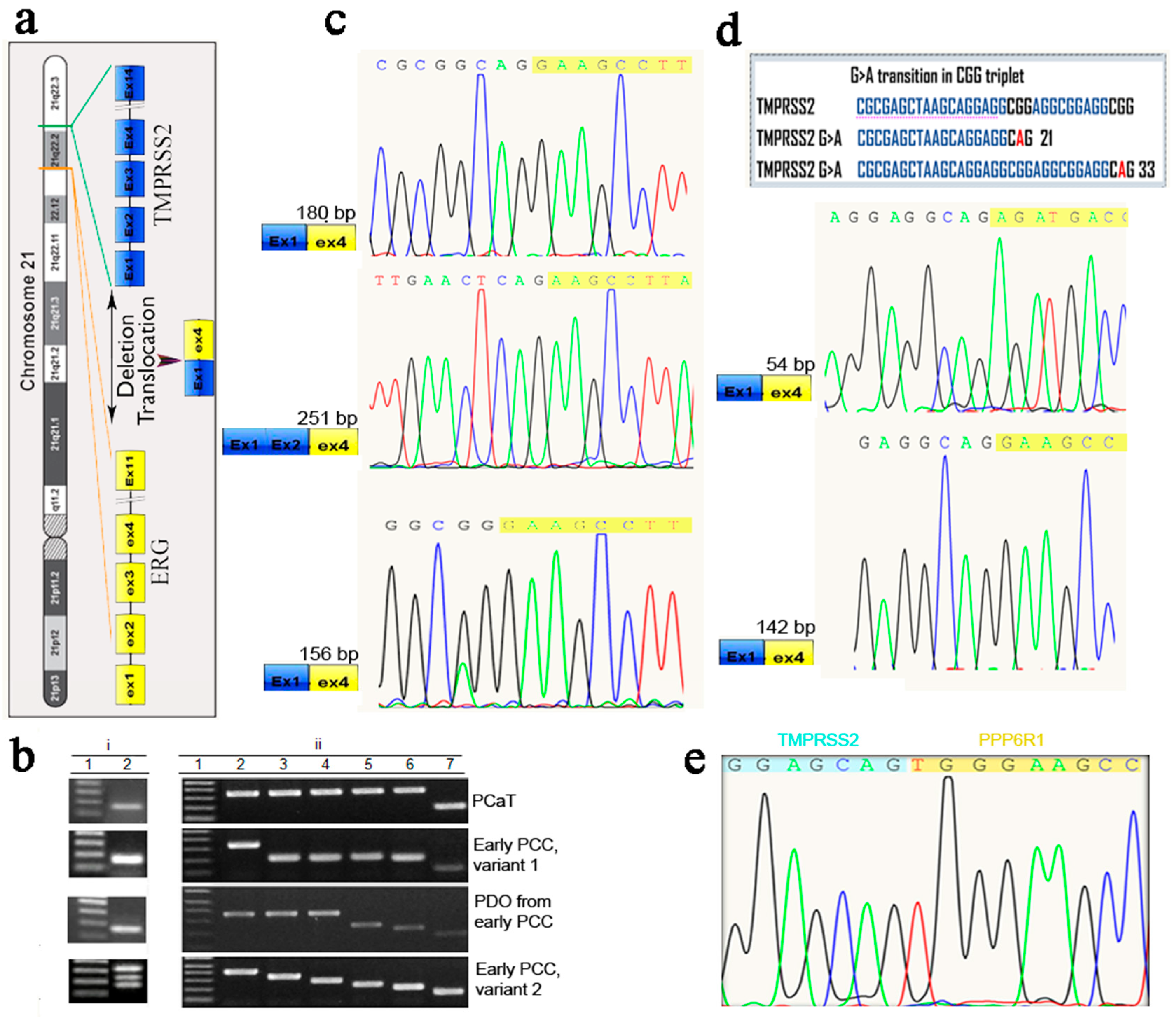
2.4. RT-PCR Evaluation of Epithelial and Cancer Markers in Prostate Tissue, Early and Late PCCs, and Derived PDOs
2.5. Cloning and Sequencing of RT-PCR Products of the Prostate Cancer Marker TMPRSS2-ERG
2.6. Expression of AMACR, EZH2 and TMPRSS2-ERG in Cancerous and Normal Prostate Tissues and Early PCCs
3. Discussion
4. Material and Methods
4.1. Prostate Tissue Samples
4.2. Establishment of Prostate Cell Cultures
4.3. Production, Culturing, and Subculturing of PDOs Derived from Cancerous and Normal Prostate Tissues
4.4. Establishment and Quantification of PDOs from Early and Late Prostate Stromal Cell Cultures
4.5. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
4.6. Sodium Dodecyl Sulfate (SDS)-Polyacrylamide Gel Electrophoresis (PAGE)
4.7. Immunoblotting
4.8. Immunofluorescent Staining
4.9. TA Cloning and DNA Sequencing
4.10. Antibodies
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Yegnasubramanian, S. Prostate cancer epigenetics and its clinical implications. Asian J. Androl. 2016, 18, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Xin, L. Cells of origin for cancer: An updated view from prostate cancer. Oncogene 2013, 32, 3655–3663. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Lee, J.K.; Phillips, J.W.; Huang, P.; Cheng, D.; Huang, J.; Witte, O.N. Prostate epithelial cell of origin determines cancer differentiation state in an organoid transformation assay. Proc. Natl. Acad. Sci. USA 2016, 113, 4482–4487. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.A.; Mitrofanova, A.; Bergren, S.K.; Abate-Shen, C.; Cardiff, R.D.; Califano, A.; Shen, M.M. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat. Cell Biol. 2013, 15, 274–283. [Google Scholar] [CrossRef]
- Karthaus, W.R.; Iaquinta, P.J.; Drost, J.; Gracanin, A.; van Boxtel, R.; Wongvipat, J.; Dowling, C.M.; Gao, D.; Begthel, H.; Sachs, N.; et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 2014, 159, 163–175. [Google Scholar] [CrossRef]
- Xin, L. Cells of Origin for Prostate Cancer. Adv. Exp. Med. Biol. 2019, 1210, 67–86. [Google Scholar]
- Wang, Y.; Revelo, M.P.; Sudilovsky, D.; Cao, M.; Chen, W.G.; Goetz, L.; Xue, H.; Sadar, M.; Shappell, S.B.; Cunha, G.R.; et al. Development and characterization of efficient xenograft models for benign and malignant human prostate tissue. Prostate 2005, 64, 149–159. [Google Scholar] [CrossRef]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef]
- Byrne, A.T.; Alférez, D.G.; Amant, F.; Annibali, D.; Arribas, J.; Biankin, A.V.; Bruna, A.; Budinská, E.; Caldas, C.; Chang, D.K.; et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat. Rev. Cancer 2017, 17, 254–268. [Google Scholar] [CrossRef]
- Kemper, K.; Krijgsman, O.; Cornelissen-Steijger, P.; Shahrabi, A.; Weeber, F.; Song, J.Y.; Kuilman, T.; Vis, D.J.; Wessels, L.F.; Voest, E.E.; et al. Intra- and inter-tumor heterogeneity in a vemurafenib-resistant melanoma patient and derived xenografts. EMBO Mol. Med. 2015, 7, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Pearson, A.; Finkel, K.A.; Warner, K.A.; Nör, F.; Tice, D.; Martins, M.D.; Jackson, T.L.; Nör, J.E. Patient-derived xenograft (PDX) tumors increase growth rate with time. Oncotarget 2016, 7, 7993–8005. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Chua, C.W.; Shibata, M.; Lei, M.; Toivanen, R.; Barlow, L.; Bergren, S.K.; Badani, K.K.; McKiernan, J.M.; Benson, M.C.; Hibshoosh, H.; et al. Single luminal epithelial progenitors can generate prostate organoids in culture. Nat. Cell Biol. 2014, 16, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.H.N.; Siu, H.C.; Law, S.; Ho, S.L.; Yue, S.S.K.; Tsui, W.Y.; Chan, D.; Chan, A.S.; Ma, S.; Lam, K.O.; et al. A Comprehensive Human Gastric Cancer Organoid Biobank Captures Tumor Subtype Heterogeneity and Enables Therapeutic Screening. Cell Stem. Cell 2018, 23, 882–897. [Google Scholar] [CrossRef] [PubMed]
- Bleijs, M.; van de Wetering, M.; Clevers, H.; Drost, J. Xenograft and organoid model systems in cancer research. EMBO J. 2019, 38, e101654. [Google Scholar] [CrossRef]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschênes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef] [PubMed]
- Van Hemelryk, A.; Mout, L.; Erkens-Schulze, S.; French, P.J.; van Weerden, W.M.; van Royen, M.E. Modeling Prostate Cancer Treatment Responses in the Organoid Era: 3D Environment Impacts Drug Testing. Biomolecules 2021, 11, 572. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, C.; Zhang, Y.; Shi, C. Application of Organoid Models in Prostate Cancer Research. Front. Oncol. 2021, 11, 736431. [Google Scholar] [CrossRef]
- Popov, B.V.; Serikov, V.B.; Petrov, N.S.; Izusova, T.V.; Gupta, N.; Matthay, M.A. Lung epithelial cells induce endodermal differentiation in mouse mesenchymal bone marrow stem cells by paracrine mechanism. Tissue Eng. 2007, 13, 2441–2450. [Google Scholar] [CrossRef] [PubMed]
- Musina, R.; Belyavski, A.V.; Tarusova, O.; Solovyova, E.; Sukhikh, G. Endometrial mesenchymal stem cells isolated from the menstrual blood. Bull. Exp. Biol. Med. 2008, 145, 539–543. [Google Scholar] [CrossRef]
- Da Meirelles, L.S.; Nardi, N.B. Methodology, biology and clinical applications of mesenchymal stem cells. Front. Biosci.-Landmrk. 2009, 14, 4281–4298. [Google Scholar] [CrossRef] [PubMed]
- Zhidkova, O.V.; Petrov, N.S.; Popov, B.V. Production and characteristics of the growth and marker properties of mesenchymal stem cells of urinary bladder. Zhurnal Evoliutsionnoi Biokhimii I Fiziol. 2013, 49, 67–77. [Google Scholar]
- Janssen, M.; Albrecht, M.; Möschler, O.; Renneberg, H.; Fritz, B.; Aumüller, G.; Konrad, L. Cell lineage characteristics of human prostatic stromal cells cultured in vitro. Prostate 2000, 43, 20–30. [Google Scholar] [CrossRef]
- Gravina, G.L.; Mancini, A.; Ranieri, G.; Di Pasquale, B.; Marampon, F.; Di Clemente, L.; Ricevuto, E.; Festuccia, C. Phenotypic characterization of human prostatic stromal cells in primary cultures derived from human tissue samples. Int. J. Oncol. 2013, 42, 2116–2122. [Google Scholar] [CrossRef]
- Brennen, W.N.; Chen, S.; Denmeade, S.R.; Isaacs, J.T. Quantification of Mesenchymal Stem Cells (MSCs) at sites of human prostate cancer. Oncotarget 2013, 4, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Brennen, W.N.; Kisteman, L.N.; Isaacs, J.T. Rapid selection of mesenchymal stem and progenitor cells in primary prostate stromal cultures. Prostate 2016, 76, 552–564. [Google Scholar] [CrossRef]
- Bartsch, G.; Rohr, H.P. Comparative light and electron microscopic study of the human, dog and rat prostate. An approach to an experimental model for human benign prostatic hyperplasia (light and electron microscopic analysis)—A review. Urol. Int. 1980, 35, 91–104. [Google Scholar] [CrossRef]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Popov, B.V.; Sutula, G.I.; Petrov, N.S.; Yang, X.J. Preparation and characterization of the antibody recognizing AMACR inside its catalytic center. Int. J. Oncol. 2018, 52, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Laxman, B.; Varambally, S.; Cao, X.; Yu, J.; Helgeson, B.E.; Cao, Q.; Prensner, J.R.; Rubin, M.A.; Shah, R.B.; et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia 2008, 10, 177–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Niu, L.; Xiao, Z.; Zheng, C.; Shen, Y.; Shi, Y.; Han, X. Whole-genome sequencing of prostate cancer reveals novel mutation-driven processes and molecular subgroups. Life Sci. 2020, 254, 117218. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Helin, K. Polycomb group proteins: Navigators of lineage pathways led astray in cancer. Nat. Rev. Cancer 2009, 9, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate cancer. Nat. Rev. Dis. Primers 2021, 7, 9. [Google Scholar] [CrossRef]
- Ryabov, V.M.; Voskresenskiy, M.A.; Popov, B.V. Role of the Tumor Suppressor RB in the Development of Localized and Castration-Resistant Prostate Cancer. Cell Tissue Biol. 2022, 16, 434–441. [Google Scholar] [CrossRef]
- Ezhkova, E.; Pasolli, H.A.; Parker, J.S.; Stokes, N.; Su, I.H.; Hannon, G.; Tarakhovsky, A.; Fuchs, E. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell 2009, 136, 1122–1135. [Google Scholar] [CrossRef]
- Chang, C.J.; Hung, M.C. The role of EZH2 in tumour progression. Br. J. Cancer 2012, 106, 243–247. [Google Scholar] [CrossRef]
- Liu, X.; Wu, Q.; Li, L. Functional and therapeutic significance of EZH2 in urological cancers. Oncotarget 2017, 8, 38044–38055. [Google Scholar] [CrossRef]
- Ryabov, V.M.; Georgieva, A.O.; Voskresensky, M.A.; Komyakov, B.K.; Rogoza, O.V.; Grozov, R.V.; Murazov, Y.G.; Prokhorov, D.G.; Popov, B.V. Organoid and Primary Epithelial Cultures of Human Prostate Show the Key Role of the Epithelial-to-Mesenchymal Transition in Generation of Tissue-Specific Stromal Cells. Cell Tissue Biol. 2021, 15, 554–562. [Google Scholar] [CrossRef]
- Pamarthy, S.; Sabaawy, H.E. Patient derived organoids in prostate cancer: Improving therapeutic efficacy in precision medicine. Mol. Cancer 2021, 20, 125. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cai, Y.; Ren, C.; Ittmann, M. Expression of variant TMPRSS2/ERG fusion messenger RNAs is associated with aggressive prostate cancer. Cancer Res. 2006, 66, 8347–8351. [Google Scholar] [CrossRef]
- Zhou, F.; Gao, S.; Han, D.; Han, W.; Chen, S.; Patalano, S.; Macoska, J.A.; He, H.H.; Cai, C. TMPRSS2-ERG activates NO-cGMP signaling in prostate cancer cells. Oncogene 2019, 38, 4397–4411. [Google Scholar] [CrossRef]
- Linxweiler, J.; Hajili, T.; Körbel, C.; Berchem, C.; Zeuschner, P.; Müller, A.; Stöckle, M.; Menger, M.D.; Junker, K.; Saar, M. Cancer-associated fibroblasts stimulate primary tumor growth and metastatic spread in an orthotopic prostate cancer xenograft model. Sci. Rep. 2020, 10, 12575. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.A.; Need, E.F.; Toivanen, R.; Trotta, A.P.; Palethorpe, H.M.; Tamblyn, D.J.; Kopsaftis, T.; England, G.M.; Smith, E.; Drew, P.A.; et al. Stromal androgen receptor regulates the composition of the microenvironment to influence prostate cancer outcome. Oncotarget 2015, 30, 16135–16150. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J.; Horton, C.; Yu, C.; Knudsen, B.; Stefanson, J.; Hu, K.; Stefanson, O.; Green, J.; Guo, C.; et al. Stromal AR inhibits prostate tumor progression by restraining secretory luminal epithelial cells. Cell Rep. 2022, 39, 10848. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Memarzadeh, S.; Stoyanova, T.; Beharry, Z.; Kraft, A.S.; Witte, O.N. Collaboration of Kras and androgen receptor signaling stimulates EZH2 expression and tumor-propagating cells in prostate cancer. Cancer Res. 2012, 72, 4672–4681. [Google Scholar] [CrossRef]
- Drost, J.; Karthaus, W.R.; Gao, D.; Driehuis, E.; Sawyers, C.L.; Chen, Y.; Clevers, H. Organoid culture systems for prostate epithelial and cancer tissue. Nat. Protoc. 2016, 11, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Baryshev, M.; Merculov, D.; Mironov, I. A new device-mediated miniprep method. AMB Express 2022, 12, 21. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Summary of the AMACR, EZH2, TMPRSS2 Production in Cancer and Control Tissue Samples | ||||||
|---|---|---|---|---|---|---|
| Cancer Tissue | Normal Tissue | |||||
| Name of the marker | AMACR | EZH2 | TMPRSS2-ERG | AMACR | EZH2 | TMPRSS2-ERG |
| Number of the tested samples | 18 | 18 | 18 | 15 | 15 | 15 |
| Number of the positive tests | 18 (100%) | 18 (100%) | 4 (22%) | 1 (6.6%) | 1 (6.6%) | 0 (0%) |
| Frequency of the AMACR, EZH2, TMPRSS2 production in cancer tissue and corresponding early PCCs | ||||||
| Number of the tested tissue samples | 8 | 8 | 4 | 8 | 8 | 4 |
| Number of the positive tissue tests | 8 (100%) | 8 (100%) | 4 (100%) | 1 (12.5%) | 1 (12.5%) | 0 (0%) |
| Number of the tested PCCs samples | 8 | 8 | 4 | 8 | 8 | ND |
| Number of the positive PCCs tests | 8 (100%) | 8 (100%) | 4 (100 %) | 0 (0%) | 0 (0%) | |
| Gene | Forward Primer, 5′-3′ | Reverse Primer, 5′-3′ | Amplicon Size, bp |
|---|---|---|---|
| CK5 | TTCATCGACAAGGTGCGGT | TGAGGTGTCAGAGACATGCG | 423 |
| TP63 | GTCCCAGAGCACACAGACAA | GAGGAGCCGTTCTGAATCTG | 267 |
| CK18 | TGGTCACCACACAGTCTGCT | CCAAGGCATCACCAAGATTA | 348 |
| AR1 | GACATGCGTTTGGAGACTGC | TTTCTTCAGCTTCCGGGCTC | 294 |
| AMACR | TGGCCACGATATCAACTATTTGG | ACTCAATTTCTGAGTTTTCCACAGAA | 247 |
| EZH2 | TGCGACTGAGACAGCTCAAG | GCGCAATGAGCTCACAGAAG | 164 |
| VIMENTIN | GAGAACTTTGCCGTTGAAGC | GCTTCCTGTAGGTGGCAATC | 163 |
| TMPRSS2-ERG | CGCGAGCTAAGCAGGAG | GTCCATAGTCGCTGGAGGAG | 180 |
| GAPDH | CCATCTTCCAGGAGCGAGA | GGCAGTGATGGCATGGACTGT | 326 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryabov, V.M.; Baryshev, M.M.; Voskresenskiy, M.A.; Popov, B.V. Early Cell Cultures from Prostate Cancer Tissue Express Tissue Specific Epithelial and Cancer Markers. Int. J. Mol. Sci. 2023, 24, 2830. https://doi.org/10.3390/ijms24032830
Ryabov VM, Baryshev MM, Voskresenskiy MA, Popov BV. Early Cell Cultures from Prostate Cancer Tissue Express Tissue Specific Epithelial and Cancer Markers. International Journal of Molecular Sciences. 2023; 24(3):2830. https://doi.org/10.3390/ijms24032830
Chicago/Turabian StyleRyabov, Vladimir M., Mikhail M. Baryshev, Mikhail A. Voskresenskiy, and Boris V. Popov. 2023. "Early Cell Cultures from Prostate Cancer Tissue Express Tissue Specific Epithelial and Cancer Markers" International Journal of Molecular Sciences 24, no. 3: 2830. https://doi.org/10.3390/ijms24032830
APA StyleRyabov, V. M., Baryshev, M. M., Voskresenskiy, M. A., & Popov, B. V. (2023). Early Cell Cultures from Prostate Cancer Tissue Express Tissue Specific Epithelial and Cancer Markers. International Journal of Molecular Sciences, 24(3), 2830. https://doi.org/10.3390/ijms24032830